Growth Hormone Treatment Promotes Remote Hippocampal Plasticity after Experimental Cortical Stroke

 , ,
, , 
Abstract
:1. Introduction
2. Results
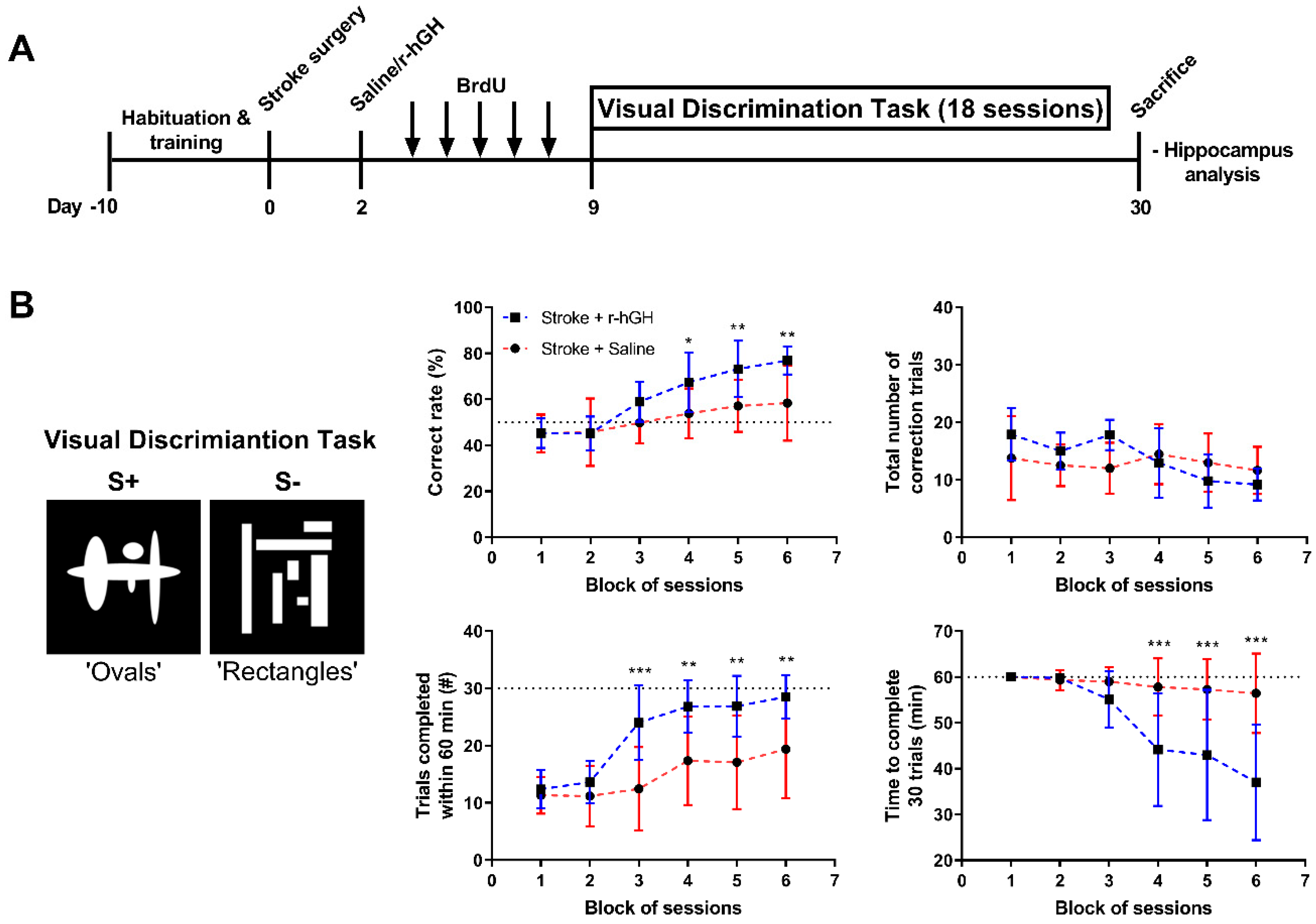
2.1. GH Treatment Improves Cognitive Function
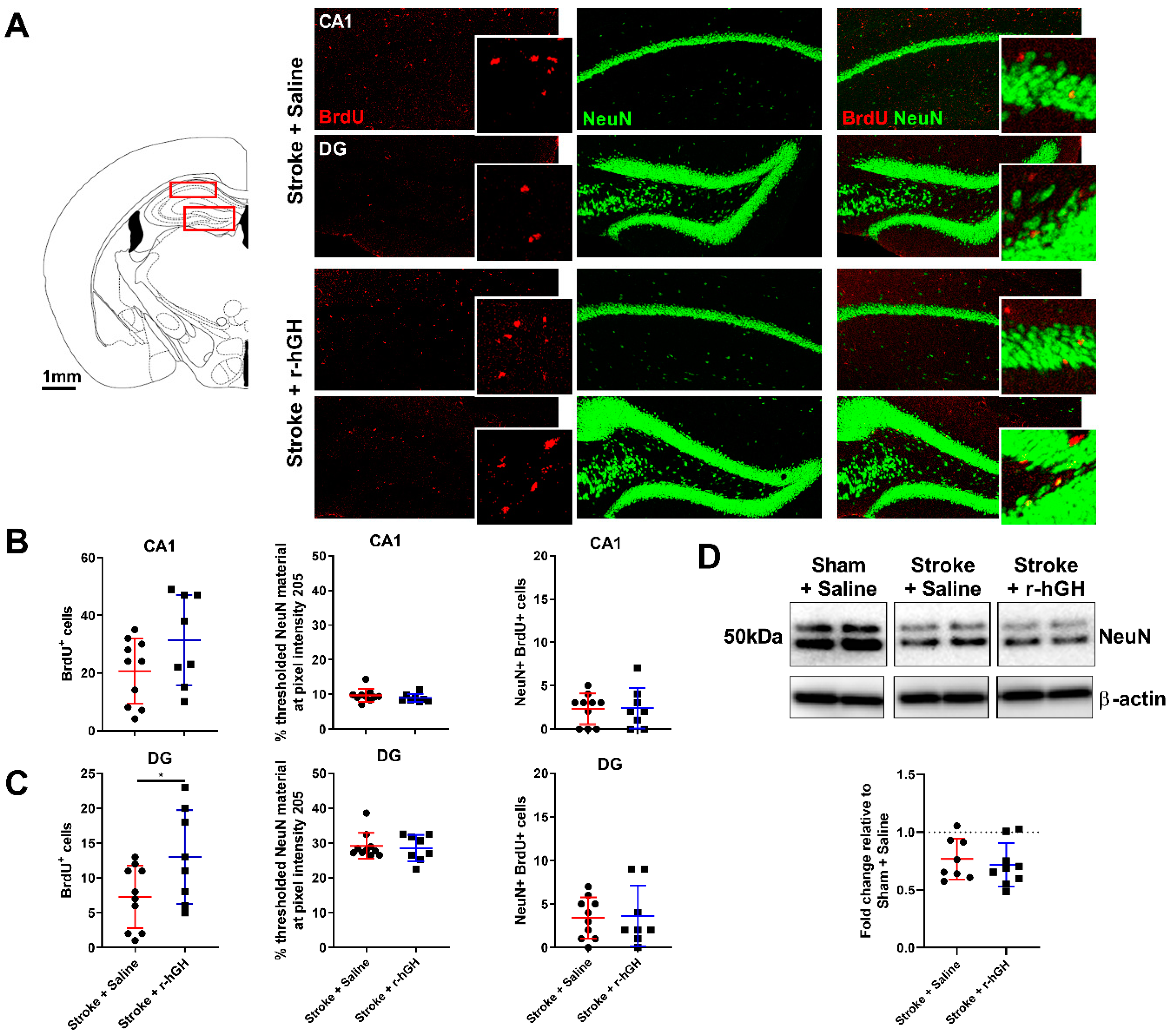
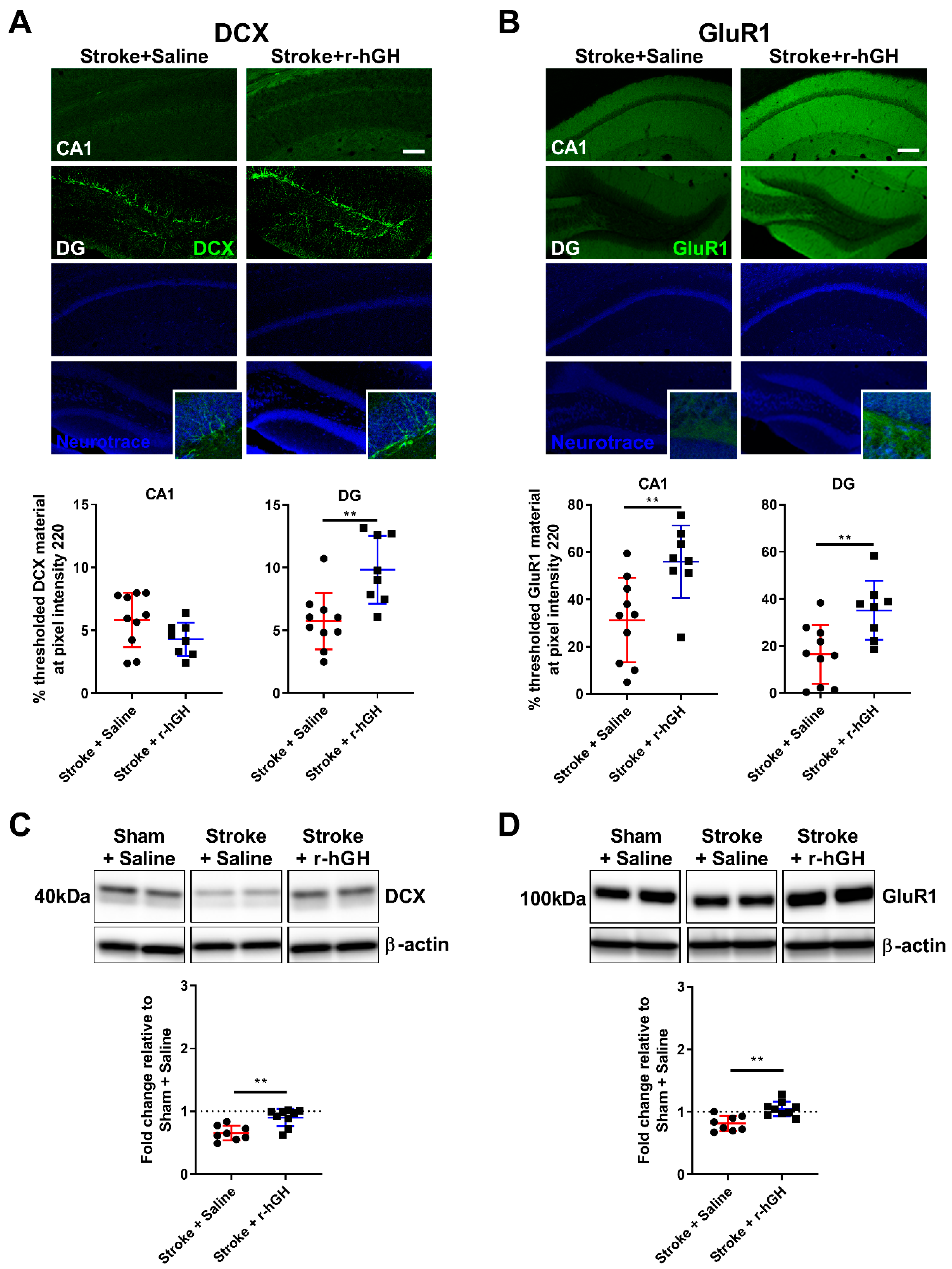
2.2. GH Treatment Promotes Cell Proliferation and Neurogenesis in the Dentate Gyrus (DG)
2.3. GH Treatment Promotes Expression of GluR1 within the Hippocampal Formation
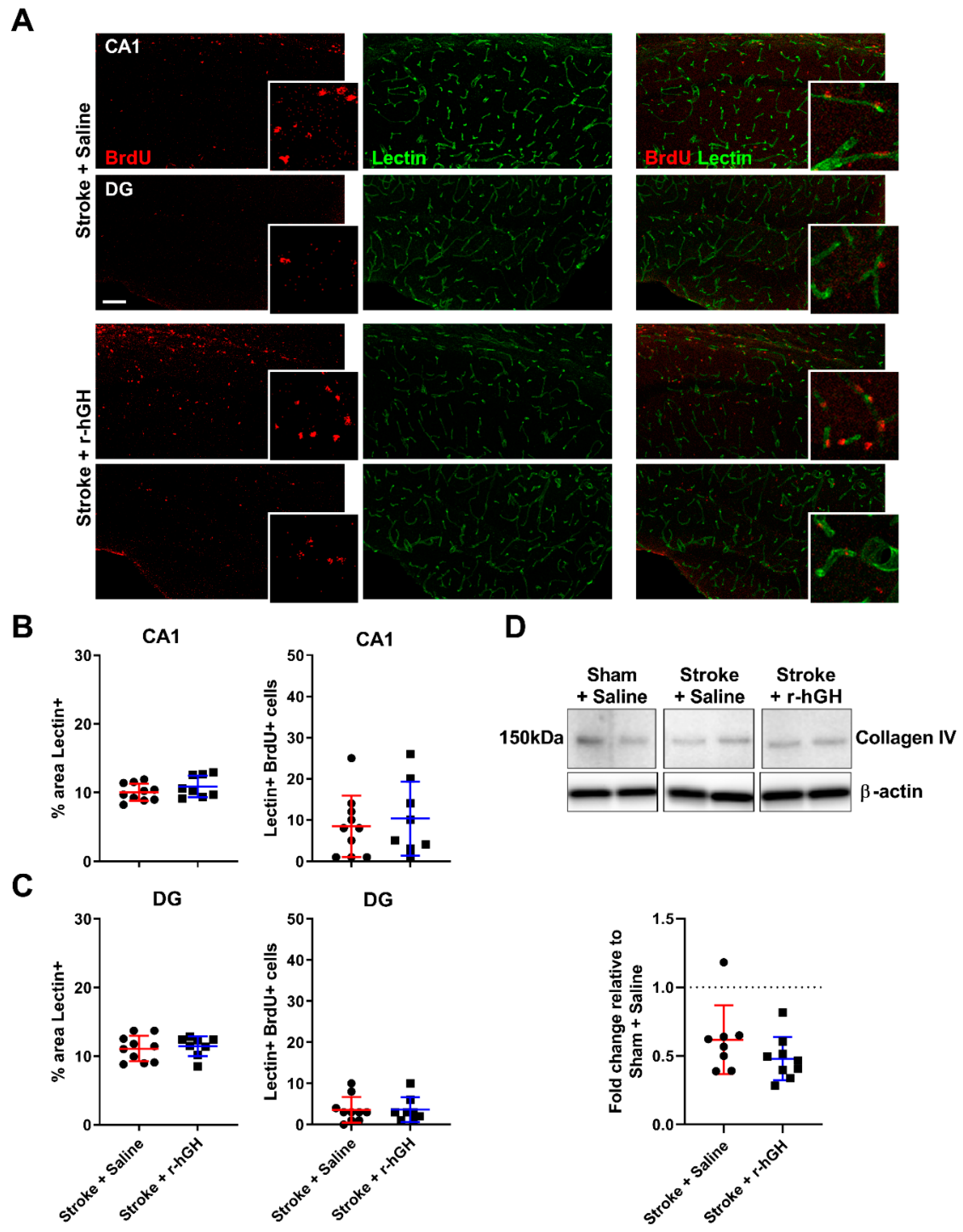
2.4. GH Treatment Had No Effect on the Formation of Cerebral Vasculature within the Hippocampal Formation
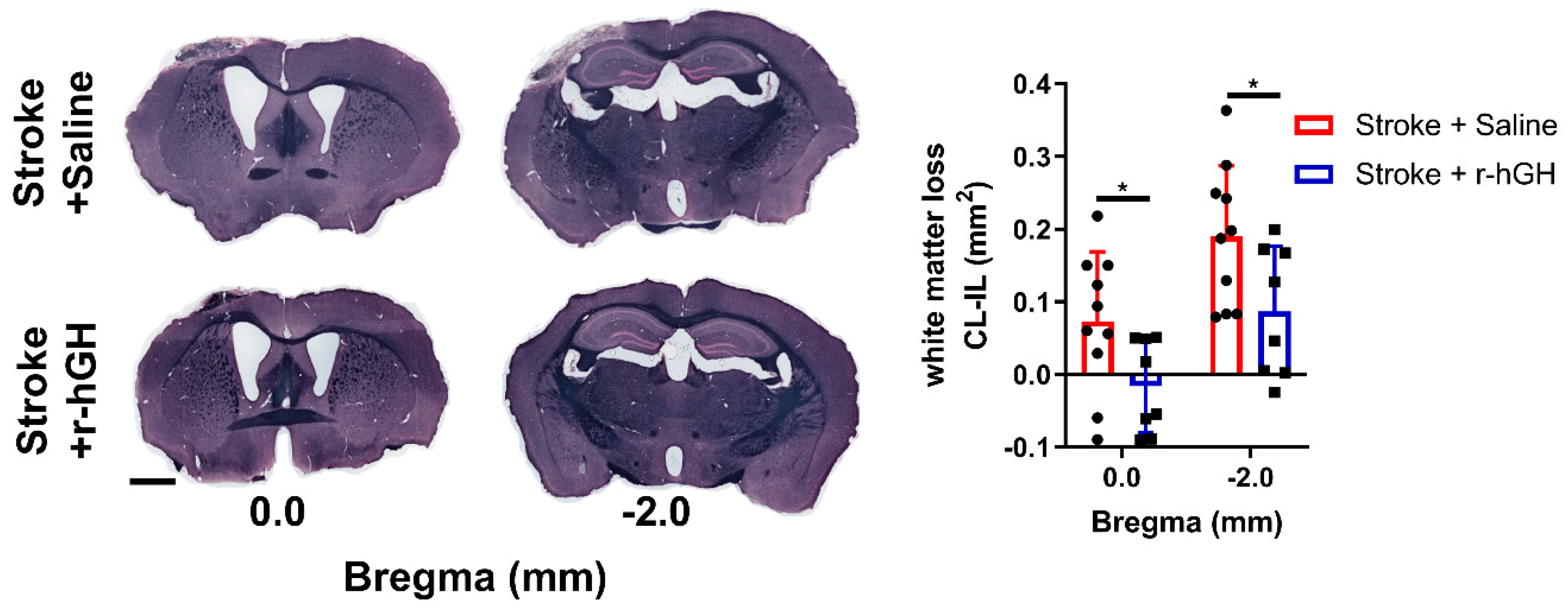
2.5. GH Treatment Promotes Restoration of White Matter Disturbances
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Animals
5.2. Sample Size Calculation
5.3. Experimental Design
5.4. Photothrombotic Occlusion
5.5. Mini-Osmotic Pump Placement
5.6. Visual Discrimination (VD) Task
5.7. Tissue Processing
5.8. Immunofluorescence
5.9. Sudan Black Staining
5.10. Image Acquisition and Analysis
5.11. Protein Extraction and Western Blotting
5.12. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| GH | Growth hormone |
| r-hGH | Recombinant human growth hormone |
| BrdU | Bromodeoxyuridine |
| NeuN | Neuronal nuclei |
| DCX | Doublecortin |
| GluR1 | AMPA Receptor 1 |
| VD | Visual Discrimination |
| DG | Dentate Gyrus |
References
- Prins, N.D.; van Dijk, E.J.; den Heijer, T.; Vermeer, S.E.; Jolles, J.; Koudstaal, P.J.; Hofman, A.; Breteler, M.M. Cerebral small-vessel disease and decline in information processing speed, executive function and memory. Brain 2005, 128, 2034–2041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blum, S.; Luchsinger, J.A.; Manly, J.J.; Schupf, N.; Stern, Y.; Brown, T.R.; DeCarli, C.; Small, S.A.; Mayeux, R.; Brickman, A.M. Memory after silent stroke: Hippocampus and infarcts both matter. Neurology 2012, 78, 38–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gemmell, E.; Bosomworth, H.; Allan, L.; Hall, R.; Khundakar, A.; Oakley, A.E.; Deramecourt, V.; Polvikoski, T.M.; O’Brien, J.T.; Kalaria, R.N. Hippocampal neuronal atrophy and cognitive function in delayed poststroke and aging-related dementias. Stroke 2012, 43, 808–814. [Google Scholar] [CrossRef] [Green Version]
- Selnes, P.; Grambaite, R.; Rincon, M.; Bjørnerud, A.; Gjerstad, L.; Hessen, E.; Auning, E.; Johansen, K.; Almdahl, I.S.; Due-Tønnessen, P.; et al. Hippocampal complex atrophy in poststroke and mild cognitive impairment. J. Cereb. Blood Flow Metab. 2015, 35, 1729–1737. [Google Scholar] [CrossRef]
- Khlif, M.S.; Werden, E.; Egorova, N.; Boccardi, M.; Redolfi, A.; Bird, L.; Brodtmann, A. Assessment of longitudinal hippocampal atrophy in the first year after ischemic stroke using automatic segmentation techniques. Neuroimage Clin. 2019, 24, 102008. [Google Scholar] [CrossRef]
- Yagita, Y.; Kitagawa, K.; Ohtsuki, T.; Takasawa, K.; Miyata, T.; Okano, H.; Hori, M.; Matsumoto, M. Neurogenesis by progenitor cells in the ischemic adult rat hippocampus. Stroke 2001, 32, 1890–1896. [Google Scholar] [CrossRef] [Green Version]
- Koh, S.-H.; Park, H.-H. Neurogenesis in Stroke Recovery. Transl. Stroke Res. 2017, 8, 3–13. [Google Scholar] [CrossRef]
- Takagi, Y.; Nozaki, K.; Takahashi, J.; Yodoi, J.; Ishikawa, M.; Hashimoto, N. Proliferation of neuronal precursor cells in the dentate gyrus is accelerated after transient forebrain ischemia in mice. Brain Res. 1999, 831, 283–287. [Google Scholar] [CrossRef]
- Kee, N.J.; Preston, E.; Wojtowicz, J.M. Enhanced neurogenesis after transient global ischemia in the dentate gyrus of the rat. Exp. Brain Res. 2001, 136, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Tonchev, A.B.; Yamashima, T.; Zhao, L.; Okano, H. Differential proliferative response in the postischemic hippocampus, temporal cortex, and olfactory bulb of young adult macaque monkeys. Glia 2003, 42, 209–224. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Solway, K.; Messing, R.O.; Sharp, F.R. Increased neurogenesis in the dentate gyrus after transient global ischemia in gerbils. J. Neurosci. 1998, 18, 7768–7778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lichtenwalner, R.J.; Parent, J.M. Adult neurogenesis and the ischemic forebrain. J. Cereb. Blood Flow Metab. 2006, 26, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Kernie, S.G.; Parent, J.M. Forebrain neurogenesis after focal Ischemic and traumatic brain injury. Neurobiol. Dis. 2010, 37, 267–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuartero, M.I.; de la Parra, J.; Pérez-Ruiz, A.; Bravo-Ferrer, I.; Durán-Laforet, V.; García-Culebras, A.; García-Segura, J.M.; Dhaliwal, J.; Frankland, P.W.; Lizasoain, I.; et al. Abolition of aberrant neurogenesis ameliorates cognitive impairment after stroke in mice. J. Clin. Investig. 2019, 129, 1536–1550. [Google Scholar] [CrossRef] [PubMed]
- Arvidsson, A.; Collin, T.; Kirik, D.; Kokaia, Z.; Lindvall, O. Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat. Med. 2002, 8, 963–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parent, J.M.; Vexler, Z.S.; Gong, C.; Derugin, N.; Ferriero, D.M. Rat forebrain neurogenesis and striatal neuron replacement after focal stroke. Ann. Neurol. 2002, 52, 802–813. [Google Scholar] [CrossRef]
- Jin, K.; Sun, Y.; Xie, L.; Peel, A.; Mao, X.O.; Batteur, S.; Greenberg, D.A. Directed migration of neuronal precursors into the ischemic cerebral cortex and striatum. Mol. Cell. Neurosci. 2003, 24, 171–189. [Google Scholar] [CrossRef]
- Zhang, R.; Zhang, Z.; Wang, L.; Wang, Y.; Gousev, A.; Zhang, L.; Ho, K.L.; Morshead, C.; Chopp, M. Activated neural stem cells contribute to stroke-induced neurogenesis and neuroblast migration toward the infarct boundary in adult rats. J. Cereb. Blood Flow Metab. 2004, 24, 441–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernal, G.M.; Peterson, D.A. Neural stem cells as therapeutic agents for age-related brain repair. Aging Cell 2004, 3, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Niv, F.; Keiner, S.; Krishna, K.; Witte, O.W.; Lie, D.C.; Redecker, C. Aberrant neurogenesis after stroke: A retroviral cell labeling study. Stroke 2012, 43, 2468–2475. [Google Scholar] [CrossRef] [Green Version]
- Woitke, F.; Ceanga, M.; Rudolph, M.; Niv, F.; Witte, O.W.; Redecker, C.; Kunze, A.; Keiner, S. Adult hippocampal neurogenesis poststroke: More new granule cells but aberrant morphology and impaired spatial memory. PLoS ONE 2017, 12, e0183463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchi, V.E.; Locatelli, V.; Rizzi, L. Neurotrophic and Neuroregenerative Effects of GH/IGF1. Int. J. Mol. Sci. 2017, 18, 2441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aberg, N.D.; Brywe, K.G.; Isgaard, J. Aspects of growth hormone and insulin-like growth factor-I related to neuroprotection, regeneration, and functional plasticity in the adult brain. ScientificWorldJournal 2006, 6, 53–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, L.K.; Chow, W.Z.; TeBay, C.; Kluge, M.; Pietrogrande, G.; Zalewska, K.; Crock, P.; Åberg, N.D.; Bivard, A.; Johnson, S.J.; et al. Growth Hormone Improves Cognitive Function After Experimental Stroke. Stroke 2018, 49, 1257–1266. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Bezanilla, S.; Åberg, N.D.; Crock, P.; Walker, F.R.; Nilsson, M.; Isgaard, J.; Ong, L.K. Growth Hormone Promotes Motor Function after Experimental Stroke and Enhances Recovery-Promoting Mechanisms within the Peri-Infarct Area. Int. J. Mol. Sci. 2020, 21, 606. [Google Scholar] [CrossRef] [Green Version]
- Heredia, M.; Palomero, J.; de la Fuente, A.; Criado, J.M.; Yajeya, J.; Devesa, J.; Devesa, P.; Vicente-Villardón, J.L.; Riolobos, A.S. Motor Improvement of Skilled Forelimb Use Induced by Treatment with Growth Hormone and Rehabilitation Is Dependent on the Onset of the Treatment after Cortical Ablation. Neural Plast. 2018, 2018, 6125901. [Google Scholar] [CrossRef] [Green Version]
- Heredia, M.; Rodríguez, N.; Sánchez Robledo, V.; Criado, J.M.; de la Fuente, A.; Devesa, J.; Devesa, P.; Sánchez Riolobos, A. Factors Involved in the Functional Motor Recovery of Rats with Cortical Ablation after GH and Rehabilitation Treatment: Cortical Cell Proliferation and Nestin and Actin Expression in the Striatum and Thalamus. Int. J. Mol. Sci. 2019, 20, 5770. [Google Scholar] [CrossRef] [Green Version]
- Heredia, M.; Fuente, A.; Criado, J.; Yajeya, J.; Devesa, J.; Riolobos, A.S. Early growth hormone (GH) treatment promotes relevant motor functional improvement after severe frontal cortex lesion in adult rats. Behav. Brain Res. 2013, 247, 48–58. [Google Scholar] [CrossRef]
- Pathipati, P.; Surus, A.; Williams, C.E.; Scheepens, A. Delayed and chronic treatment with growth hormone after endothelin-induced stroke in the adult rat. Behav. Brain Res. 2009, 204, 93–101. [Google Scholar] [CrossRef]
- Horner, A.E.; Heath, C.J.; Hvoslef-Eide, M.; Kent, B.A.; Kim, C.H.; Nilsson, S.R.O.; Alsiö, J.; Oomen, C.A.; Holmes, A.; Saksida, L.M.; et al. The touchscreen operant platform for testing learning and memory in rats and mice. Nat. Protoc 2013, 8, 1961–1984. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Bezanilla, S.; TeBay, C.; Nilsson, M.; Walker, F.R.; Ong, L.K. Visual discrimination impairment after experimental stroke is associated with disturbances in the polarization of the astrocytic aquaporin-4 and increased accumulation of neurotoxic proteins. Exp. Neurol. 2019, 318, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, P.; El Amki, M.; Bracko, O.; Luft, A.R.; Wegener, S. Sensorimotor stroke alters hippocampo-thalamic network activity. Sci. Rep. 2018, 8, 15770. [Google Scholar] [CrossRef]
- Ong, L.K.; Walker, F.R.; Nilsson, M. Is Stroke a Neurodegenerative Condition? A Critical Review of Secondary Neurodegeneration and Amyloid-beta Accumulation after Stroke. AIMS Med. Sci. 2017, 4, 1–16. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Y.; Xing, S.; Liang, Z.; Zeng, J. Secondary neurodegeneration in remote regions after focal cerebral infarction: A new target for stroke management? Stroke 2012, 43, 1700–1705. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Minami, M.; Lan, J.Q.; Mao, X.O.; Batteur, S.; Simon, R.P.; Greenberg, D.A. Neurogenesis in dentate subgranular zone and rostral subventricular zone after focal cerebral ischemia in the rat. Proc. Natl. Acad. Sci. USA 2001, 98, 4710–4715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, X.; Li, G.; Wu, W.; Xu, Y.; Lin, H.; Fan, J. Recombinant Human Growth Hormone Ameliorates Cognitive Impairment in Stroke Patients. J. Comput. Assist. Tomogr. 2020, 44, 255–261. [Google Scholar] [CrossRef]
- Nyberg, F.; Hallberg, M. Growth hormone and cognitive function. Nat. Rev. Endocrinol. 2013, 9, 357–365. [Google Scholar] [CrossRef]
- Pan, W.; Yu, Y.; Cain, C.M.; Nyberg, F.; Couraud, P.O.; Kastin, A.J. Permeation of growth hormone across the blood-brain barrier. Endocrinology 2005, 146, 4898–4904. [Google Scholar] [CrossRef]
- López-Fernández, J.; Sánchez-Franco, F.; Velasco, B.; Tolón, R.M.; Pazos, F.; Cacicedo, L. Growth hormone induces somatostatin and insulin-like growth factor I gene expression in the cerebral hemispheres of aging rats. Endocrinology 1996, 137, 4384–4391. [Google Scholar] [CrossRef] [Green Version]
- Ye, P.; Umayahara, Y.; Ritter, D.; Bunting, T.; Auman, H.; Rotwein, P.; D’Ercole, A.J. Regulation of insulin-like growth factor I (IGF-I) gene expression in brain of transgenic mice expressing an IGF-I-luciferase fusion gene. Endocrinology 1997, 138, 5466–5475. [Google Scholar] [CrossRef]
- Reinhardt, R.R.; Bondy, C.A. Insulin-like growth factors cross the blood-brain barrier. Endocrinology 1994, 135, 1753–1761. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, C.S.; Wuarin, L.; Ishii, D.N. Uptake of circulating insulin-like growth factor-I into the cerebrospinal fluid of normal and diabetic rats and normalization of IGF-II mRNA content in diabetic rat brain. J. Neurosci. Res. 2000, 59, 649–660. [Google Scholar] [CrossRef]
- Brown, J.P.; Couillard-Després, S.; Cooper-Kuhn, C.M.; Winkler, J.; Aigner, L.; Kuhn, H.G. Transient expression of doublecortin during adult neurogenesis. J. Comp. Neurol. 2003, 467, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Pekna, M.; Pekny, M.; Nilsson, M. Modulation of neural plasticity as a basis for stroke rehabilitation. Stroke 2012, 43, 2819–2828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollmann, M.; Heinemann, S. Cloned glutamate receptors. Annu. Rev. Neurosci. 1994, 17, 31–108. [Google Scholar] [CrossRef] [PubMed]
- Dingledine, R.; Borges, K.; Bowie, D.; Traynelis, S.F. The glutamate receptor ion channels. Pharmacol. Rev. 1999, 51, 7–61. [Google Scholar]
- Lu, W.; Isozaki, K.; Roche, K.W.; Nicoll, R.A. Synaptic targeting of AMPA receptors is regulated by a CaMKII site in the first intracellular loop of GluA1. Proc. Natl. Acad. Sci. USA 2010, 107, 22266–22271. [Google Scholar] [CrossRef] [Green Version]
- Chater, T.E.; Goda, Y. The role of AMPA receptors in postsynaptic mechanisms of synaptic plasticity. Front. Cell. Neurosci. 2014, 8, 401. [Google Scholar] [CrossRef]
- Mustafa, A.; Adem, A.; Roos, P.; Nyberg, F. Sex differences in binding of human growth hormone to rat brain. Neurosci. Res. 1994, 19, 93–99. [Google Scholar] [CrossRef]
- Furigo, I.C.; Metzger, M.; Teixeira, P.D.; Soares, C.R.; Donato, J., Jr. Distribution of growth hormone-responsive cells in the mouse brain. Brain Struct. Funct. 2017, 222, 341–363. [Google Scholar] [CrossRef]
- Nilsson, A.; Carlsson, B.; Isgaard, J.; Isaksson, O.G.; Rymo, L. Regulation by GH of insulin-like growth factor-I mRNA expression in rat epiphyseal growth plate as studied with in-situ hybridization. J. Endocrinol. 1990, 125, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Walser, M.; Schiöler, L.; Oscarsson, J.; Åberg, M.A.; Wickelgren, R.; Svensson, J.; Isgaard, J.; Åberg, N.D. Mode of GH administration and gene expression in the female rat brain. J. Endocrinol. 2017, 233, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Walser, M.; Schiöler, L.; Oscarsson, J.; Aberg, M.A.; Svensson, J.; Aberg, N.D.; Isgaard, J. Different modes of GH administration influence gene expression in the male rat brain. J. Endocrinol. 2014, 222, 181–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.; Park, K.; Lee, H.; Kim, M. The effect of recombinant human growth hormone therapy in patients with completed stroke: A pilot trial. Ann. Rehabil. Med. 2012, 36, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Lillicrap, T.; Garcia-Esperon, C.; Walker, F.R.; Ong, L.K.; Nilsson, M.; Spratt, N.; Levi, C.R.; Parsons, M.; Isgaard, J.; Bivard, A. Growth Hormone Deficiency Is Frequent After Recent Stroke. Front. Neurol 2018, 9, 713. [Google Scholar] [CrossRef]
- Kreber, L.A.; Ashley, M.J.; Masel, B.E.; Singh, C.K.; Randle, K.D.; Johnson, C.; Helvie, R.; Ashley, M.J.; Griesbach, G.S. Prevalence of growth hormone deficiency in middle-age adults recovering from stroke. Brain Inj. 2020, 34, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Zalewska, K.; Ong, L.K.; Johnson, S.J.; Nilsson, M.; Walker, F.R. Oral administration of corticosterone at stress-like levels drives microglial but not vascular disturbances post-stroke. Neuroscience 2017, 352, 30–38. [Google Scholar] [CrossRef]
- Zhao, Z.; Ong, L.K.; Johnson, S.; Nilsson, M.; Walker, F.R. Chronic stress induced disruption of the peri-infarct neurovascular unit following experimentally induced photothrombotic stroke. J. Cereb. Blood Flow Metab. 2017, 37, 3709–3724. [Google Scholar] [CrossRef] [Green Version]
- Walser, M.; Hansén, A.; Svensson, P.-A.; Jernås, M.; Oscarsson, J.; Isgaard, J.; Åberg, N.D. Peripheral administration of bovine GH regulates the expression of cerebrocortical beta-globin, GABAB receptor 1, and the Lissencephaly-1 protein (LIS-1) in adult hypophysectomized rats. Growth Horm. IGF Res. 2011, 21, 16–24. [Google Scholar] [CrossRef]
- Kluge, M.G.; Abdolhoseini, M.; Zalewska, K.; Ong, L.K.; Johnson, S.J.; Nilsson, M.; Walker, F.R. Spatiotemporal analysis of impaired microglia process movement at sites of secondary neurodegeneration post-stroke. J. Cereb. Blood Flow Metab. 2019, 39, 2456–2470. [Google Scholar] [CrossRef]
- Kluge, M.G.; Kracht, L.; Abdolhoseini, M.; Ong, L.K.; Johnson, S.J.; Nilsson, M.; Walker, F.R. Impaired microglia process dynamics post-stroke are specific to sites of secondary neurodegeneration. Glia 2017, 65, 1885–1899. [Google Scholar] [CrossRef] [PubMed]
- Pietrogrande, G.; Zalewska, K.; Zhao, Z.; Abdolhoseini, M.; Chow, W.Z.; Sanchez-Bezanilla, S.; Ong, L.K.; Johnson, S.J.; Nilsson, M.; Walker, F.R. Low oxygen post conditioning prevents thalamic secondary neuronal loss caused by excitotoxicity after cortical stroke. Sci. Rep. 2019, 9, 4841. [Google Scholar] [CrossRef] [PubMed]
- Ong, L.K.; Zhao, Z.; Kluge, M.; Walker, F.R.; Nilsson, M. Chronic stress exposure following photothrombotic stroke is associated with increased levels of Amyloid beta accumulation and altered oligomerisation at sites of thalamic secondary neurodegeneration in mice. J. Cereb. Blood Flow Metab. 2017, 37, 1338–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Bezanilla, S.; Nilsson, M.; Walker, F.R.; Ong, L.K. Can We Use 2,3,5-Triphenyltetrazolium Chloride-Stained Brain Slices for Other Purposes? The Application of Western Blotting. Front. Mol. Neurosci. 2019, 12, 181. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Targets | Sources of Antibodies | Application | Dilution |
|---|---|---|---|
| BrdU | Sigma-Aldrich, mouse anti-BrdU, #B8434 | IF | 1:1000 |
| NeuN | Cell Signaling, rabbit anti-NeuN (D3S31), #12943 | WB | 1:2000 |
| IF | 1:1000 | ||
| DCX | abcam, rabbit anti-doublecortin, #ab18723 | WB | 1:1000 |
| IF | 1:1000 | ||
| GluR1 | Cell Signaling, rabbit anti-AMPA Receptor 1 (GluA1), #13185 | WB | 1:2000 |
| IF | 1:1000 | ||
| Collagen IV | Abcam, rabbit anti-collagen IV, #ab6586 | WB | 1:1000 |
| β-actin | Sigma-Aldrich, Monoclonal Anti-β-actin-HRP antibody, A3854 | WB | 1:50,000 |
| NeuroTrace | ThermoFisher Scientific, NeuroTrace™ 640/660 Deep-Red Fluorescent Nissl Stain, #N21483 | IF | 1:1000 |
| Tomato Lectin | Vector Laboratories, DyLight 649 Lycopersicon esculentum (Tomato) Lectin #DL-1178 | IF | 1:1000 |
| Rabbit IgG | Biorad, Anti-Rabbit-HRP antibody, #170-6515 | WB | 1:7500 |
| ThermoFisher Scientific, anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488, #A21206 | IF | 1:400 | |
| Mouse IgG | Biorad, Anti-Mouse-HRP antibody, #170-6516 | WB | 1:10,000 |
| ThermoFisher Scientific, anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 594, #A21203 | IF | 1:400 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanchez-Bezanilla, S.; Åberg, N.D.; Crock, P.; Walker, F.R.; Nilsson, M.; Isgaard, J.; Ong, L.K. Growth Hormone Treatment Promotes Remote Hippocampal Plasticity after Experimental Cortical Stroke. Int. J. Mol. Sci. 2020, 21, 4563. https://doi.org/10.3390/ijms21124563
Sanchez-Bezanilla S, Åberg ND, Crock P, Walker FR, Nilsson M, Isgaard J, Ong LK. Growth Hormone Treatment Promotes Remote Hippocampal Plasticity after Experimental Cortical Stroke. International Journal of Molecular Sciences. 2020; 21(12):4563. https://doi.org/10.3390/ijms21124563
Chicago/Turabian StyleSanchez-Bezanilla, Sonia, N. David Åberg, Patricia Crock, Frederick R. Walker, Michael Nilsson, Jörgen Isgaard, and Lin Kooi Ong. 2020. "Growth Hormone Treatment Promotes Remote Hippocampal Plasticity after Experimental Cortical Stroke" International Journal of Molecular Sciences 21, no. 12: 4563. https://doi.org/10.3390/ijms21124563
APA StyleSanchez-Bezanilla, S., Åberg, N. D., Crock, P., Walker, F. R., Nilsson, M., Isgaard, J., & Ong, L. K. (2020). Growth Hormone Treatment Promotes Remote Hippocampal Plasticity after Experimental Cortical Stroke. International Journal of Molecular Sciences, 21(12), 4563. https://doi.org/10.3390/ijms21124563