Complete Mitogenomes of Three Carangidae (Perciformes) Fishes: Genome Description and Phylogenetic Considerations



Abstract
:
1. Introduction
2. Results and Discussion
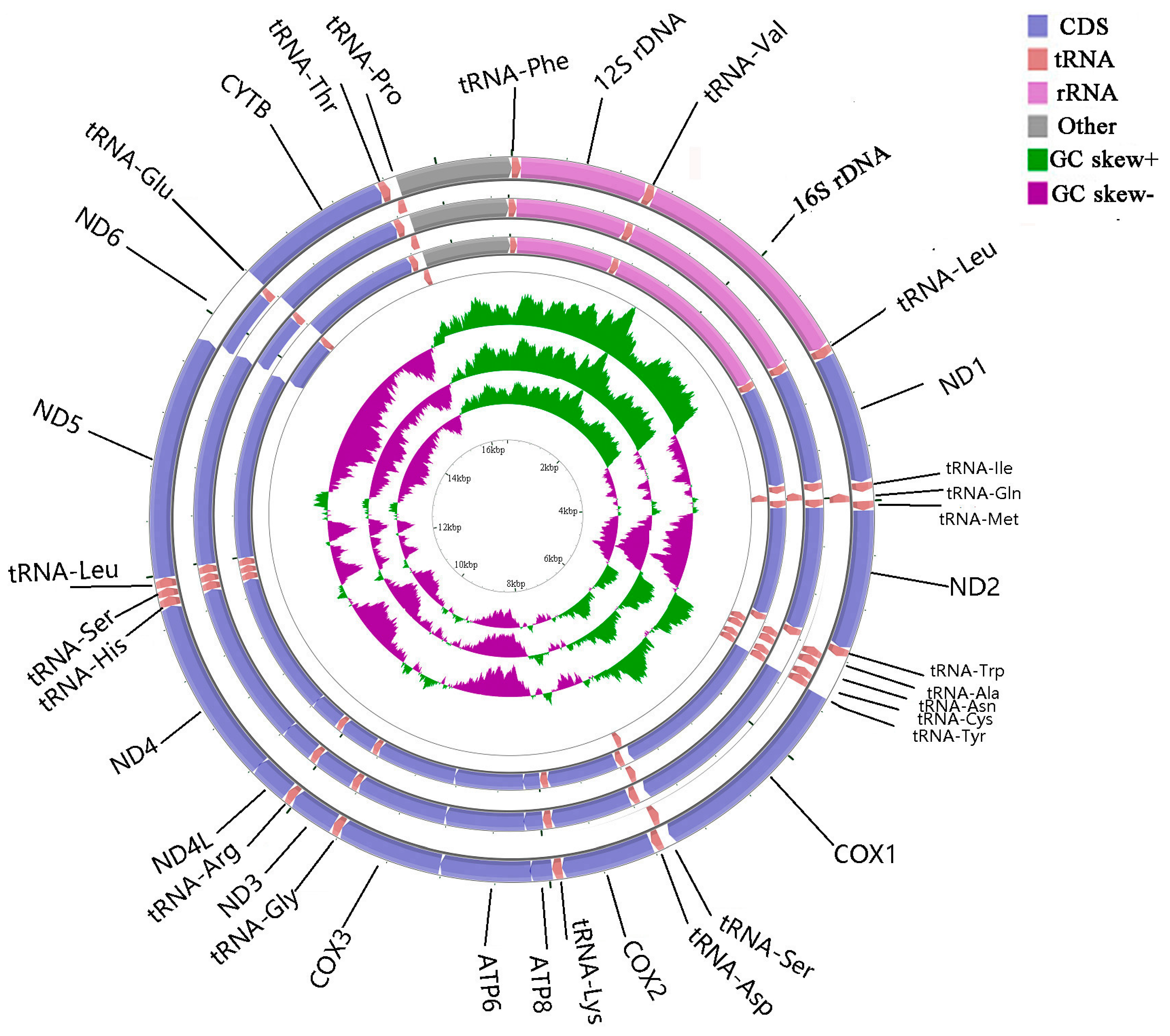
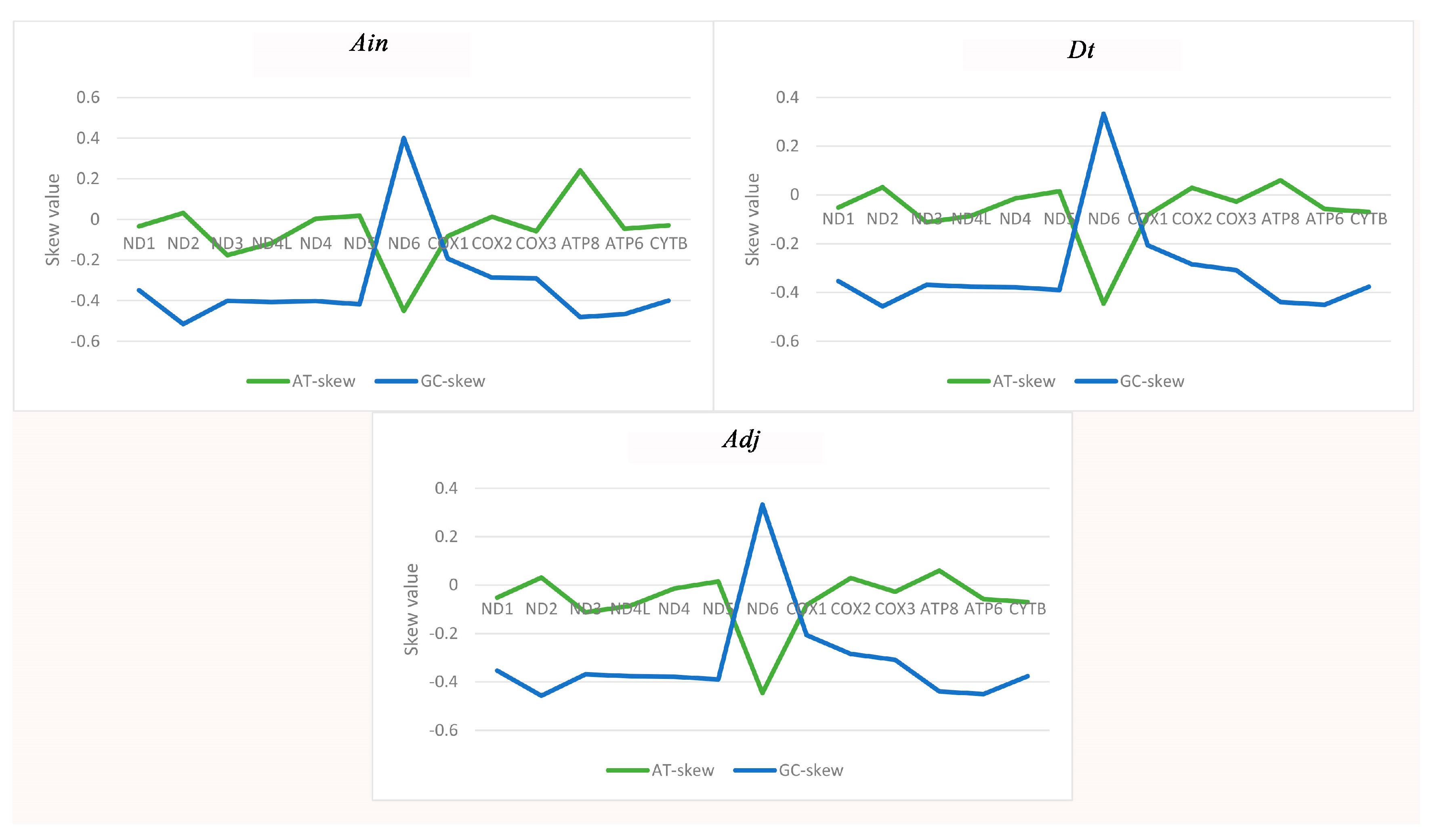
2.1. Genome Organization and Base Composition
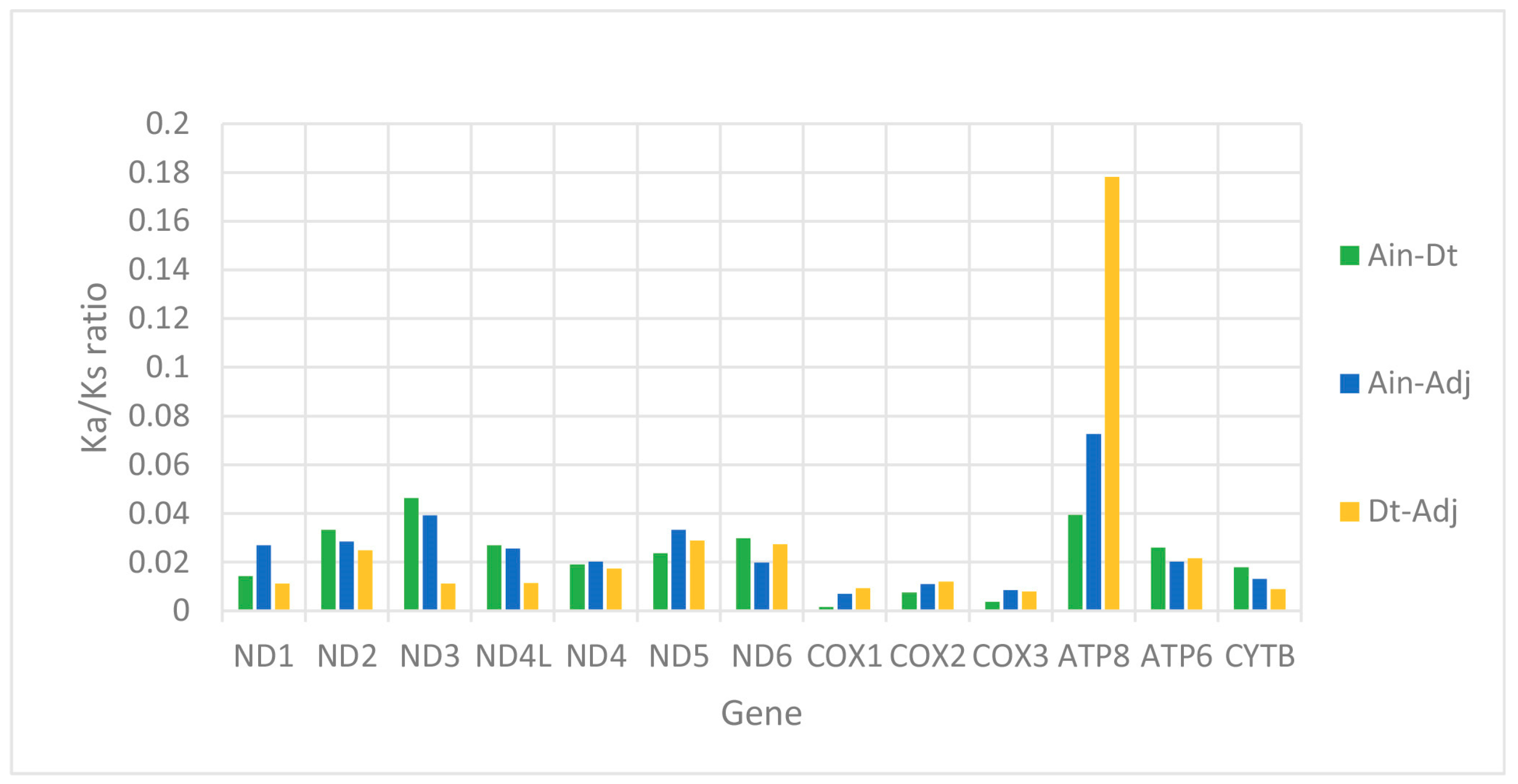
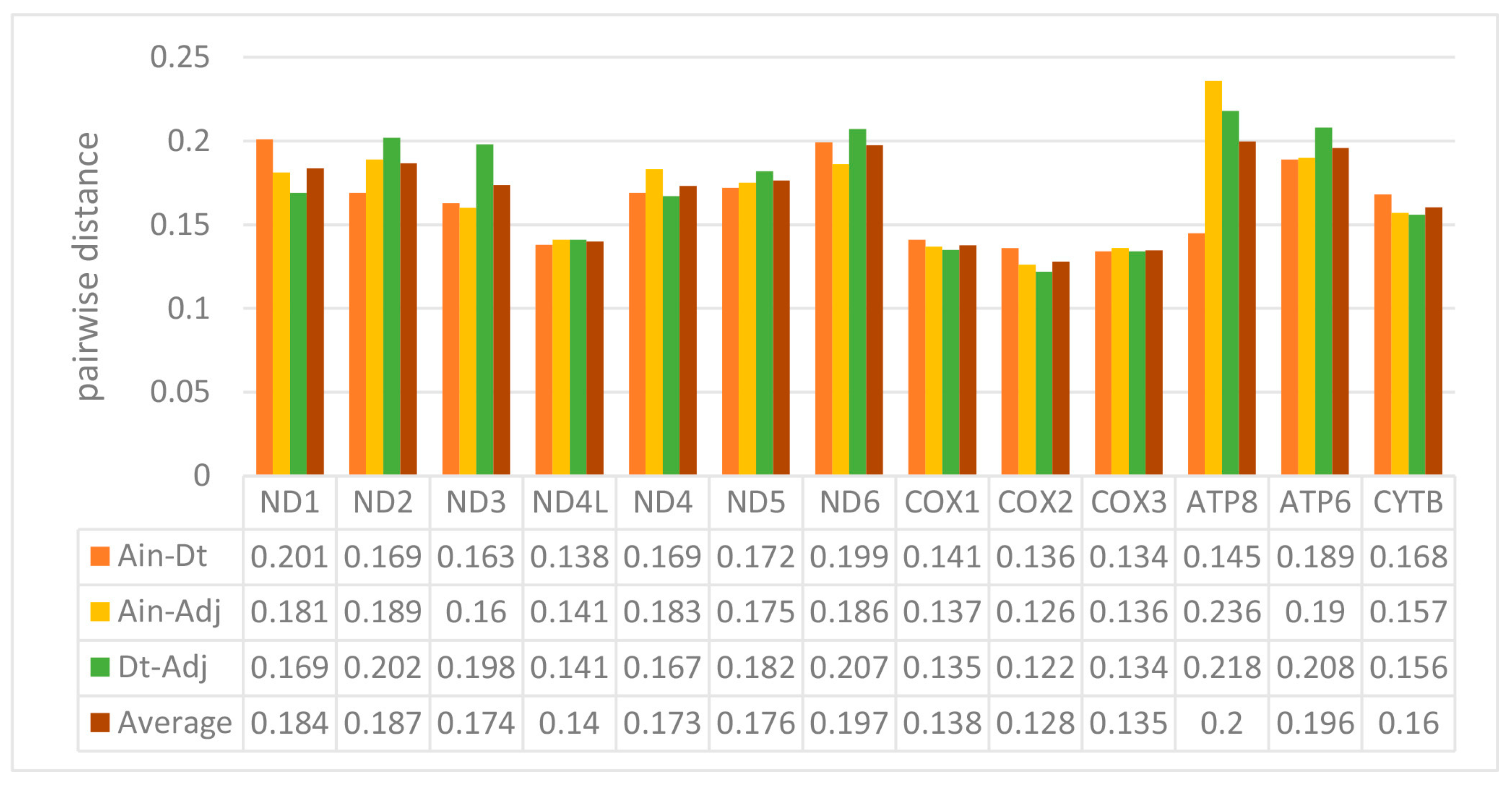
2.2. Protein-Coding Genes (PCGs)
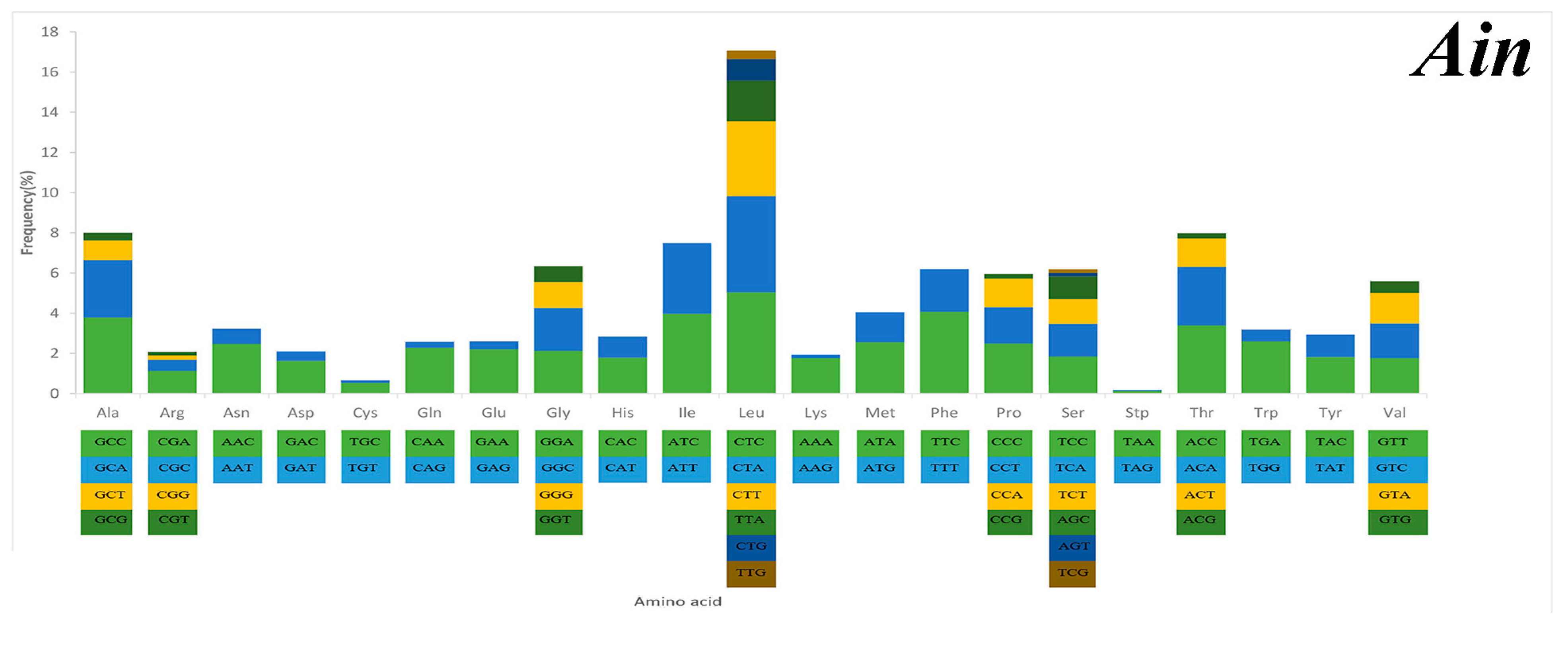
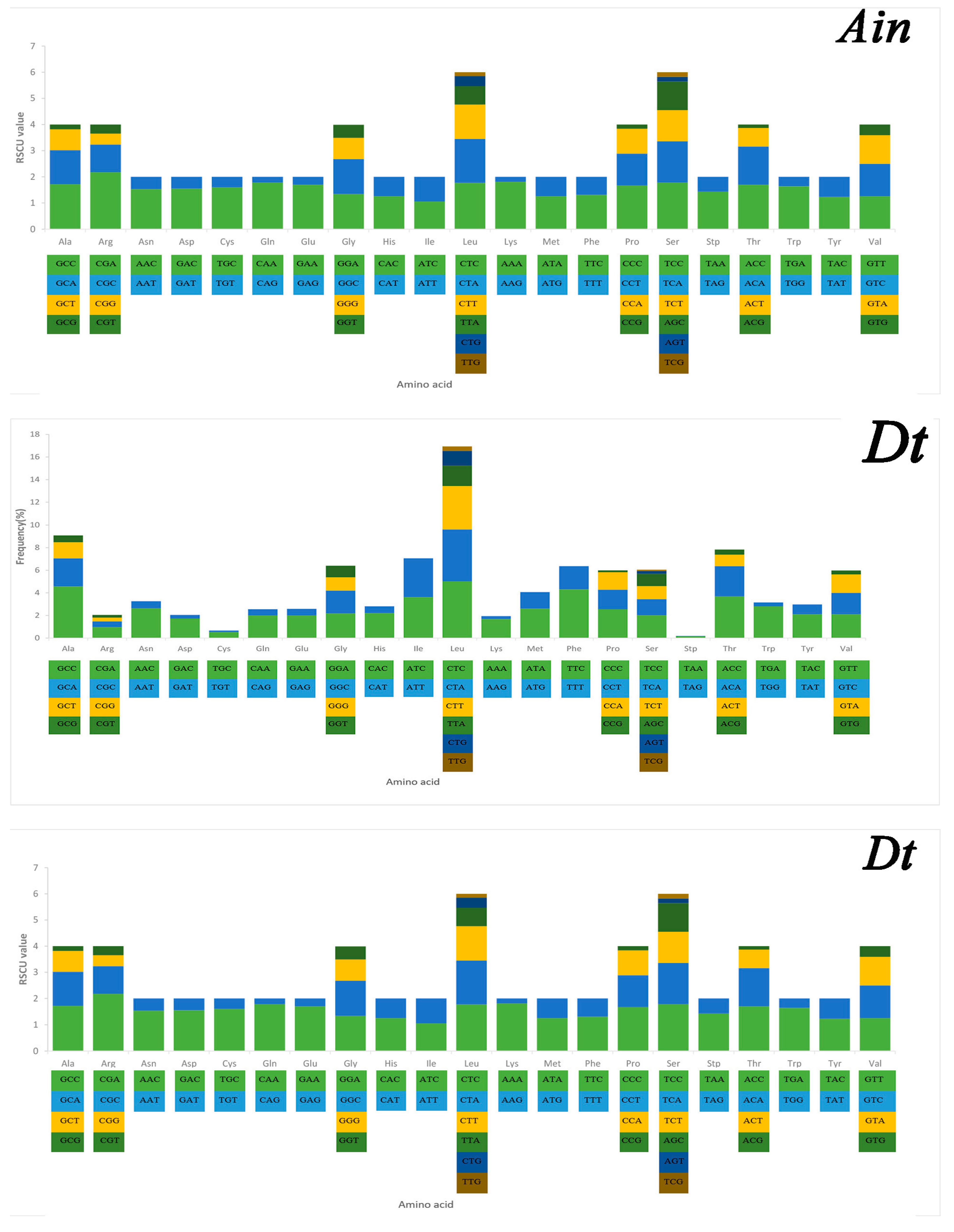
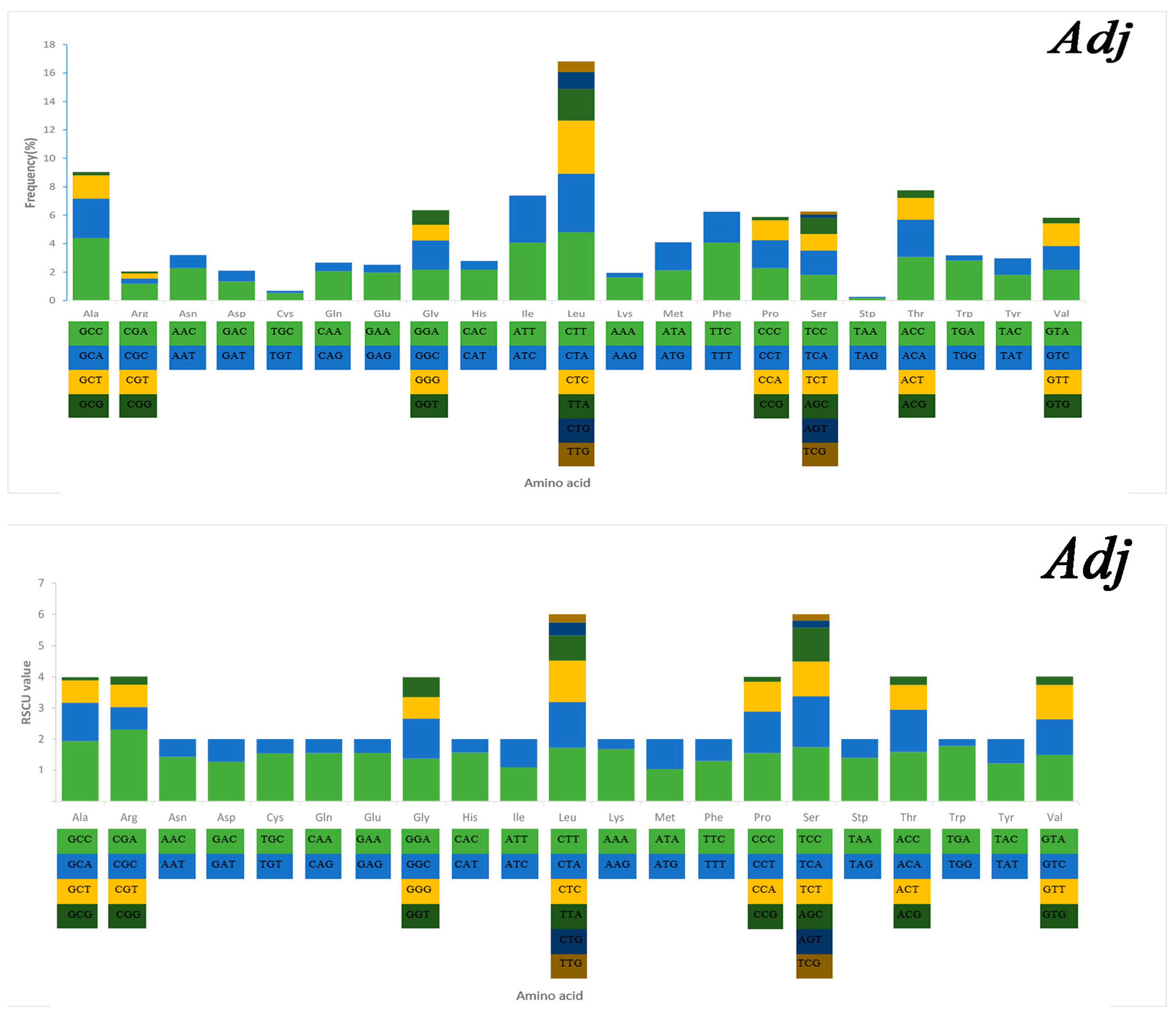
2.3. Mitochondrial Gene Codon Usage
2.4. Ribosomal and Transfer RNA Genes
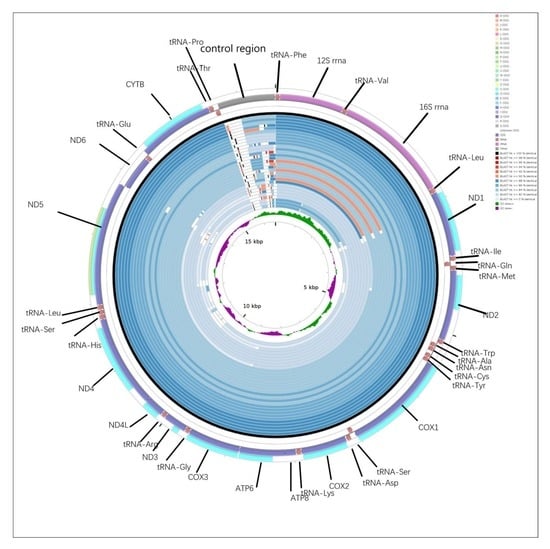
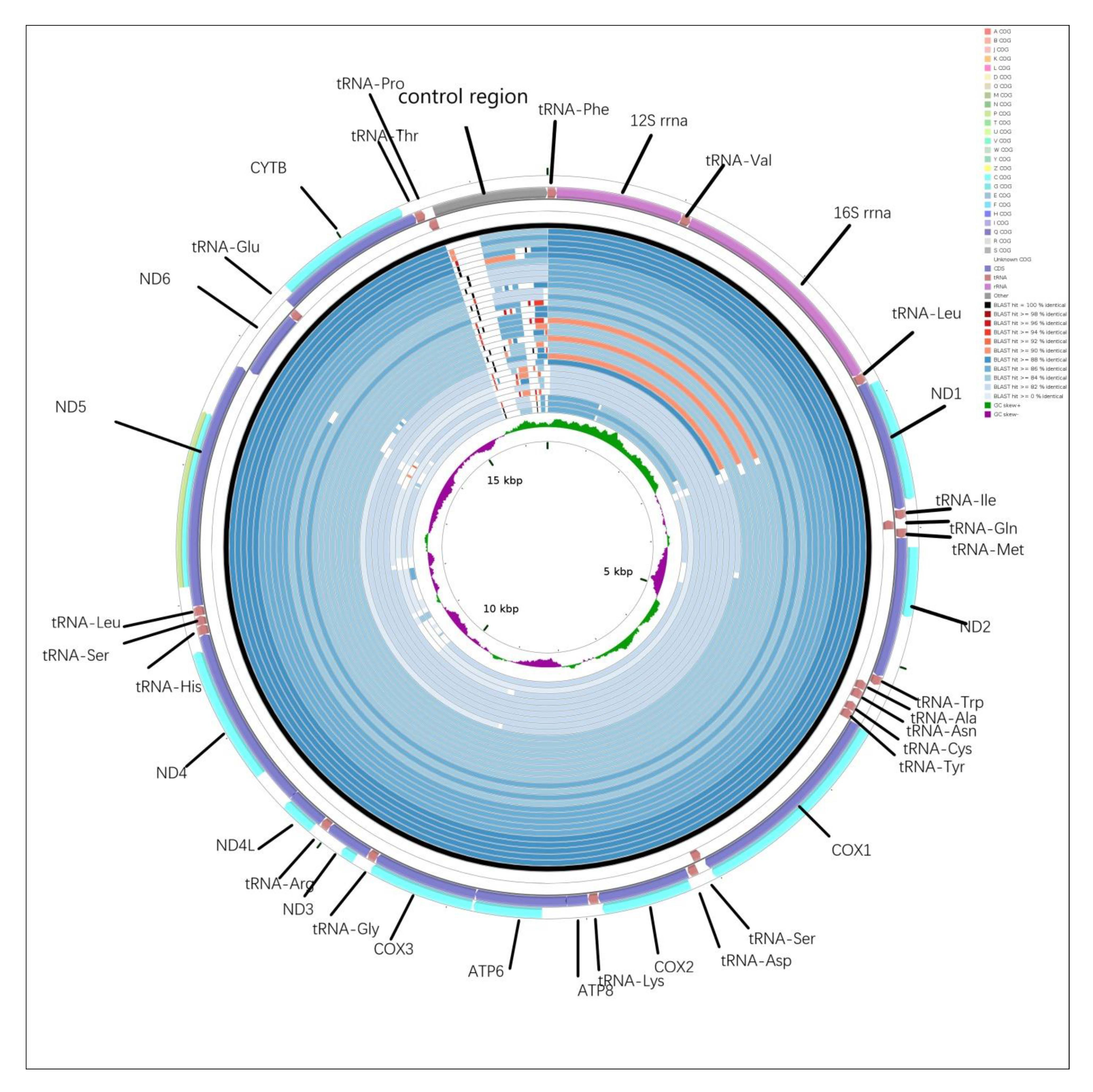
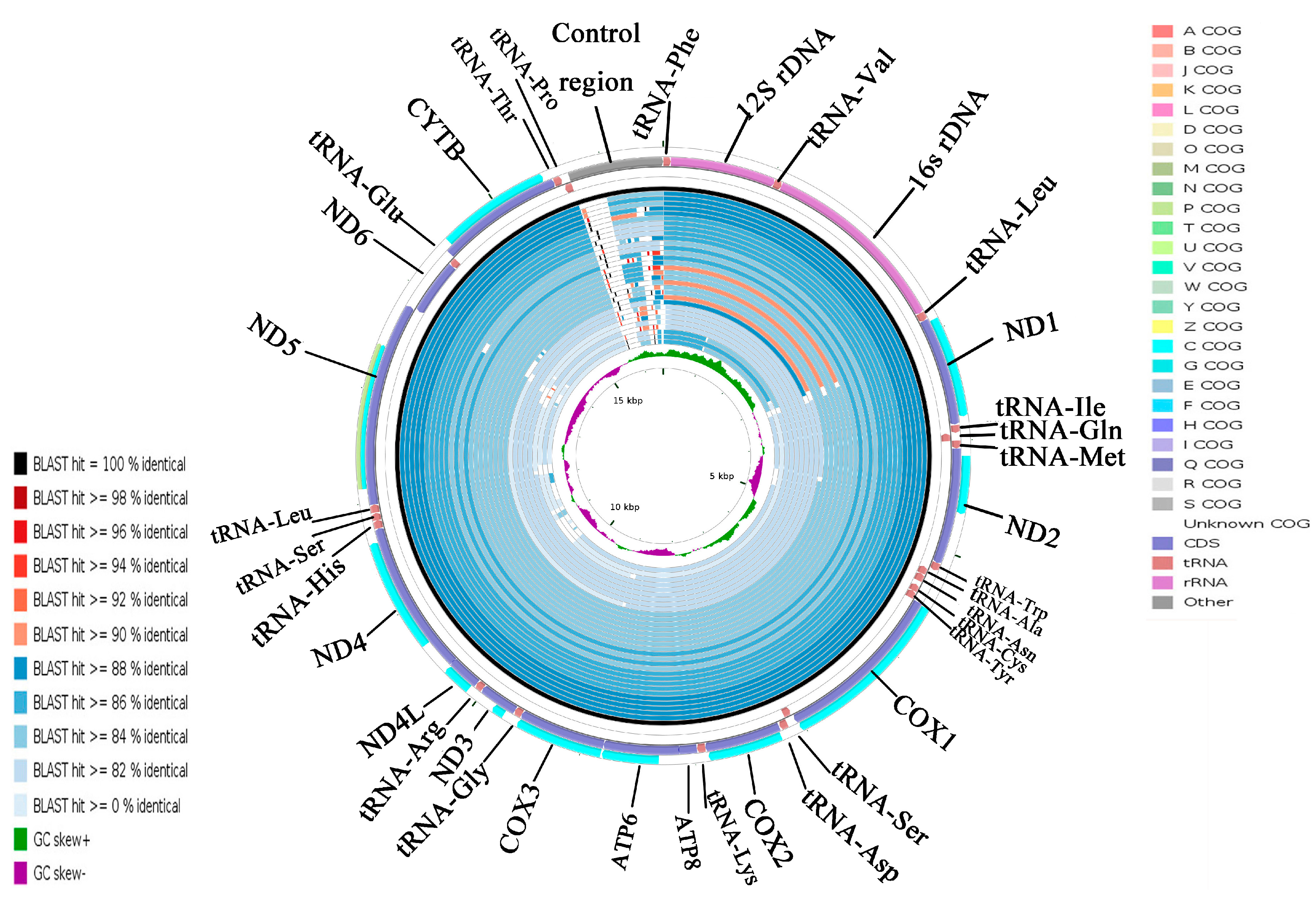
2.5. CGView Comparison Tool (CCT) Map
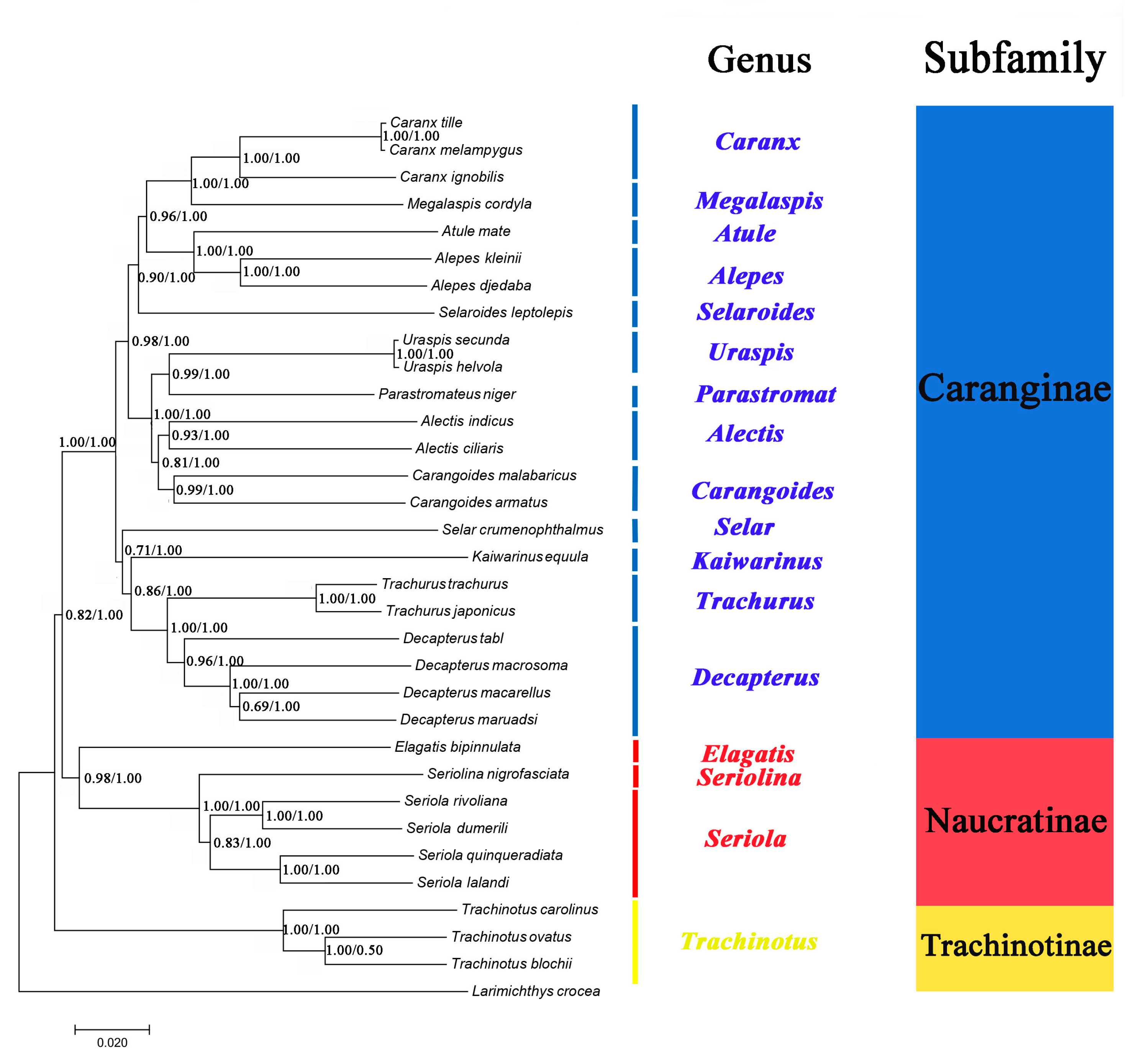
2.6. Phylogenetic Analyses
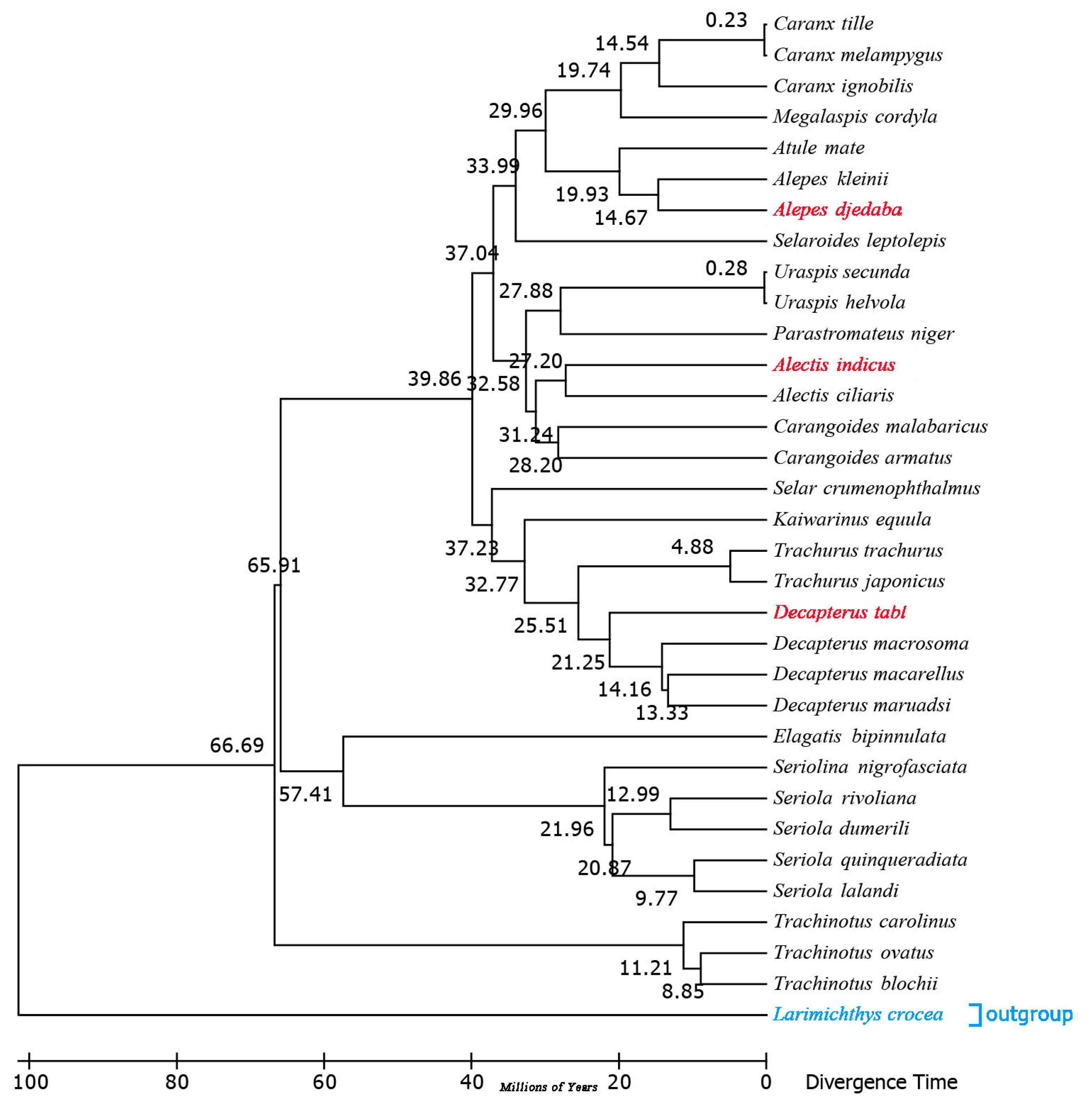
2.7. Estimation of Divergence Times
3. Materials and Methods
3.1. Sample Collection and DNA Extraction
3.2. Sequencing, Assembly, and Annotation
3.3. Sequence Analyses
3.4. Phylogenetic Analyses
3.5. Divergence Times
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolstenholme, D.R. Animal Mitochondrial DNA: Structure and Evolution. Int. Rev. Cytol. 1992, 141, 173–216. [Google Scholar] [CrossRef]
- Ceruso, M.; Mascolo, C.; Anastasio, A.; Pepe, T.; Sordino, P. Frauds and fish species authentication: Study of the complete mitochondrial genome of some Sparidae to provide specific barcode markers. Food Control 2019, 103, 36–47. [Google Scholar] [CrossRef]
- Sousa-Santos, C.; Pereira, A.; Branco, P.; Costa, G.; Santos, J.; Ferreira, M.; Lima, C.; Doadrio, I.; Robalo, J. Mito-nuclear sequencing is paramount to correctly identify sympatric hybridizing fishes. Acta Ichthyol. Piscat. 2018, 48, 123–141. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.-C.; Zakaria, D.; Altarawneh, H.; Andrews, G.N.; Ganesan, G.S.; John, K.M.; Khan, S.; Ladumor, H. DNA barcoding of fish species reveals low rate of package mislabeling in Qatar. Genome 2019, 62, 69–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeter, J.C.; Maloy, A.P.; Rees, C.B.; Bartron, M.L. Fish mitochondrial genome sequencing: Expanding genetic resources to support species detection and biodiversity monitoring using environmental DNA. Conserv. Genet. Resour. 2019, 1–14. [Google Scholar] [CrossRef]
- Rincón-Sandoval, M.; Betancur-R, R.; Maldonado-Ocampo, J.A. Comparative phylogeography of trans-Andean freshwater fishes based on genome-wide nuclear and mitochondrial markers. Mol. Ecol. 2019, 28, 1096–1115. [Google Scholar] [CrossRef]
- Saad, Y. Analysis of 16S mitochondrial ribosomal DNA sequence variations and phylogenetic relations among some Serranidae fishes. South Afr. J. Anim. Sci. 2019, 49, 80. [Google Scholar] [CrossRef] [Green Version]
- Mirande, J.M. Morphology, molecules and the phylogeny of Characidae (Teleostei, Characiformes). Cladistics 2018, 35, 282–300. [Google Scholar] [CrossRef]
- Betancur-R, R.; Broughton, R.E.; Wiley, E.O.; Carpenter, K.; López, J.A.; Li, C.; Holcroft, N.I.; Arcila, D.; Sanciangco, M.; Ii, J.C.C.; et al. The Tree of Life and a New Classification of Bony Fishes. PLoS Curr. 2013, 5. [Google Scholar] [CrossRef]
- Betancur-R, R.; Wiley, E.O.; Arratia, G.; Acero, A.; Bailly, N.; Miya, M.; Lecointre, G.; Orti, G. Phylogenetic classification of bony fishes. BMC Evol. Boil. 2017, 17, 162. [Google Scholar] [CrossRef] [Green Version]
- Cárdenas, L.; Hernández, C.E.; Poulin, E.; Magoulas, A.; Kornfield, I.; Ojeda, F.P. Origin, diversification, and historical biogeography of the genus Trachurus (Perciformes: Carangidae). Mol. Phylogenet. Evol. 2005, 35, 496–507. [Google Scholar] [CrossRef] [PubMed]
- Olusola, O. Technological properties and proximate composition of two imported fish species in Nigeria. EJEAFChe Electron. J. Environ. Agric. Food Chem. 2011, 10, 2398–2403. [Google Scholar]
- Hosseinzadeh Sahafi, H. Identification of marine ornamental fishes in northern part of the Persian Gulf. Iran. J. Fish. Sci. 2000, 2, 21–36. [Google Scholar]
- Edwards, R.R.C.; Bakhader, A.; Shaher, S. Growth, mortality, age composition and fisheries yields of fish from the Gulf of Aden. J. Fish Boil. 1985, 27, 13–21. [Google Scholar] [CrossRef]
- Ohshimo, S.; Shiraishi, T.; Tanaka, H.; Yasuda, T.; Yoda, M.; Ishida, H.; Tomiyasu, S. Growth and Reproductive Characteristics of the Roughear Scad Decapterus tabl in the East China Sea. Jpn. Agric. Res. Q. JARQ 2014, 48, 245–252. [Google Scholar] [CrossRef] [Green Version]
- Siwat, V.; Ambariyanto, A.; Widowati, I. Biometrics of bigeye scad, Selar crumenophthalmus and shrimp scad, Alepes djedaba from Semarang waters, Indonesia. Aquacult. Aquarium Conserv. Legis. 2016, 9, 915–922. [Google Scholar]
- Reed, D.L.; E Carpenter, K.; Degravelle, M.J. Molecular systematics of the Jacks (Perciformes: Carangidae) based on mitochondrial cytochrome b sequences using parsimony, likelihood, and Bayesian approaches. Mol. Phylogenet. Evol. 2002, 23, 513–524. [Google Scholar] [CrossRef]
- Damerau, M.; Freese, M.; Hanel, R. Multi-gene phylogeny of jacks and pompanos (Carangidae), including placement of monotypic vadigo Campogramma glaycos. J. Fish Boil. 2017, 92, 190–202. [Google Scholar] [CrossRef]
- Tomkova, M.; Tomek, J.; Kriaucionis, S.; Schuster-Böckler, B. Mutational signature distribution varies with DNA replication timing and strand asymmetry. Genome Boil. 2018, 19, 129. [Google Scholar] [CrossRef] [Green Version]
- Lü, Z.; Zhu, K.; Jiang, H.; Lu, X.; Liu, B.; Ye, Y.; Jiang, L.; Liu, L.; Gong, L. Complete mitochondrial genome of Ophichthus brevicaudatus reveals novel gene order and phylogenetic relationships of Anguilliformes. Int. J. Boil. Macromol. 2019, 135, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Mu, X.; Liu, Y.; Lai, M.; Song, H.; Wang, X.; Hu, Y.; Luo, J. Characterization of the Macropodus opercularis complete mitochondrial genome and family Channidae taxonomy using Illumina-based de novo transcriptome sequencing. Gene 2015, 559, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Xia, J.; Zhang, J.-E.; Yang, J.; Zhao, H.; Wang, Q.; Sun, J.; Xue, H.; Wu, Y.; Chen, J.; et al. Characterization of the Complete Mitochondrial Genome Sequences of Three Croakers (Perciformes, Sciaenidae) and Novel Insights into the Phylogenetics. Int. J. Mol. Sci. 2018, 19, 1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, R.; Yang, Z. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 1998, 148, 929–936. [Google Scholar] [PubMed]
- García, G.O.; Oteo, J.A. Evolutionary distances corrected for purifying selection and ancestral polymorphisms. J. Theor. Boil. 2019, 483, 110004. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, Q.; Chen, C.; Jin, X.; Chen, Z.; Xiong, C.; Li, P.; Zhao, J.; Huang, W. Characterization and comparative mitogenomic analysis of six newly sequenced mitochondrial genomes from ectomycorrhizal fungi (Russula) and phylogenetic analysis of the Agaricomycetes. Int. J. Boil. Macromol. 2018, 119, 792–802. [Google Scholar] [CrossRef]
- Huang, J.; Ma, T. Comparative analysis of two mitochondrial genomes of flesh flies (Sarcophaga antilope and Sarcophaga dux) with phylogeny and evolutionary timescale for Sarcophagidae. Int. J. Boil. Macromol. 2018, 120, 1955–1964. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, J.-E.; Xia, J.; Yang, J.; Guo, J.; Deng, Z.; Luo, M. Comparative Characterization of the Complete Mitochondrial Genomes of the Three Apple Snails (Gastropoda: Ampullariidae) and the Phylogenetic Analyses. Int. J. Mol. Sci. 2018, 19, 3646. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.-L.; Gao, J.; Li, M.; Yuan, J.; Liang, J.; Yang, H.; Bu, W. The complete mitochondrial genome of Tetraphleps aterrimus (Hemiptera: Anthocoridae): Genomic comparisons and phylogenetic analysis of Cimicomorpha. Int. J. Boil. Macromol. 2019, 130, 369–377. [Google Scholar] [CrossRef]
- Lavrov, D.; Brown, W.M.; Boore, J.L. A novel type of RNA editing occurs in the mitochondrial tRNAs of the centipede Lithobius forficatus. Proc. Natl. Acad. Sci. USA 2000, 97, 13738–13742. [Google Scholar] [CrossRef] [Green Version]
- Cui, P.; Jirimutu, Z.; Ding, F.; Qi, D.; Gao, H.; Menghe, B.; Yu, J.; Hu, S.; Zhang, H. A complete mitochondrial genome sequence of the wild two-humped camel (Camelus bactrianus ferus): An evolutionary history of camelidae. BMC Genom. 2007, 8, 241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Zhang, J.-Y.; Zheng, R.; Yu, B.-G.; Yang, G. Complete nucleotide sequence and gene organization of the mitochondrial genome of Paa spinosa (Anura: Ranoidae). Gene 2009, 447, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuki, T.; Kawai, G.; Watanabe, K. The minimal tRNA: Unique structure of Ascaris suum mitochondrial tRNASer UCU having a short T arm and lacking the entire D arm. FEBS Lett. 2002, 514, 37–43. [Google Scholar] [CrossRef] [Green Version]
- Remm, M.; Storm, C.E.; Sonnhammer, E. Automatic clustering of orthologs and in-paralogs from pairwise species comparisons. J. Mol. Boil. 2001, 314, 1041–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- A Natale, D.; Shankavaram, U.T.; Galperin, M.Y.; I Wolf, Y.; Aravind, L.; Koonin, E.V. Towards understanding the first genome sequence of a crenarchaeon by genome annotation using clusters of orthologous groups of proteins (COGs). Genome Boil. 2000, 1, research0009-1. [Google Scholar] [CrossRef] [Green Version]
- Meyers, R. Children’s Oncology Group (COG) Surgery Discipline Committee: Research Agenda and Collaborations. Pediatr. Blood Cancer 2019, 66, S2. [Google Scholar]
- Gushiken, S. Phylogenetic Relationships of the Perciform Genera of the Family Carangidae. Ichthyol. Res. 1988, 34, 443–461. [Google Scholar] [CrossRef]
- FishBase. Available online: https://www.fishbase.org.version (accessed on 2 June 2020).
- Miya, M.; Friedman, M.; Satoh, T.P.; Takeshima, H.; Sado, T.; Iwasaki, W.; Yamanoue, Y.; Nakatani, M.; Mabuchi, K.; Inoue, J.; et al. Evolutionary Origin of the Scombridae (Tunas and Mackerels): Members of a Paleogene Adaptive Radiation with 14 Other Pelagic Fish Families. PLoS ONE 2013, 8, e73535. [Google Scholar] [CrossRef]
- Zinsmeister, W.J. Discovery of fish mortality horizon at the K-T Boundary on Seymour Island: Re-evaluation of events at the end of the Cretaceous. J. Paléontol. 1998, 72, 556–571. [Google Scholar] [CrossRef]
- Swart, B.L.; Von Der Heyden, S.; Der Merwe, A.B.-V.; Roodt-Wilding, R. Molecular systematics and biogeography of the circumglobally distributed genus Seriola (Pisces: Carangidae). Mol. Phylogenet. Evol. 2015, 93, 274–280. [Google Scholar] [CrossRef]
- Mak, S.S.T.; Gopalakrishnan, S.; Carøe, C.; Geng, C.; Liu, S.; Sinding, M.-H.S.; Kuderna, L.F.K.; Zhang, W.; Fu, S.; Vieira, F.G.; et al. Comparative performance of the BGISEQ-500 vs Illumina HiSeq2500 sequencing platforms for palaeogenomic sequencing. GigaScience 2017, 6, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-Y.; Li, Y.-C.; Chen, N.-C.; Huang, X.-X.; Lu, Y.-C. In A special processor design for nucleotide basic local alignment search tool with a new banded two-hit method. In Proceedings of the 2016 IEEE Nordic Circuits and Systems Conference (NORCAS), Copenhagen, Denmark, 1–2 November 2016. [Google Scholar]
- Zhang, Q.-L.; Yang, X.-Z.; Zhang, L.; Feng, R.-Q.; Zhu, Q.-H.; Chen, J.-Y.; Yuan, M.-L. Adaptive evidence of mitochondrial genomes in Dolycoris baccarum (Hemiptera: Pentatomidae) to divergent altitude environments. Mitochondrial DNA Part A 2018, 30, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, W.; Fukunaga, T.; Isagozawa, R.; Yamada, K.; Maeda, Y.; Satoh, T.P.; Sado, T.; Mabuchi, K.; Takeshima, H.; Miya, M.; et al. MitoFish and MitoAnnotator: A mitochondrial genome database of fish with an accurate and automatic annotation pipeline. Mol. Boil. Evol. 2013, 30, 2531–2540. [Google Scholar] [CrossRef] [PubMed]
- Junqueira, A.C.M.; Lessinger, A.C.; Torres, T.T.; Da Silva, F.R.; Vettore, A.L.; Arruda, P.; Espin, A.M.L.A. The mitochondrial genome of the blowfly Chrysomya chloropyga (Diptera: Calliphoridae). Gene 2004, 339, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Genovese, L.M.; Mosca, M.M.; Pellegrini, M.; Geraci, F. Dot2dot: Accurate whole-genome tandem repeats discovery. Bioinformatics 2019, 35, 914–922. [Google Scholar] [CrossRef]
- Kumar, V.; Dey, A.; Singh, A. MEGA: A Bio Computational Software for Sequence and Phylogenetic Analysis. Lect. Notes Eng. Comput. Sci. 2009, 1, 1863–1865. [Google Scholar]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Vadimirov, N.V.; Kochetov, A.V.; Grigorovich, D.A.; Matushkin, Y.G. RSCU_comparer: A new statistical tool for practical analysis of codon usage. Proceedings of the Fifth International Conference on Bioinformatics of Genome Regulation And Structure. Comp. Evol. Genom. Proteom. 2006, 3, 241–244. [Google Scholar]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, S.; Stones, R.J.; Ren, M.; Liu, X.-G.; Wang, G.; Xia, H.-J.; Wu, H.-Y.; Liu, Y.; Xie, Q. GPU MrBayes V3.1: MrBayes on Graphics Processing Units for Protein Sequence Data: Table 1. Mol. Boil. Evol. 2015, 32, 2496–2497. [Google Scholar] [CrossRef] [Green Version]
- Tan, M.; Armbruster, J.W. Phylogenetic classification of extant genera of fishes of the order Cypriniformes (Teleostei: Ostariophysi). Zootaxa 2018, 4476, 6–39. [Google Scholar] [CrossRef] [Green Version]
- Sadjjadi, S.M.; Ebrahimipour, M.; Sadjjadi, S.M. Comparison between Echinococcus granulosus sensu stricto (G1) and E. canadensis (G6) mitochondrial genes (cox1 and nad1) and their related protein models using experimental and bioinformatics analysis. Comput. Boil. Chem. 2019, 79, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Mello, B. Estimating TimeTrees with MEGA and the TimeTree Resource. Mol. Boil. Evol. 2018, 35, 2334–2342. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Suleski, M.; Hedges, S.B. TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol. Boil. Evol. 2017, 34, 1812–1819. [Google Scholar] [CrossRef]
- Grant, J.R.; Arantes, A.S.; Stothard, P. Comparing thousands of circular genomes using the CGView Comparison Tool. BMC Genom. 2012, 13, 202. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Element | Strand | Size (bp) | GC Content (%) | Amino Acids (aa) | Inferred Initiation Codon | Inferred Termination Codon | One Letter Code | Anti-Codon | Intergenic Nucleotide*(bp) |
|---|---|---|---|---|---|---|---|---|---|
| tRNA-Phe | H | 68 | 45.59–51.47 | F | GAA | 0 | |||
| 12S rDNA | H | 951–954 | 47.79–49.21 | 0 | |||||
| tRNA-Val | H | 72 | 47.22–50.00 | V | TAC | 0 | |||
| 16S rDNA | H | 1716–1728 | 45.37–46.50 | 0 | |||||
| tRNA-Leu | H | 74–75 | 48.00–48.65 | L | TAA | 0 | |||
| ND1 | H | 975 | 48.82–51.08 | 324 | ATG | TAG, TAA | 0 | ||
| tRNA-Ile | H | 70 | 50.00–52.86 | I | GAT | 5–6 | |||
| tRNA-Gln | L | 71 | 40.85 | Q | TTG | −1 | |||
| tRNA-Met | H | 70–71 | 45.71–47.14 | M | CAT | −1 | |||
| ND2 | H | 1045–1047 | 49.00–50.43 | 348 | ATG | T, TAG | 0 | ||
| tRNA-Trp | H | 69–71 | 50.70–52.17 | W | TCA | 0 | |||
| tRNA-Ala | L | 69 | 39.13–43.48 | A | TGC | 1 | |||
| tRNA-Asn | L | 73 | 47.95–52.05 | N | GTT | 1 | |||
| tRNA-Cys | L | 66–67 | 43.28–45.45 | C | GCA | 37–38 | |||
| tRNA-Tyr | L | 70 | 45.71–47.14 | Y | GTA | 0 | |||
| COX1 | H | 1551 | 45.07–47.58 | 516 | GTG | TAA | 1 | ||
| tRNA-Ser | L | 71 | 46.48–47.89 | S | TGA | 0 | |||
| tRNA-Asp | H | 71 | 43.66–46.48 | D | GTC | 3 | |||
| COX2 | H | 691 | 41.68–44.86 | 230 | ATG | T | 6–7 | ||
| tRNA-Lys | H | 74–75 | 45.67–48.00 | K | TTT | 0 | |||
| ATP8 | H | 165–168 | 44.64–49.70 | 55 | ATG | TAA, TAG | 1 | ||
| ATP6 | H | 678–684 | 44.84–50.00 | 225–227 | ATG or ATA | TAA | (−10)–(−4) | ||
| COX3 | H | 785–786 | 47.46–49.17 | 260–261 | ATG | TA, TAA | −1 | ||
| tRNA-Gly | H | 69–70 | 32.86–36.23 | G | TCC | 0 | |||
| ND3 | H | 349–351 | 47.85–49.00 | 115–116 | ATG | T, TAG | 0 | ||
| tRNA-Arg | H | 67-69 | 29.85-37.68 | R | TCG | 0 | |||
| ND4L | H | 297 | 48.82–52.86 | 98 | ATG | TAA | 1 | ||
| ND4 | H | 1381 | 46.85–49.17 | 460 | ATG | T | −7 | ||
| tRNA-His | H | 71–73 | 36.99–39.44 | H | GTG | 0 | |||
| tRNA-Ser | H | 67–68 | 50.75–57.35 | S | GCT | 0 | |||
| tRNA-Leu | H | 73 | 45.21 | L | TAG | 4 | |||
| ND5 | H | 1839 | 44.54–47.80 | 612 | ATG | TAA, TAG | 0 | ||
| ND6 | L | 522 | 44.44–48.85 | 173 | ATG | TAG, TAA | −4 | ||
| tRNA-Glu | L | 69 | 40.58–43.48 | E | TTC | 0 | |||
| CYTB | H | 1141 | 46.71–49.69 | 380 | ATG | T | 4 | ||
| tRNA-Thr | H | 72 | 52.78–58.33 | T | TGT | 0 | |||
| tRNA-Pro | L | 71 | 38.03–45.07 | P | TGG | −1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Li, M.; Xu, S.; Liu, L.; Chen, Z.; Zou, K. Complete Mitogenomes of Three Carangidae (Perciformes) Fishes: Genome Description and Phylogenetic Considerations. Int. J. Mol. Sci. 2020, 21, 4685. https://doi.org/10.3390/ijms21134685
Li Z, Li M, Xu S, Liu L, Chen Z, Zou K. Complete Mitogenomes of Three Carangidae (Perciformes) Fishes: Genome Description and Phylogenetic Considerations. International Journal of Molecular Sciences. 2020; 21(13):4685. https://doi.org/10.3390/ijms21134685
Chicago/Turabian StyleLi, Zhenhai, Min Li, Shannan Xu, Li Liu, Zuozhi Chen, and Keshu Zou. 2020. "Complete Mitogenomes of Three Carangidae (Perciformes) Fishes: Genome Description and Phylogenetic Considerations" International Journal of Molecular Sciences 21, no. 13: 4685. https://doi.org/10.3390/ijms21134685
APA StyleLi, Z., Li, M., Xu, S., Liu, L., Chen, Z., & Zou, K. (2020). Complete Mitogenomes of Three Carangidae (Perciformes) Fishes: Genome Description and Phylogenetic Considerations. International Journal of Molecular Sciences, 21(13), 4685. https://doi.org/10.3390/ijms21134685