Scalable High-Performance Production of Recombinant Horseradish Peroxidase from E. coli Inclusion Bodies


Abstract
:1. Introduction
2. Results and Discussion
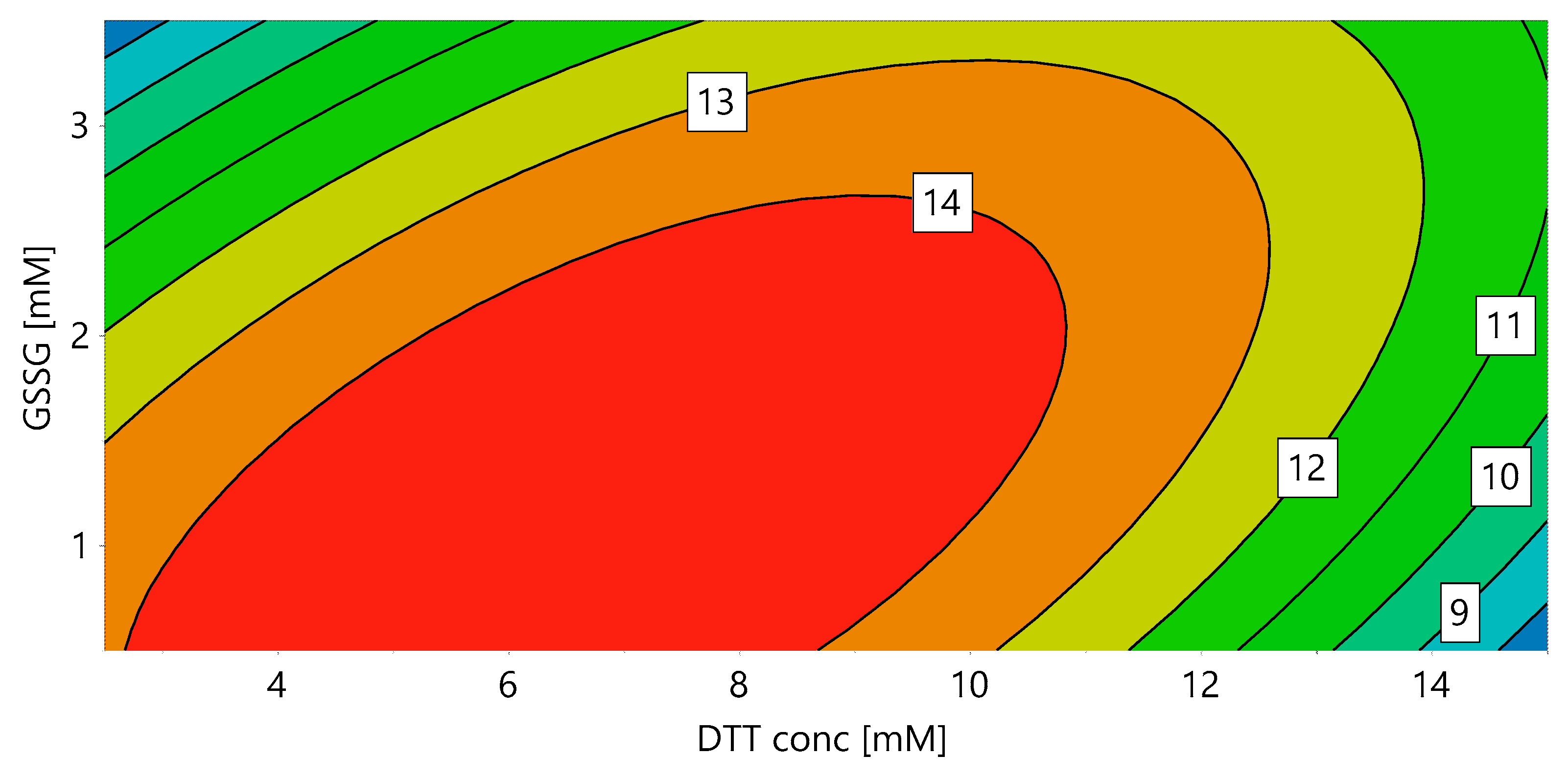
2.1. Solubilization and Refolding
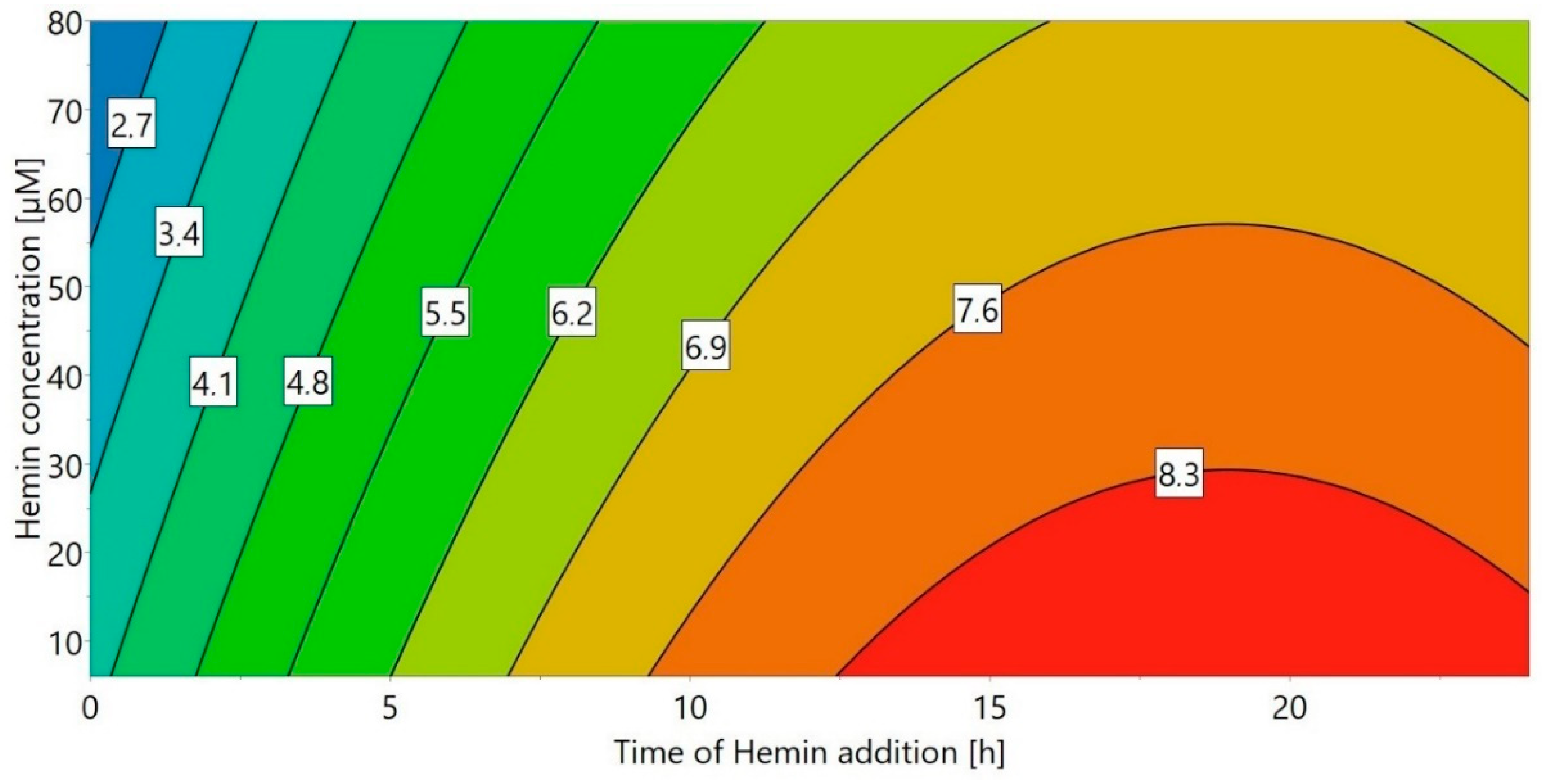
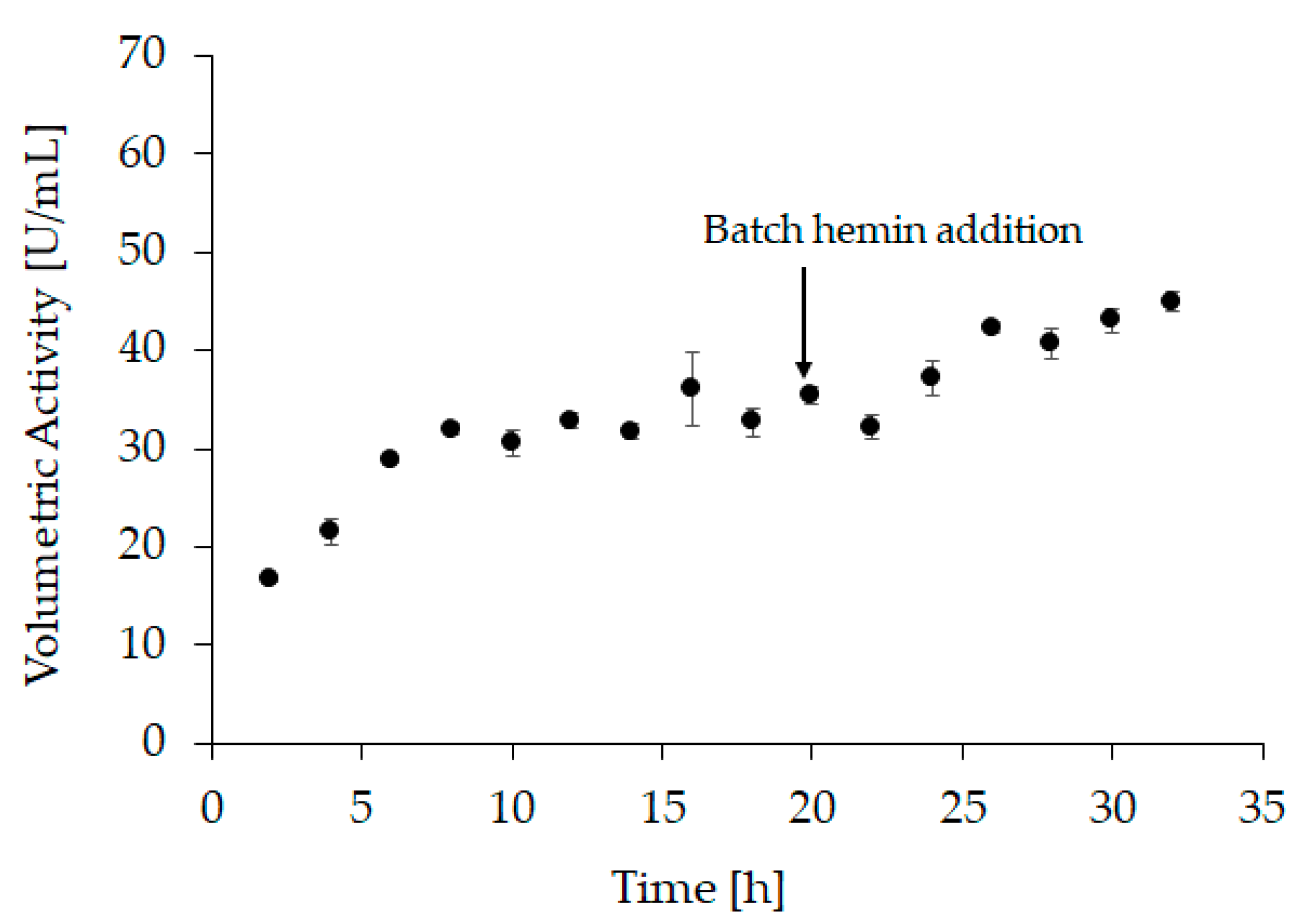
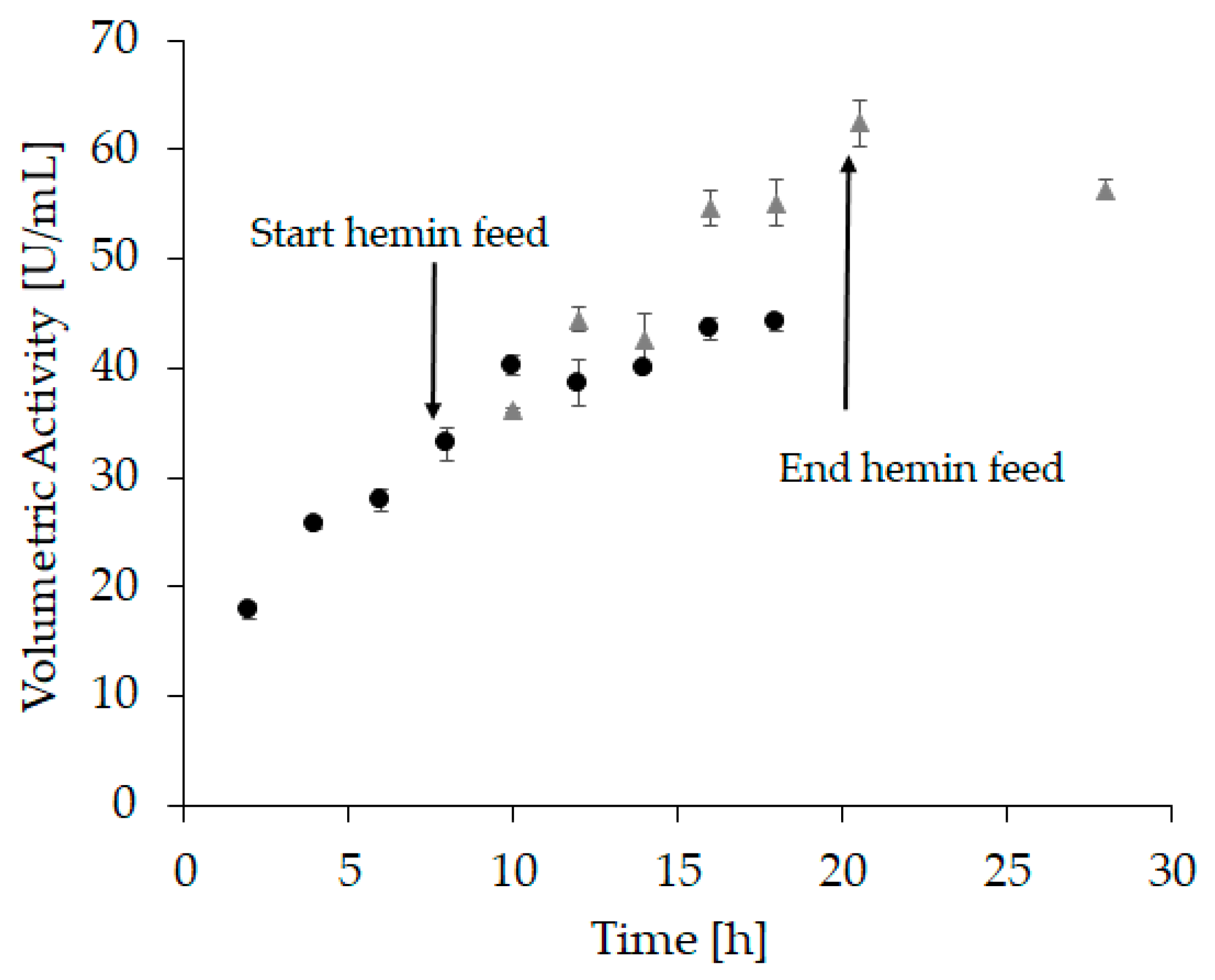
2.2. Hemin Addition
2.3. Capture and Concentration
2.4. pH 8.5 vs. pH 10
2.5. Characterization of Refolded HRP
2.6. Final HRP Production Process
3. Materials and Methods
3.1. Chemicals
3.2. Strain and Growth Conditions
3.3. Homogenization and Wash
3.4. Solubilization
3.5. Refolding
3.5.1. Small-Scale
3.5.2. DoE 1: Redox Conditions
3.5.3. DoE 2: Protein Concentration during Refolding
3.5.4. DoE 3: Redox Conditions and pH
3.5.5. DoE 4: Hemin Addition
3.6. Refolding Vessel
3.6.1. Refolding Vessel Set-Up
3.6.2. Refolding Vessel Experiment 1
3.6.3. Refolding Vessel Experiment 2
3.6.4. Refolding Vessel Experiment 3
3.7. Capture and Concentration
3.7.1. HIC Experiment 4 (Small-Scale)
3.7.2. HIC Experiment 5 (Scale-Up)
3.7.3. Analytics
- Vtotal … total volume in cuvette in (μL)
- ∆A/min … change in absorption (∆Abs 420 nm/min)
- Dilution … dilution of the sample
- Vsample … volume of sample (µL)
- d … length of the beam path through the cuvette (d = 0.58 cm)
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABTS | 2,2′-azino-bis (3-ethylbenzothiazoline-6-sulphonic acid) |
| CaCl2 | Calcium chloride |
| CCF | Central composite face centered |
| CEX | Cation exchange chromatography |
| CV | Column volumes |
| dO2 | Dissolved oxygen |
| DoE | Design of experiments |
| DSP | Downstream process |
| DTT | Dithiothreitol |
| GSSG | Glutathione disulfide |
| GuHCl | Guanidine hydrochloride |
| HIC | Hydrophobic interaction chromatography |
| HPLC | High-performance liquid chromatography |
| IB | Inclusion body |
| IMAC | Immobilized metal affinity chromatography |
| pHRP | Plant horseradish peroxidase |
| rHRP | Recombinant Horseradish Peroxidase |
| RP | Reversed-phase |
| RT | Room temperature |
| Rz | Reinheitszahl |
| SEC | Size exclusion chromatography |
| TFA | Trifluoroacetic acid |
| TMB | 3,3′,5,5′-tetramethylbenzidine |
| USP | Upstream process |
| wIB | Wet inclusion body |
References
- Krainer, F.W.; Glieder, A. An updated view on horseradish peroxidases: Recombinant production and biotechnological applications. Appl. Microbiol. Biotechnol. 2015, 99, 1611–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azevedo, A.M.; Martins, V.C.; Prazeres, D.M.; Vojinovic, V.; Cabral, J.M.; Fonseca, L.P. Horseradish peroxidase: A valuable tool in biotechnology. Biotechnol. Annu. Rev. 2003, 9, 1387–2656. [Google Scholar] [CrossRef]
- Grigorenko, V.; Chubar, T.; Kapeliuch, Y.; Börchers, T.; Spener, F.; Egorova, A. New approaches for functional expression of recombinant horseradish peroxidase C in Escherichia coli. Biocatal. Biotransform. 1999, 17, 359–379. [Google Scholar] [CrossRef]
- Grigorenko, V.G.; Andreeva, I.P.; Rubtsova, M.Y.; Egorov, A.M. Recombinant horseradish peroxidase: Production and analytical applications. Biochem. (Mosc.) 2015, 80, 408–416. [Google Scholar] [CrossRef]
- Spadiut, O.; Herwig, C. Production and purification of the multifunctional enzyme horseradish peroxidase. Pharm. Bioprocess 2013, 1, 283–295. [Google Scholar] [CrossRef] [Green Version]
- Veitch, N.C. Horseradish peroxidase: A modern view of a classic enzyme. Phytochemistry 2004, 65, 249–259. [Google Scholar] [CrossRef]
- Veitch, N.C.; Smith, A.T. Horseradish peroxidase. Adv. Inorg. Chem. 2000, 51, 107–162. [Google Scholar]
- Welinder, K.G. Covalent structure of the glycoprotein horseradish peroxidase (EC 1.11. 1.7). FEBS Lett. 1976, 72, 19–23. [Google Scholar] [CrossRef] [Green Version]
- Wuhrer, M.; Hokke, C.H.; Deelder, A.M. Glycopeptide analysis by matrix-assisted laser desorption/ionization tandem time-of-flight mass spectrometry reveals novel features of horseradish peroxidase glycosylation. Rapid Commun. Mass Spectrom. 2004, 18, 1741–1748. [Google Scholar] [CrossRef]
- Wuhrer, M.; Balog, C.I.; Koeleman, C.A.; Deelder, A.M.; Hokke, C.H. New features of site-specific horseradish peroxidase (HRP) glycosylation uncovered by nano-LC-MS with repeated ion-isolation/fragmentation cycles. Biochim. Biophys. Acta 2005, 1723, 229–239. [Google Scholar] [CrossRef]
- Berglund, G.I.; Carlsson, G.H.; Smith, A.T.; Szöke, H.; Henriksen, A.; Hajdu, J. The catalytic pathway of horseradish peroxidase at high resolution. Nature 2002, 417, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Van Gijlswijk, R.P.; Talman, E.G.; Peekel, I.; Bloem, J.; Van Velzen, M.A.; Heetebrij, R.J.; Tanke, H.J. Use of horseradish peroxidase-and fluorescein-modified cisplatin derivatives for simultaneous labeling of nucleic acids and proteins. Clin. Chem. 2002, 48, 1352–1359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yakovleva, J.; Davidsson, R.; Lobanova, A.; Bengtsson, M.; Eremin, S.; Laurell, T.; Emnéus, J. Microfluidic enzyme immunoassay using silicon microchip with immobilized antibodies and chemiluminescence detection. Anal. Chem. 2002, 74, 2994–3004. [Google Scholar] [CrossRef] [PubMed]
- Moody, M.; Arsdell, S.V.; Murphy, K.; Orencole, S.; Burns, C. Array-based ELISAs for high-throughput analysis of human cytokines. Biotechniques 2001, 31, 186–194. [Google Scholar] [CrossRef]
- Lomillo, M.A.A.; Ruiz, J.G.; Pascual, F.J.M. Biosensor based on platinum chips for glucose determination. Anal. Chim. Acta 2005, 547, 209–214. [Google Scholar] [CrossRef]
- Azevedo, A.M.; Prazeres, D.M.F.; Cabral, J.M.; Fonseca, L.P. Ethanol biosensors based on alcohol oxidase. Biosens. Bioelectron. 2005, 21, 235–247. [Google Scholar] [CrossRef]
- Raghu, P.; Reddy, T.M.; Reddaiah, K.; Jaidev, L.; Narasimha, G. A novel electrochemical biosensor based on horseradish peroxidase immobilized on Ag-nanoparticles/poly (l-arginine) modified carbon paste electrode toward the determination of pyrogallol/hydroquinone. Enzyme Microb. Technol. 2013, 52, 377–385. [Google Scholar] [CrossRef]
- Kubota, K.; Mizukoshi, T.; Miyano, H. A new approach for quantitative analysis of L-phenylalanine using a novel semi-sandwich immunometric assay. Anal. Bioanal. Chem. 2013, 405, 8093–8103. [Google Scholar] [CrossRef]
- Yang, H. Enzyme-based ultrasensitive electrochemical biosensors. Curr. Opin. Chem. Biol. 2012, 16, 422–428. [Google Scholar] [CrossRef]
- Litescu, S.C.; Eremia, S.; Radu, G.L. Biosensors for the determination of phenolic metabolites. In Bio-Farms for Nutraceuticals, 1st ed.; Giardi, M.T., Rea, G., Berra, B., Eds.; Springer: New York, NY, USA, 2010; Volume 698, pp. 234–240. [Google Scholar] [CrossRef]
- Vasileva, N.; Godjevargova, T.; Ivanova, D.; Gabrovska, K. Application of immobilized horseradish peroxidase onto modified acrylonitrile copolymer membrane in removing of phenol from water. Int. J. Biol. Macromol. 2009, 44, 190–194. [Google Scholar] [CrossRef]
- Tatsumi, K.; Wada, S.; Ichikawa, H. Removal of chlorophenols from wastewater by immobilized horseradish peroxidase. Biotechnol. Bioeng. 1996, 51, 126–130. [Google Scholar] [CrossRef]
- Bhunia, A.; Durani, S.; Wangikar, P.P. Horseradish peroxidase catalyzed degradation of industrially important dyes. Biotechnol. Bioeng. 2001, 72, 562–567. [Google Scholar] [CrossRef]
- Colonna, S.; Gaggero, N.; Carrea, G.; Pasta, P. Horseradish peroxidase catalysed sulfoxidation is enantioselective. J. Chem. Soc. Chem. Commun. 1992, 357–358. [Google Scholar] [CrossRef]
- Colonna, S.; Gaggero, N.; Richelmi, C.; Pasta, P. Recent biotechnological developments in the use of peroxidases. Trends Biotechnol. 1999, 17, 163–168. [Google Scholar] [CrossRef]
- Kalliney, S.; Zaks, A. An efficient peroxidase-catalyzed oxidation of hydroxylaminoeverninomicin in aqueous-organic media. Tetrahedron Lett. 1995, 36, 4163–4166. [Google Scholar] [CrossRef]
- Kalra, B.; Gross, R.A. Horseradish peroxidase mediated free radical polymerization of methyl methacrylate. Biomacromolecules 2000, 1, 501–505. [Google Scholar] [CrossRef]
- Singh, A.; Ma, D.; Kaplan, D.L. Enzyme-mediated free radical polymerization of styrene. Biomacromolecules 2000, 1, 592–596. [Google Scholar] [CrossRef]
- Sakai, S.; Khanmohammadi, M.; Khoshfetrat, A.B.; Taya, M. Horseradish peroxidase-catalyzed formation of hydrogels from chitosan and poly (vinyl alcohol) derivatives both possessing phenolic hydroxyl groups. Carbohydr. Polym. 2014, 111, 404–409. [Google Scholar] [CrossRef]
- Saikrishnan, D.; Goyal, M.; Rossiter, S.; Kukol, A. A cellulose-based bioassay for the colorimetric detection of pathogen DNA. Anal. Bioanal. Chem. 2014, 406, 7887–7898. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Wang, M.; Li, B.; Yang, Z.; Zhou, Y.; Ai, S. A sensitive electrochemical biosensor for detection of protein kinase A activity and inhibitors based on Phos-tag and enzymatic signal amplification. Biosens. Bioelectron. 2015, 63, 26–32. [Google Scholar] [CrossRef]
- Wardman, P. Indole-3-acetic acids and horseradish peroxidase: A new prodrug/enzyme combination for targeted cancer therapy. Curr. Pharm. Des. 2002, 8, 1363. [Google Scholar] [CrossRef] [PubMed]
- Tupper, J.; Tozer, G.M.; Dachs, G.U. Use of horseradish peroxidase for gene-directed enzyme prodrug therapy with paracetamol. Br. J. Cancer 2004, 90, 1858–1862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, S.A. Appropriate glycosylation of recombinant proteins for human use. Mol. Biotechnol. 2004, 28, 241–255. [Google Scholar] [CrossRef]
- Kaur, J.; Kumar, A.; Kaur, J. Strategies for optimization of heterologous protein expression in E. coli: Roadblocks and reinforcements. Int. J. Biol. Macromol. 2017, 106, 803–822. [Google Scholar] [CrossRef]
- Slouka, C.; Kopp, J.; Hutwimmer, S.; Strahammer, M.; Strohmer, D.; Eitenberger, E.; Schwaighofer, A.; Herwig, C. Custom made inclusion bodies: Impact of classical process parameters and physiological parameters on inclusion body quality attributes. Microb. Cell Fact. 2018, 17, 148. [Google Scholar] [CrossRef] [PubMed]
- Eggenreich, B.; Willim, M.; Wurm, D.J.; Herwig, C.; Spadiut, O. Production strategies for active heme-containing peroxidases from E. coli inclusion bodies–a review. Biotechnol. Rep. 2016, 10, 75–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinas, U.; Garcia-Fruitós, E.; Corchero, J.L.; Vázquez, E.; Seras-Franzoso, J.; Villaverde, A. Bacterial inclusion bodies: Discovering their better half. Trends Biochem. Sci. 2017, 42, 726–737. [Google Scholar] [CrossRef]
- Humer, D.; Spadiut, O. Wanted: More monitoring and control during inclusion body processing. World J. Microb. Biot. 2018, 34, 158. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.T.; Santama, N.; Dacey, S.; Edwards, M.; Bray, R.C.; Thorneley, R.; Burke, J.F. Expression of a synthetic gene for horseradish peroxidase C in Escherichia coli and folding and activation of the recombinant enzyme with Ca2+ and heme. J. Biol. Chem. 1990, 265, 13335–13343. [Google Scholar]
- Asad, S.; Dabirmanesh, B.; Ghaemi, N.; Etezad, S.M.; Khajeh, K. Studies on the refolding process of recombinant horseradish peroxidase. Mol. Biotechnol. 2013, 54, 484–492. [Google Scholar] [CrossRef]
- Gazaryan, I.G.; Doseeva, V.V.; Galkin, A.G.; Tishkov, V.I. Effect of single-point mutations Phe41→ His and Phe143→ Glu on folding and catalytic properties of recombinant horseradish peroxidase expressed in E. coli. FEBS Lett. 1994, 354, 248–250. [Google Scholar] [CrossRef] [Green Version]
- Gazaryan, I.; Chubar, T.; Ignatenko, O.; Mareeva, E.; Orlova, M.; Kapeliuch, Y.L.; Savitsky, P.; Rojkova, A.; Tishkov, V. Tryptophanless recombinant horseradish peroxidase: Stability and catalytic properties. Biochem. Biophys. Res. Commun. 1999, 262, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Gundinger, T.; Spadiut, O. A comparative approach to recombinantly produce the plant enzyme horseradish peroxidase in Escherichia coli. J. Biotechnol. 2017, 248, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Song, B.K.; Kim, Y.H. Optimized refolding and characterization of S-peroxidase (CWPO_C of Populus alba) expressed in E. coli. Protein Expr. Purif. 2011, 80, 268–273. [Google Scholar] [CrossRef]
- Shigeto, J.; Itoh, Y.; Tsutsumi, Y.; Kondo, R. Identification of Tyr74 and Tyr177 as substrate oxidation sites in cationic cell wall-bound peroxidase from Populus alba L. FEBS J. 2012, 279, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Shigeto, J.; Nagano, M.; Fujita, K.; Tsutsumi, Y. Catalytic profile of Arabidopsis peroxidases, AtPrx-2, 25 and 71, contributing to stem lignification. PLoS ONE 2014, 9, e105332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakharova, G.; Poloznikov, A.; Chubar, T.; Gazaryan, I.; Tishkov, V. High-yield reactivation of anionic tobacco peroxidase overexpressed in Escherichia coli. Protein Expression Purif. 2015, 113, 85–93. [Google Scholar] [CrossRef]
- Fattahian, Y.; Riahi-Madvar, A.; Mirzaee, R.; Torkzadeh-Mahani, M.; Asadikaram, G. Heterologous expression, purification and characterization of a peroxidase isolated from Lepidium draba. Protein J. 2017, 36, 461–471. [Google Scholar] [CrossRef]
- Hushpulian, D.; Savitski, P.; Rojkova, A.; Chubar, T.; Fechina, V.; Sakharov, I.Y.; Lagrimini, L.; Tishkov, V.; Gazaryan, I. Expression and refolding of tobacco anionic peroxidase from E. coli inclusion bodies. Biochem. (Mosc) 2003, 68, 1189–1194. [Google Scholar] [CrossRef]
- Teilum, K.; Østergaard, L.; Welinder, K.G. Disulfide Bond Formation and Folding of Plant Peroxidases Expressed as Inclusion Body Protein in Escherichia coli Thioredoxin Reductase Negative Strains. Protein Expr. Purif. 1999, 15, 77–82. [Google Scholar] [CrossRef]
- Doyle, W.A.; Smith, A.T. Expression of lignin peroxidase H8 in Escherichia coli: Folding and activation of the recombinant enzyme with Ca2+ and haem. Biochem. J. 1996, 315, 15–19. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Boada, M.; Doyle, W.; Ruiz-Dueñas, F.; Martınez, M.; Martınez, A.; Smith, A. Expression of Pleurotus eryngii versatile peroxidase in Escherichia coli and optimisation of in vitro folding. Enzyme Microb. Technol. 2002, 30, 518–524. [Google Scholar] [CrossRef]
- Nie, G.; Reading, N.S.; Aust, S.D. Expression of the lignin peroxidase h2 gene fromphanerochaete chrysosporiuminescherichia coli. Biochem. Biophys. Res. Commun. 1998, 249, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Coupe, E.; Smyth, M.; Fosberry, A.; Hall, R.; Littlechild, J. The dodecameric vanadium-dependent haloperoxidase from the marine algae Corallina officinalis: Cloning, expression, and refolding of the recombinant enzyme. Protein Expression Purif. 2007, 52, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Miki, Y.; Morales, M.; Ruiz-Dueñas, F.J.; Martínez, M.J.; Wariishi, H.; Martínez, A.T. Escherichia coli expression and in vitro activation of a unique ligninolytic peroxidase that has a catalytic tyrosine residue. Protein Expression Purif. 2009, 68, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Linde, D.; Coscolín, C.; Liers, C.; Hofrichter, M.; Martínez, A.T.; Ruiz-Dueñas, F.J. Heterologous expression and physicochemical characterization of a fungal dye-decolorizing peroxidase from Auricularia auricula-judae. Protein Expression Purif. 2014, 103, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Ren, K.; Jia, R.; Chen, W.; Sun, R. Expression of a fungal manganese peroxidase in Escherichia coli: A comparison between the soluble and refolded enzymes. BMC Biotech. 2016, 16, 87. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.-H.; Kim, D.-H. Heterologous expression of lignin peroxidase H2 in Escherichia coli: In vitro refolding and activation. BMB Rep. 1999, 32, 486–491. [Google Scholar]
- Whitwam, R.; Tien, M. Heterologous expression and reconstitution of fungal Mn peroxidase. Arch. Biochem. Biophys. 1996, 333, 439–446. [Google Scholar] [CrossRef]
- Rodríguez-Cabrera, N.A.; Regalado, C.; García-Almendárez, B.E. Cloning, heterologous expression and properties of a recombinant active turnip peroxidase. J. Agric. Food Chem. 2011, 59, 7120–7126. [Google Scholar] [CrossRef]
- Dong, X.-Y.; Huang, Y.; Sun, Y. Refolding kinetics of denatured-reduced lysozyme in the presence of folding aids. J. Biotechnol. 2004, 114, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Pappa, H.S.; Cass, A.E. A step towards understanding the folding mechanism of horseradish peroxidase: Tryptophan fluorescence and circular dichroism equilibrium studies. Eur. J. Biochem. 1993, 212, 227–235. [Google Scholar] [CrossRef] [PubMed]
- DeLisa, M.P.; Li, J.; Rao, G.; Weigand, W.A.; Bentley, W.E. Monitoring GFP-operon fusion protein expression during high cell density cultivation of Escherichia coli using an on-line optical sensor. Biotechnol. Bioeng. 1999, 65, 54–64. [Google Scholar] [CrossRef]
- Childs, R.E.; Bardsley, W.G. The steady-state kinetics of peroxidase with 2, 2′-azino-di-(3-ethyl-benzthiazoline-6-sulphonic acid) as chromogen. Biochem. J. 1975, 145, 93–103. [Google Scholar] [CrossRef]
- Humer, D.; Spadiut, O. Improving the Performance of Horseradish Peroxidase by Site-Directed Mutagenesis. Int. J. Mol. Sci. 2019, 20, 916. [Google Scholar] [CrossRef] [Green Version]
- Josephy, P.D.; Eling, T.; Mason, R.P. The horseradish peroxidase-catalyzed oxidation of 3, 5, 3’, 5’-tetramethylbenzidine. Free radical and charge-transfer complex intermediates. J. Biol. Chem. 1982, 257, 3669–3675. [Google Scholar]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Yield (% or mg/L Culture) | Specific Activity (U/mg) | Reference |
|---|---|---|---|
| HRP | 3% | 630 U/mg (ABTS) 14500 U/mg (pyrogallol) | Smith et al. [40] |
| HRP | 6-8 mg/L | 1160 U/mg (ABTS) | Grigorenko et al. [3] |
| HRP | 24% | 10 U/mg (4-aminoantipyrine) | Asad et al. [41] |
| HRP | 16.7 mg/L | 4000 U/mg (ABTS) | Gazaryan et al. [42] |
| HRP | 20 mg/L | 2000 U/mg (ABTS) | Gazaryan et al. [43] |
| HRP | 15 mg/L | 62.5 U/mg (ABTS) | Gundinger et al. [44] |
| CWPO_C 1 | 0.2 mg/L | 1000 U/mg (ABTS) | Kim et al. [45] |
| CWPO_C 1 | 27.3% | 1066 U/mg (syringaldazine) 120 U/mg (guaiacol) | Shigeto et al. [46] |
| rAtPrx71 | 28% | 1291 U/mg (syringaldazine) | Shigeto et al. [47] |
| rAtPrx25 | 30.3% | 270 U/mg (guaiacol) | Shigeto et al. [47] |
| TOP 2 | 79 mg/L | 2950 U/mg (ABTS) | Zakharova et al. [48] |
| LDP 3 | 16.8 mg/L | 70.7 U/mg (TMB) 580.7 U/mg (H2O2) | Fattahian et al. [49] |
| TOP 2 | 4.6 mg/L | 1100 U/mg (ABTS) | Hushpulian et al. [50] |
| ATP N 4 | 13 mg/L | n.m. | Teilum et al. [51] |
| BP1 5 | 9.4 mg/L | n.m. | Teilum et al. [51] |
| LiP H8 6 | 1% | 39 µmol of veratryl alcohol ox/min/mg of protein | Doyle et al. [52] |
| VPL2 7 | 5.5 mg/L | n.m. | Pérez-Boada et al. [53] |
| LiP H2 6 | 3.4 mg/L | n.m. | Nie et al. [54] |
| VBPO 8 | 40 mg/L | 550 U/mg (bromination of monochlorodimedone) | Coupe et al. [55] |
| LiP 6 | 0.38 mg/L | 16,300 U/mg (ABTS) | Miki et al. [56] |
| DyP 9 | 1.5 mg/L | 247 U/mg (ABTS) | Linde et al. [57] |
| MnP 10 | 2.4% | 12.9 U/mg (oxidation of Mn2+ to Mn3+) | Wang et al. [58] |
| LiP H2 6 | 2.4% | 55.6 U/mg (veratryl alcohol) | Lee et al. [59] |
| MnP 10 | 0.275 mg/L | 140 U/mg (oxidation of Mn2+ to Mn3+) | Whitwam et al. [60] |
| BnPA 11 | 29 mg/L | 981 U/mg (ABTS) | Rodríguez-Cabrera et al. [61] |
| Unit Operation | Parameters | Range |
|---|---|---|
| Solubilization | DTT | 2.5 mM–28.44 mM |
| Protein concentration | 20 g/L–80 g/L | |
| pH | 7–10 | |
| Refolding | GSSG | 0.4 mM–3.5 mM |
| Protein concentration | 0.5 g/L–2 g/L | |
| pH | 7–10 | |
| Time of hemin addition | 0 h–24 h after refolding start | |
| Hemin concentration | 6 µM–80 µM | |
| Salt precipitation | Type of salt | NaCl, (NH4)2SO4 |
| Salt concentration | 0 M–4 M | |
| Capture step HIC | Hydrophobicity of resin | Octyl, Butyl, Phenyl |
| pH value (load) | 8.5, 10 | |
| Type of elution | Step gradient, linear gradient |
| Protein Conc. (g/L) | Optimized DTT (mM) | Optimized GSSG (mM) | Specific Activity (U/mg) | Volumetric Activity (U/mL) |
|---|---|---|---|---|
| 0.5 (93.2%) | 17.19 (1.5%) | 2.16 (5.3%) | 45.1 | 22.5 |
| 1 (91.5%) | 13.49 (1.8%) | 2.17 (6.7%) | 26.6 | 26.6 |
| 1.5 (88.5%) | 9.83 (2.5%) | 2.18 (9.0%) | 18.9 | 28.4 |
| 2 (81.0%) | 7.11 (6.0%) | 2.21 (13.0%) | 13.7 | 27.3 |
| Salt | Volumetric Activity (U/mL) | Protein Conc. (g/L) | Specific Activity (U/mg) | Purification Factor |
|---|---|---|---|---|
| (NH4)2SO4 1 M | 38.0 | 0.12 | 317 | 2.5 |
| NaCl 4 M | 44.3 | 0.09 | 492 | 4.5 |
| Volume (mL) | Protein Conc. (mg/mL) | Specific Activity (U/mg) | Purification Factor | |
|---|---|---|---|---|
| Refolding end | n.a. | 0.51 | 126 | 1 |
| Load (after salt precipitation) | 50 | 0.09 | 726 | 5.8 |
| Active HRP fraction | 4 | 0.50 | 1176 | 9.4 |
| Process Variables | pH 8.5 | pH 10 |
|---|---|---|
| Specific activity (U/mg) | 1507 ± 13 | 1468 ± 24 |
| Purity SEC-HPLC (%) | ≥99 | ≥99 |
| Refolding yield (%) | 44 | 74 |
| Pure HRP/L culture medium (mg) | 562 | 959 |
| Rz | 3.7 | 4.3 |
| Total Units/refolding vessel (7 mM ABTS) | 146700 | 209500 |
| Overall yield active HRP per 100 mg expressed protein (mg) | 19 | 28 |
| ABTS | ||||
| Vmax (U/mg) | Km (mM) | kcat (s−1) | kcat/Km (mM−1·s−1) | |
| pHRP | 1285 ± 70 | 0.70 ± 0.14 | 734 ± 41 | 1043 ± 215 |
| rHRP | 1411 ± 43 | 0.49 ± 0.06 | 823 ± 25 | 1677 ± 205 |
| TMB | ||||
| Vmax (U/mg) | Km (mM) | kcat (s−1) | kcat/Km (mM−1·s−1) | |
| pHRP | 7446 ± 528 | 0.101 ± 0.020 | 4343 ± 308 | 42830 ± 8864 |
| rHRP | 7146 ± 355 | 0.105 ± 0.014 | 4169 ± 207 | 39582 ± 5661 |
| Unit operation. | Parameters | Final Conditions |
|---|---|---|
| Solubilization | DTT | 7.11 mM |
| Protein concentration | 20 g/L | |
| pH | 10 | |
| Refolding | GSSG | 1.27 mM |
| Protein concentration | 0.5 g/L | |
| pH | 10 | |
| Time of hemin addition | 8 h after refolding start | |
| Hemin concentration | 20 µM | |
| Salt precipitation | pH | Adjust to pH 8.5 with 2 M HCl |
| Type of salt | NaCl | |
| Salt concentration | 4 M (pH 8.5) | |
| Capture step HIC | Hydrophobicity of resin | Butyl |
| pH value (load) | 8.5 | |
| Type of elution | Step gradient |
| DTT conc. (mM in Solubilizate) | GSSG (mM in Refolding Buffer) |
|---|---|
| 2.5 | 0.5 |
| 8.75 | 2 |
| 15 | 3.5 |
| DTT (mM in Solubilizate) | GSSG (mM in Refolding Buffer) | Protein in the Refolding Mix (g/L) |
|---|---|---|
| 7.11 | 1.27 | 0.5 |
| 14.22 | 2.54 | 1 |
| 28.44 | 5.08 | 2 |
| DTT (mM in Solubilizate) | GSSG (mM in Refolding Buffer) | pH |
|---|---|---|
| 2.5 | 0.4 | 7 |
| 7.11 | 1.27 | 8.5 |
| 11.72 | 3.01 | 10 |
| Hemin Addition (Time after Refolding Start) (h) | Final Hemin Concentration (µM) |
|---|---|
| 0 | 6 |
| 6 | 20 |
| 12 | 40 |
| 24 | 80 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Humer, D.; Ebner, J.; Spadiut, O. Scalable High-Performance Production of Recombinant Horseradish Peroxidase from E. coli Inclusion Bodies. Int. J. Mol. Sci. 2020, 21, 4625. https://doi.org/10.3390/ijms21134625
Humer D, Ebner J, Spadiut O. Scalable High-Performance Production of Recombinant Horseradish Peroxidase from E. coli Inclusion Bodies. International Journal of Molecular Sciences. 2020; 21(13):4625. https://doi.org/10.3390/ijms21134625
Chicago/Turabian StyleHumer, Diana, Julian Ebner, and Oliver Spadiut. 2020. "Scalable High-Performance Production of Recombinant Horseradish Peroxidase from E. coli Inclusion Bodies" International Journal of Molecular Sciences 21, no. 13: 4625. https://doi.org/10.3390/ijms21134625
APA StyleHumer, D., Ebner, J., & Spadiut, O. (2020). Scalable High-Performance Production of Recombinant Horseradish Peroxidase from E. coli Inclusion Bodies. International Journal of Molecular Sciences, 21(13), 4625. https://doi.org/10.3390/ijms21134625