Familial Infertility (Azoospermia and Cryptozoospermia) in Two Brothers—Carriers of t(1;7) Complex Chromosomal Rearrangement (CCR): Molecular Cytogenetic Analysis

 ,
,  , , , ,
, , , , 
Abstract
:1. Introduction
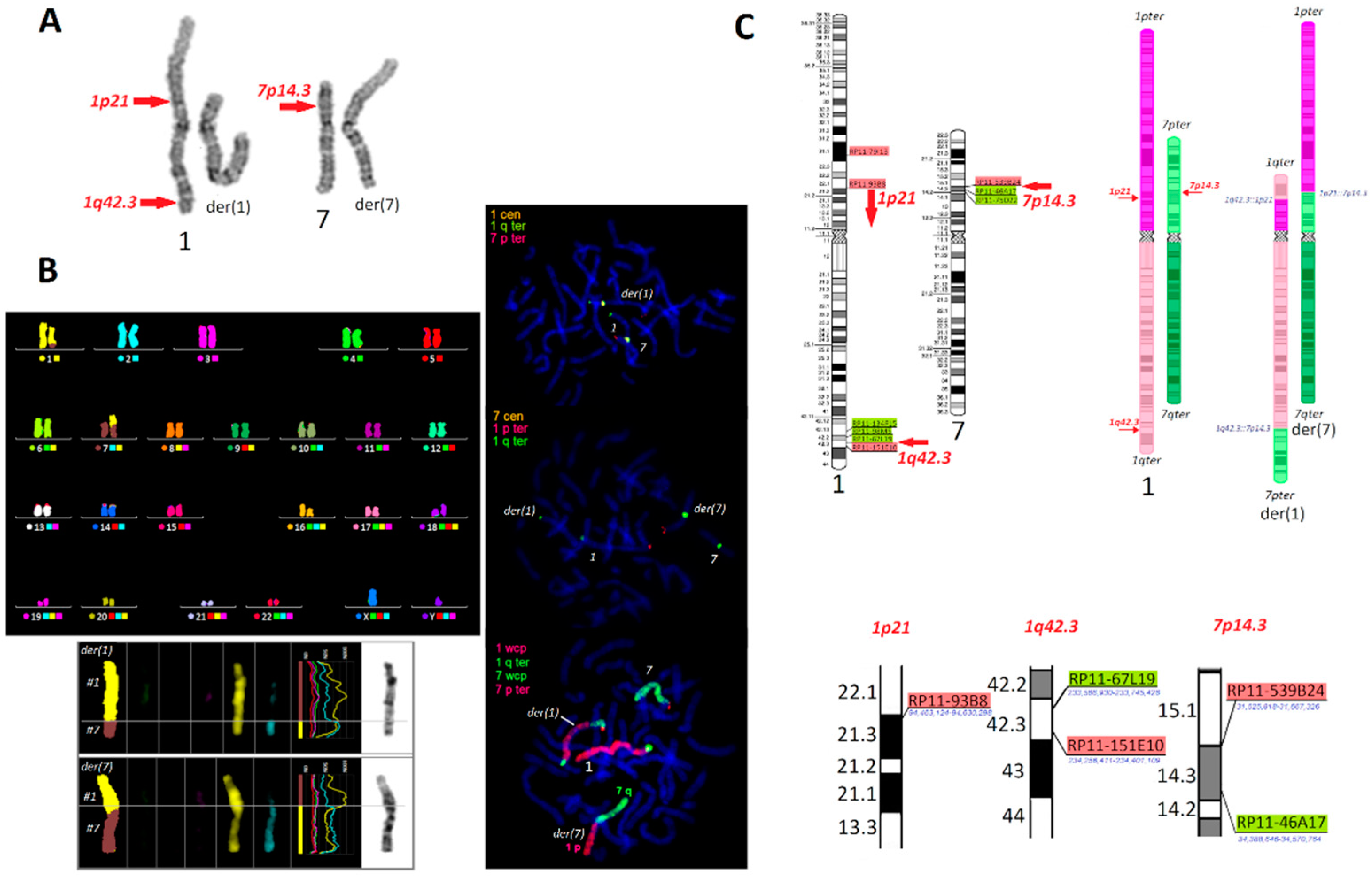
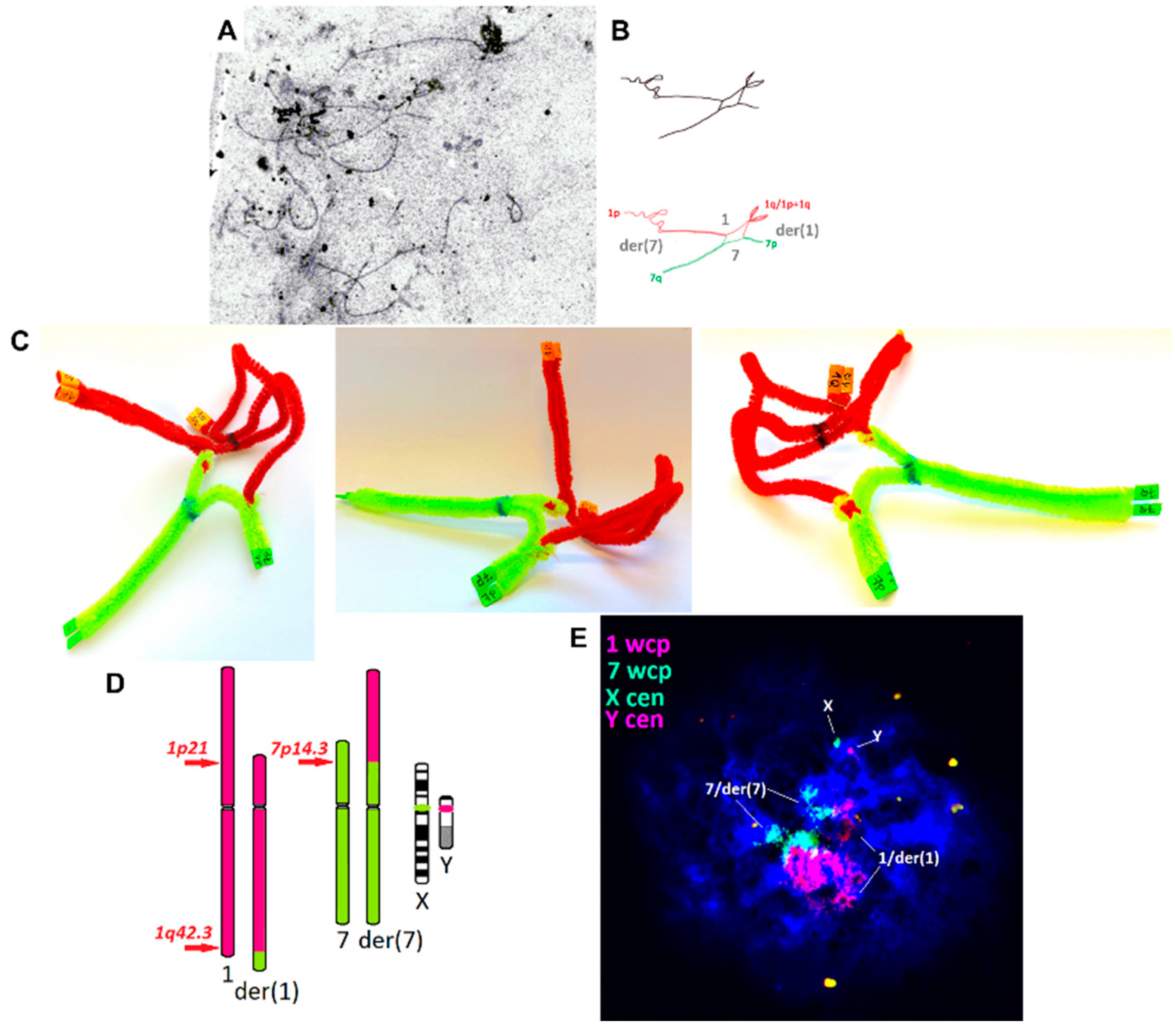
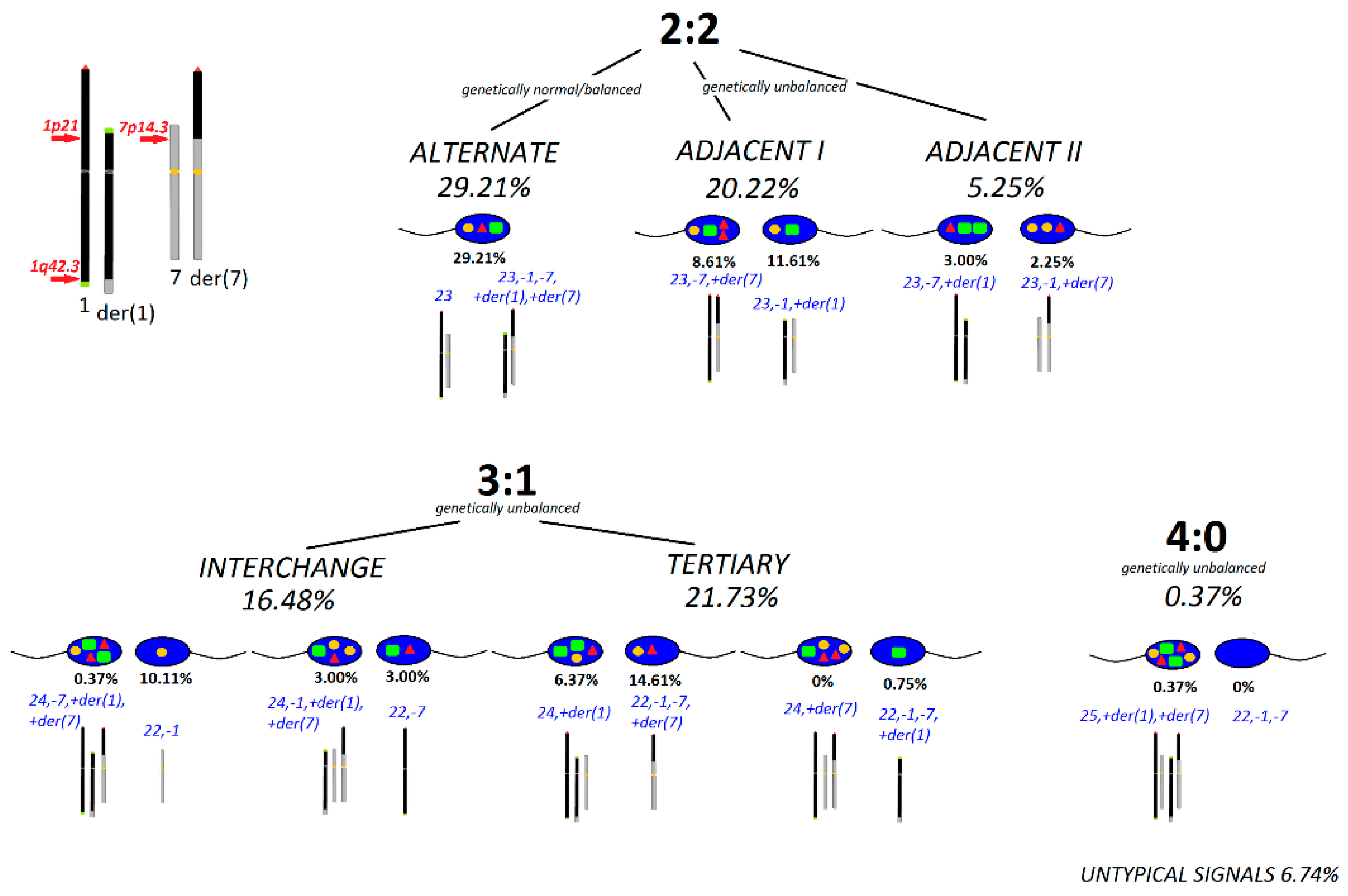
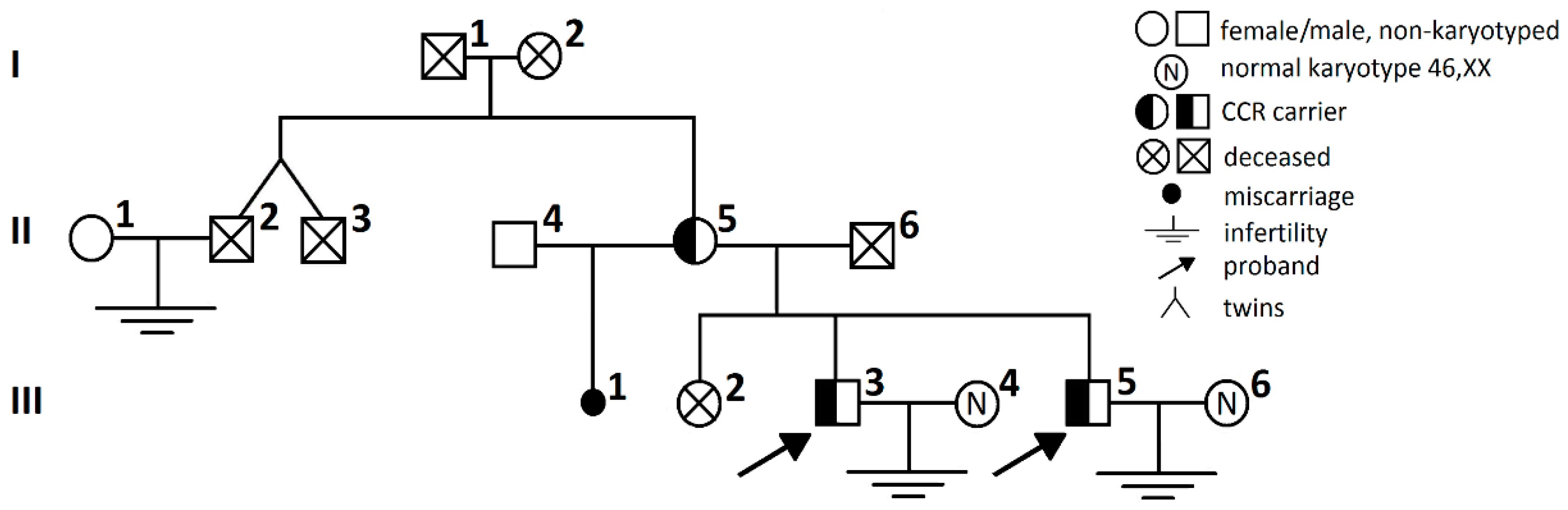
2. Results
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Fluorescence In Situ Hybridization (FISH)
- (i)
- The characterization of chromosomes involved in CCR. Centromere-specific: 1cen (locus D1Z1, catalogue number LPE01R/G) and 7 cen (D7Z1, LPE07R/G). Subtelomere: 1pter (clone CEB108, LPT01pR), 1qter (clone 160H23, LPT01qG), and 7pter (clone 109a6, LPT07pR, Cytocell, UK). Whole chromosome painting: 1wcp (catalogue number XCP1R), 7wcp (XCP7G), and mFISH (multicolour FISH) (MetaSystems, Altlussheim, Germany). For each combination, at least 50 metaphase plates were analyzed, and 1000 interphase cells (FISH with cen/subtel probes) were counted to exclude the mosaicism.
- (ii)
- The translocation breakpoint analysis—BAC probes for the proper chromosomal regions are listed in Supplementary Table S6 and in Figure 1. BAC clones were chosen from the RPCI-11 library collection (BACPAC Resource Center, the Children’s Hospital Oakland Research Institute) based on hg19 (human genome reference GRCh37 h37). At least 10 metaphase plates were analyzed for each round of FISH.
- (iii)
- The analysis of the association between the chromosomes involved in CCR and sex chromosome bivalent (spermatocytes II from the testicular oligobiopsy of brother-1). wcp and cen-specific probes: 1wcp (XCP1R), 7wcp (XCP7G), Xcen (DXZ1, LPE0XG), and Ycen (DYZ3, LPE0YcR) (MetaSystems, Germany; Cytocell, UK); n = 24.
- (iv)
- The meiotic segregation pattern in the sperm cells from the ejaculate of brother-2. A 3-colour combination of cen-specific and subtel-specific probes: 1pter (LPT01pR), 1qter (LPT01qG), and 7cen (D7Z1, LPE07R/G) (Cytocell, UK); n = 267.
4.3. BAC Preparation
4.4. Hybridization
4.5. Genomic Microarray CGH
4.6. Synaptonemal Complex Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sharma, A. Male Infertility; Evidences, Risk Factors, Causes, Diagnosis and Management in Human. Ann. Clin. Lab. Res. 2017, 5, 188. [Google Scholar] [CrossRef]
- Matzuk, M.M.; Lamb, D.J. The biology of infertility: Research advances and clinical challenges. Nat. Med. 2008, 14, 1197–1213. [Google Scholar] [CrossRef] [PubMed]
- Mau-Holzmann, U.A. Somatic chromosomal abnormalities in infertile men and women. Cytogenet. Genome Res. 2005, 111, 317–336. [Google Scholar] [CrossRef] [PubMed]
- Tüttelmann, F.; Ruckert, C.; Röpke, A. Disorders of spermatogenesis: Perspectives for novel genetic diagnostics after 20 years of unchanged routine. Med. Genet. 2018, 30, 12–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieschlag, E.; Behre, H.M.; Nieschlag, S. Andrology. Male Reproductive Health and Dysfunction, 3rd ed.; Nieschlag, E., Behre, H.M., Nieschlag, S., Eds.; Springer-Verlag: Berlin/Heidelberg, Germany, 2010. [Google Scholar]
- Krausz, C.; Riera-Escamilla, A. Monogenic Forms of Male Infertility. In Genetics of Endocrine Diseases and Syndromes. Experientia Supplementum; Igaz, P., Patócs, A., Eds.; Springer Nature: Cham, Switzerland, 2020; Volume 111, pp. 341–366. [Google Scholar]
- Cannarella, R.; Condorelli, R.A.; Mongioì, L.M.; La Vignera, S.; Calogero, A.E. Molecular Biology of Spermatogenesis: Novel Targets of Apparently Idiopathic Male Infertility. Int. J. Mol. Sci. 2020, 21, 1728. [Google Scholar] [CrossRef] [Green Version]
- Hwang, K.; Yatsenko, A.N.; Jorgez, C.J.; Mukherjee, S.; Nalam, R.L.; Matzuk, M.M.; Lamb, D.J. Mendelian genetics of male infertility. Ann. N. Y. Acad. Sci. 2010, 1214, E1–E17. [Google Scholar] [CrossRef] [Green Version]
- Templado, C.; Bosch, M.; Benet, J. Frequency and distribution of chromosome abnormalities in human spermatozoa. Cytogenet. Genome Res. 2005, 111, 199–205. [Google Scholar] [CrossRef]
- Harton, G.L.; Tempest, H.G. Chromosomal disorders and male infertility. Asian J. Androl. 2012, 14, 32–39. [Google Scholar] [CrossRef] [Green Version]
- Kefer, J.C.; French, D.B. Azoospermia: Diagnosis and Management. In Male Infertility. Current Clinical Urology; Sabanegh, E.S., Ed.; Humana Press: Totowa, NJ, USA, 2011; pp. 23–30. [Google Scholar]
- Lee, J.Y.; Dada, R.; Sabanegh, E.; Carpi, A.; Agarwal, A. Role of genetics in azoospermia. Urology 2011, 77, 598–601. [Google Scholar] [CrossRef]
- Ergul, E.; Liehr, T.; Mrasek, K.; Sazci, A. A de novo complex chromosome rearrangement involving three chromosomes (2, 13, and 18) in an oligozoospermic male. Fertil. Steril. 2009, 92, 391.e9–391.e12. [Google Scholar] [CrossRef]
- Madan, K. Balanced complex chromosome rearrangements: Reproductive aspects. A review. Am. J. Med. Genetics 2012, 158A, 947–963. [Google Scholar] [CrossRef] [PubMed]
- Trpchevska, N.; Dimova, I.; Arabadji, T.; Milachich, T.; Angelova, S.; Dimitrova, M.; Hristova-Savova, M.; Andreeva, P.; Timeva, T.; Shterev, A. A family study of complex chromosome rearrangement involving chromosomes 1, 8, and 11 and its reproductive consequences. J. Assist. Reprod. Genet. 2017, 34, 659–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruchy, N.; Barreau, M.; Kessler, K.; Gourdier, D.; Leporrier, N. A paternally transmitted complex chromosomal rearrangement (CCR) involving chromosomes 2, 6, and 18 includes eight breakpoints and five insertional translocations (ITs) through three generations. Am. J. Med. Genet. 2010, 152A, 185–190. [Google Scholar] [CrossRef]
- Pellestor, F.; Anahory, T.; Lefort, G.; Puechberty, J.; Liehr, T.; Hedon, B.; Sarda, P. Complex chromosomal rearrangements: Origin and meiotic behavior. Hum. Reprod. Update 2011, 17, 479–494. [Google Scholar] [CrossRef] [PubMed]
- Pellestor, F.; Puechberty, J.; Weise, A.; Lefort, G.; Anahory, T.; Liehr, T.; Sarda, P. Meiotic segregation of complex reciprocal translocations: Direct analysis of the spermatozoa of a t(5;13;14) carrier. Fertil. Steril. 2011, 95, e17–e22. [Google Scholar] [CrossRef]
- Kang, S.-H.L.; Shaw, C.; Ou, Z.; Eng, P.A.; Cooper, M.L.; Pursley, A.N.; Sahoo, T.; Bacino, C.A.; Chinaut, C.; Stankiewicz, P.; et al. Insertional translocation detected using FISH confirmation of array-comparative genomic hybridization (aCGH) results. Am. J. Med. Genet. 2010, 152A, 1111–1126. [Google Scholar] [CrossRef] [Green Version]
- Poot, M.; Haaf, T. Mechanisms of origin, phenotypic effects and diagnostic implications of complex chromosome rearrangements. Mol. Syndromol. 2015, 6, 110–134. [Google Scholar] [CrossRef] [Green Version]
- Houge, G.; Liehr, T.; Schoumans, J.; Ness, G.O.; Solland, K.; Starke, H.; Claussen, U.; Strømme, P.; Akre, B.; Vermeulen, S. Ten years follow up of a boy with a complex chromosomal rearrangement: Going from a > 5 to 15-breakpoint CCR. Am. J. Med. Genet. A 2003, 118A, 235–240. [Google Scholar] [CrossRef]
- Gorski, J.L.; Kistenmacher, M.N.; Punnett, H.H.; Zackai, E.H.; Emanuel, B.S. Reproductive risks for carriers of complex chromosome rearrangements: Analysis of 25 families. Am. J. Med. Genet. 1988, 29A, 247–261. [Google Scholar] [CrossRef]
- Madan, K.; Nieuwint, A.W.M.; van Bever, Y. Recombination in a balanced complex translocation of a mother leading to a balanced reciprocal translocation in the child. Review of 60 cases of balanced complex translocations. Hum. Genet. 1997, 99, 806–815. [Google Scholar] [CrossRef]
- Lee, I.W.; Su, M.T.; Hsu, C.C.; Lin, Y.H.; Chen, P.Y.; Kuo, P.L. Constitutional Complex Chromosomal Rearrangements In Azoospermic Men—Case Report And Literature Review. Urology 2006, 68, 1343.E5–1343.E8. [Google Scholar] [CrossRef] [PubMed]
- Bartels, I.; Starke, H.; Argyriou, L.; Sauter, S.M.; Zoll, B.; Liehr, T. An exceptional complex chromosomal rearrangement (CCR) with eight breakpoints involving four chromosomes (1;3;9;14) in an azoospermic male with normal phenotype. Eur. J. Med. Genet. 2007, 50, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Hunt, P.A.; Hassold, T.J. Sex matters in meiosis. Science 2002, 296, 2181–2183. [Google Scholar] [CrossRef] [PubMed]
- Oliver-Bonet, M.; Benet, J.; Sun, F.; Navarro, J.; Abad, C.; Liehr, T.; Starke, H.; Greene, C.; Ko, E.; Martin, R.H. Meiotic studies in two human reciprocal translocations and their association with spermatogenic failure. Hum. Reprod. 2005, 20, 683–688. [Google Scholar] [CrossRef]
- Faraut, T.; Mermet, M.A.; Demongeot, J.; Cohen, O. Cooperation of selection and meiotic mechanisms in the production of imbalances in reciprocal translocations. Cytogenet. Cell Genet. 2000, 88, 15–21. [Google Scholar] [CrossRef]
- Batista, D.A.; Pai, G.S.; Stetten, G. Molecular analysis of a complex chromosomal rearrangement and a review of familial cases. Am. J. Med. Genet. 1994, 53, 255–263. [Google Scholar] [CrossRef]
- Kim, J.W.; Chang, E.M.; Song, S.H.; Park, S.H.; Yoon, T.K.; Shim, A.H. Complex chromosomal rearrangements in infertile males: Complexity of rearrangement affects spermatogenesis. Fertil. Steril. 2011, 95, 349–352. [Google Scholar] [CrossRef]
- Walker, S.; Howard, P.J.; Hunter, D. Familial complex autosomal translocations involving chromosomes 7, 8, and 9 exhibiting male and female transmission with segregation and recombination. J. Med. Genet. 1985, 22, 484–491. [Google Scholar] [CrossRef]
- Zahed, L.; Der Kaloustian, V.; Batanian, J.R. Familial Complex Chromosome Rearrangement Giving Rise to Balanced and Unbalanced Recombination Products. Am. J. Med. Genet. 1998, 79, 30–34. [Google Scholar] [CrossRef]
- Soler, A.; Sanchez, A.; Carrio, A.; Badenas, C.; Mila, M.; Margarit, E.; Borrell, A. 2005 Recombination in a male carrier of two reciprocal translocations involving chromosomes 14, 140, 15, and 21 leading to balanced and unbalanced rearrangements in offspring. Am. J. Med. Genet. 2005, 134A, 309–314. [Google Scholar] [CrossRef]
- Ferfouri, F.; Boitrelle, F.; Clement, P.; Gomes, D.M.; Selva, J.; Vialard, F. Can one translocation impact the meiotic segregation of another translocation? A sperm-FISH analysis of a 46,XY,t(1;16)(q21;p11.2), t(8;9)(q24.3;p24) patient and his 46,XY,t(8;9)(q24.3;p24) brother and cousin. Mol. Hum. Reprod. 2013, 19, 109–117. [Google Scholar] [CrossRef] [Green Version]
- Bass, H.N.; Sparkes, R.S.; Lessner, M.M.; Fox, M.; Phoenix, B.; Bernar, J. A family with three independent autosomal translocations associated with 7q32->7qter syndrome. J. Med. Genet. 1985, 22, 59–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferfouri, F.; Boitrelle, F.; Clement, P.; Molina, G.D.; Selva, J.; Vialard, F. 2014 Sperm FISH analysis of a 44,X,der(Y),t(Y;15)(q12;q10)pat, rob (13;14)(q10;q10)mat complex chromosome rearrangement. Andrologia 2014, 46, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Johannisson, R.; Lohrs, U.; Passarge, E. Pachytene analysis in males heterozygous for a familial translocation (9;12;13)(q22;q22;q32) ascer- tained through a child with partial trisomy 9. Cytogenet. Cell. Genet. 1988, 47, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Johannisson, R.; Lohrs, U.; Wolff, H.H.; Schwinger, E. Two different XY- quadrivalent associations and impairment of fertility in men. Cytogenet. Cell. Genet. 1987, 45, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Estop, A.M.; Levinson, F.; Cieply, K.; Vankirk, V. The segregation of a translocation t(1;4) in two male carriers heterozygous for the translocation. Hum. Genet. 1992, 89, 425–429. [Google Scholar] [CrossRef]
- Rousseaux, S.; Chevret, E.; Monteli, M.; Cozzi, J.; Pelletier, R.; Devillard, F.; Lespinasse, J.; Sèle, B. Meiotic segregation in males heterozygote for reciprocal translocations: Analysis of sperm nuclei by two and three colour fluorescence in situ hybridization. Cytogenet. Cell. Genet. 1995, 71, 240–246. [Google Scholar] [CrossRef]
- Cora, T.; Acar, H.; Kaynak, M. Molecular cytogenetic detection of meiotic segregation patterns in sperm nuclei of carriers of 46,XY,t(15;17)(q21; q25). J. Androl. 2002, 23, 793–798. [Google Scholar]
- Anton, E.; Vidal, F.; Egozcue, J.; Blanco, J. Preferential alternate segregation in the common t(11;22)(q23;q11) reciprocal translocation: Sperm FISH analysis in two brothers. Reprod. BioMed. Online. 2004, 9, 637–644. [Google Scholar] [CrossRef]
- Morel, F.; Douet-Guilbert, N.; Roux, C.; Tripogney, C.; Le Bris, M.J.; De Braekeleer, M.; Bresson, J.-L. Meiotic segregation of a t(7;8)(q11.21;cen) translocation in two carrier brothers. Fertil. Steril. 2004, 81, 682–685. [Google Scholar] [CrossRef]
- Wiland, E.; Midro, A.T.; Panasiuk, B.; Kurpisz, M. The analysis of meiotic segregation patterns and aneuploidy in the spermatozoa of father and son with translocation t(4;5)(p15.1;p12) and the prediction of the individual probability rate for unbalanced progeny at birth. J. Androl. 2007, 28, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Vozdova, M.; Oracova, E.; Horinova, V.; Rubes, J. Sperm fluorescence in situhybridization study of meiotic segregation and an interchromosomal effect in carriers of t(11;18). Hum. Reprod. 2008, 23, 581–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olszewska, M.; Wiland, E.; Huleyuk, N.; Fraczek, M.; Midro, A.T.; Zastavna, D.; Kurpisz, M. Chromosome (re)positioning in spermatozoa of fathers and sons—Carriers of reciprocal chromosome translocation (RCT). BMC Med. Genom. 2019, 12, 30. [Google Scholar] [CrossRef]
- Li, L.; Heng, X.; Yun, W.; Zheng, S.; Zhang, J.; Fan, W. Familial complex chromosome rearrangement (CCR) involving 5 breakpoints on chromosomes 1, 3 and 13 in a severe oligozoospermic patient. J. Assist. Reprod. Genet. 2013, 30, 423–429. [Google Scholar] [CrossRef] [Green Version]
- Hornak, M.; Vozdova, M.; Musilova, P.; Prinosilova, P.; Oracova, E.; Linkova, V.; Vesela, K.; Rubes, J. Comprehensive meiotic segregation analysis of a 4-breakpoint t(1;3;6) complex chromosome rearrangement using single sperm array comparative genomic hybridization and FISH. Reprod. BioMed. Online 2014, 29, 499–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeMaire-Adkins, R.; Radke, K.; Hunt, P.A. Lack of checkpoint control at the metaphase/anaphase transition: A mechanism of meiotic nondisjunction in mammalian females. J. Cell Biol. 1997, 139, 1611–1619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurahashi, H.; Bolor, H.; Kato, T.; Kogo, H.; Tsutsumi, M.; Inagaki, H.; Ohye, T. Recent advance in our understanding of the molecular nature of chromosomal abnormalities. J. Hum. Genet. 2009, 54, 253–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steuerwald, N. Meiotic spindle checkpoints for assessment of aneuploid oocytes. Cytogenet. Genome. Res. 2005, 111, 256–259. [Google Scholar] [CrossRef]
- Roeder, G.S.; Bailis, J.M. The pachytene checkpoint. Trends. Genet. 2000, 16, 395–403. [Google Scholar] [CrossRef]
- Ku, C.S.; Cooper, D.N.; Polychronakos, C.; Naidoo, N.; Wu, M.; Soong, R. Exome sequencing: Dual role as a discovery and diagnostic tool. Ann. Neurol. 2012, 71, 5–14. [Google Scholar] [CrossRef]
- Botstein, D.; Risch, N. Discovering genotypes underlying human phenotypes: Past successes for mendelian disease, future approaches for complex disease. Nat. Genet. 2003, 33, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Eisfeldt, J.; Pettersson, M.; VezziI, F.; Wincent, J.; Kaller, M.; Gruselius, J.; Nilsson, D.; Syk Lundberg, E.; Carvalho, C.M.B.; Lindstrand, A. Comprehensive structural variation genome map of individuals carrying complex chromosomal rearrangements. PLoS Genet. 2019, 15, e1007858. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Reference values and semen nomen- clature. In WHO Laboratory Manual for the Examination and Processing of Human Semen, 5th ed.; WHO Press: Geneva, Switzerland, 2010; pp. 223–225. [Google Scholar]
- Olszewska, M.; Fraczek, M.; Huleyuk, N.; Czernikiewicz, A.; Wiland, E.; Boksa, M.; Zastavna, D.; Panasiuk, B.; Midro, A.T.; Kurpisz, M. Chromatin structure analysis of spermatozoa from reciprocal chromosome translocation carriers (RCT) with known meiotic segregation patterns. Reprod. Biol. 2013, 13, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Howell, W.M.; Black, D.A. Controlled silver-staining of nucleolus organizer regions with a protective colloidal developer: A 1-step method. Experientia 1980, 36, 1014–1015. [Google Scholar] [CrossRef]
- Latos-Bielenska, A. Badanie chromosomów mitotycznych i mejotycznych techniką immunocytochemicznej detekcji bromodezoksyurydyny wbudowanej do chromosomowego DNA. Praca Habilitacyjna (D.Sc. Thesis), Wydawnictwo Akademii Medycznej im., K. Marcinkowskiego w Poznaniu, 1991. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Event | Chr | Cytoband | #Probes | Log Ratio | Annotations | Gene Effect | Inherited from Mother |
|---|---|---|---|---|---|---|---|
| 1 | chr1:152556449–152581944 | q21.3 | 6 | 0.617541 | LCE3C | pol | Yes |
| 2 | chr1:196742735–196796220 | q31.3 | 5 | −0.978775 | CFHR3, CFHR1 | pol | No |
| 3 | chr2:34697718–34730142 | p22.3 | 7 | −0.636087 | pol | Yes | |
| 4 | chr2:78709955–78721280 | p12 | 4 | −0.852013 | pol | No | |
| 5 | chr2:87392136–87801877 | p11.2 | 11 | 0.508958 | NCRNA00152 | pol | Yes |
| 6 | chr2:132205346–132217492 | q21.1 | 3 | −0.809717 | pol | No | |
| 7 | chr2:180069605–180070733 | q31.2 | 3 | 1.002822 | SESTD1, 1MB dup | rare | Yes |
| 8 | chr3:162556223–162619141 | q26.1 | 7 | 0.471521 | pol | No | |
| 9 | chr4:69387056–69483277 | q13.2 | 12 | −0.742031 | UGT2B17, UGT2B15 | pol | No |
| 10 | chr5:17345455–17353452 | p15.1 | 3 | −0.926486 | pol | Yes | |
| 11 | chr5:140223256–140236399 | q31.3 | 4 | −0.878128 | PCDHA1, PCDHA2, PCDHA3 | pol | Yes |
| 12 | chr6:259881–287425 | p25.3 | 6 | 0.523226 | pol | Yes | |
| 13 | chr6:32450699–32493043 | p21.32 | 6 | −1.853271 | HLA-DRB5 | pol | No |
| 14 | chr6:165725547–165737665 | q27 | 3 | 0.948835 | rare | No | |
| 15 | chr7:141750430–141792094 | q34 | 9 | −0.511123 | MGAM, | pol | Yes |
| 16 | chr8:39234992–39386158 | p11.22 | 28 | 0.821278 | ADAM5P, ADAM3A, 151 kb dup | pol | Yes |
| 17 | chr9:130041553–130145721 | q33.3 | 23 | −0.981719 | GARNL3, 104 kb del, het | rare | No |
| 18 | chr11:18949929–18960666 | p15.1 | 3 | 0.804263 | MRGPRX1, 10 kb dup het | pol | No |
| 19 | chr11:25635357–25764082 | p14.3 | 9 | 0.689271 | rare | Yes | |
| 20 | chr11:55368154–55450788 | q11 | 16 | 0.436620 | OR4C11, OR4P4, OR4S2 | pol | Yes |
| 21 | chr12:9637323–9718846 | p13.31 | 11 | 1.538928 | pol | Yes | |
| 22 | chr12:10583558–10593748 | p13.2 | 3 | −0.864549 | KLRC2,10 kb del het, gene cluster | pol | No |
| 23 | chr12:11218244–11225675 | p13.2 | 3 | −1.376538 | PRR4, PRH1 | pol | No |
| 24 | chr12:11230835–11249210 | p13.2 | 3 | −4.773687 | PRR4, PRH1, TAS2R43 | pol | Yes |
| 26 | chr14:74001651–74012568 | q24.3 | 3 | −5.04754 | HEATR4, ACOT1, 1.1MB del | pol | Yes |
| 27 | chr16:70174866–70193942 | q22.1 | 4 | −0.996207 | PDPR | pol | No |
| 28 | chr16:74394080–74407341 | q23.1 | 3 | −0.943902 | LOC283922 | pol | No |
| 29 | chr16:78372097–78381281 | q23.1 | 3 | −5.242076 | WWOX | pol | No |
| 30 | chr17:34437475–34475514 | q12 | 7 | 0.847688 | pol | Yes | |
| 31 | chr19:53522243–53550020 | q13.41 | 6 | 0.783781 | pol | No | |
| 32 | chr20:1563715–1577359 | p13 | 4 | −3.864894 | SIRPB1, 13 kb homo del | pol | Yes |
| 33 | chr22:18889039–19010562 | q11.21 | 20 | 0.522787 | DGCR6, PRODH, DGCR5… | pol | Yes |
| 34 | chr22:24347959–24395353 | q11.23 | 10 | 0.607834 | LOC391322, GSTT1, GSTTP2 | pol | No |
| 35 | chr22:25664618–25919542 | q11.23–q12.1 | 54 | 0.578645 | IGLL3, LRP5L | pol | No |
| 36 | chrX:1731610–1752284 | p22.33 | 10 | 0.513510 | ASMT | rare | No |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olszewska, M.; Stokowy, T.; Pollock, N.; Huleyuk, N.; Georgiadis, A.; Yatsenko, S.; Zastavna, D.; Yatsenko, A.N.; Kurpisz, M. Familial Infertility (Azoospermia and Cryptozoospermia) in Two Brothers—Carriers of t(1;7) Complex Chromosomal Rearrangement (CCR): Molecular Cytogenetic Analysis. Int. J. Mol. Sci. 2020, 21, 4559. https://doi.org/10.3390/ijms21124559
Olszewska M, Stokowy T, Pollock N, Huleyuk N, Georgiadis A, Yatsenko S, Zastavna D, Yatsenko AN, Kurpisz M. Familial Infertility (Azoospermia and Cryptozoospermia) in Two Brothers—Carriers of t(1;7) Complex Chromosomal Rearrangement (CCR): Molecular Cytogenetic Analysis. International Journal of Molecular Sciences. 2020; 21(12):4559. https://doi.org/10.3390/ijms21124559
Chicago/Turabian StyleOlszewska, Marta, Tomasz Stokowy, Nijole Pollock, Nataliya Huleyuk, Andrew Georgiadis, Svetlana Yatsenko, Danuta Zastavna, Alexander N. Yatsenko, and Maciej Kurpisz. 2020. "Familial Infertility (Azoospermia and Cryptozoospermia) in Two Brothers—Carriers of t(1;7) Complex Chromosomal Rearrangement (CCR): Molecular Cytogenetic Analysis" International Journal of Molecular Sciences 21, no. 12: 4559. https://doi.org/10.3390/ijms21124559
APA StyleOlszewska, M., Stokowy, T., Pollock, N., Huleyuk, N., Georgiadis, A., Yatsenko, S., Zastavna, D., Yatsenko, A. N., & Kurpisz, M. (2020). Familial Infertility (Azoospermia and Cryptozoospermia) in Two Brothers—Carriers of t(1;7) Complex Chromosomal Rearrangement (CCR): Molecular Cytogenetic Analysis. International Journal of Molecular Sciences, 21(12), 4559. https://doi.org/10.3390/ijms21124559