Clinical Assessment, Genetics, and Treatment Approaches in Autism Spectrum Disorder (ASD)



Abstract
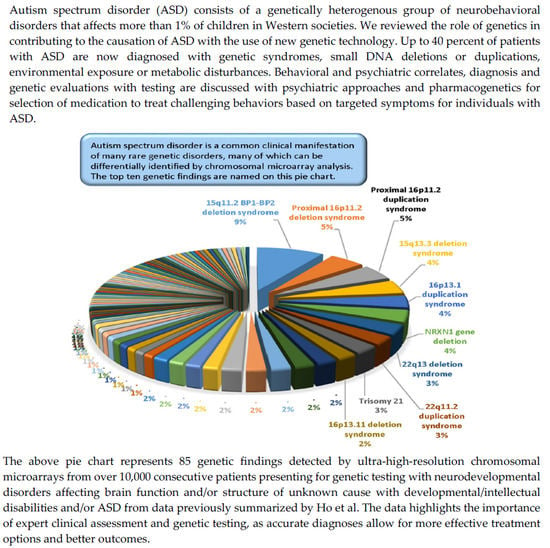
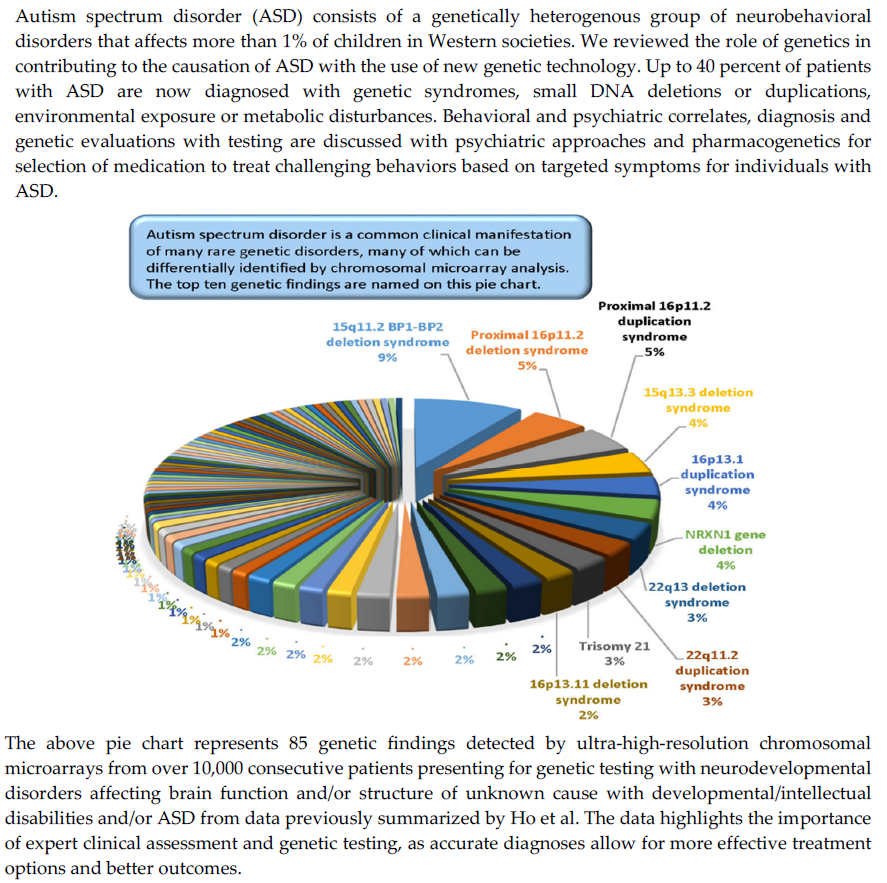
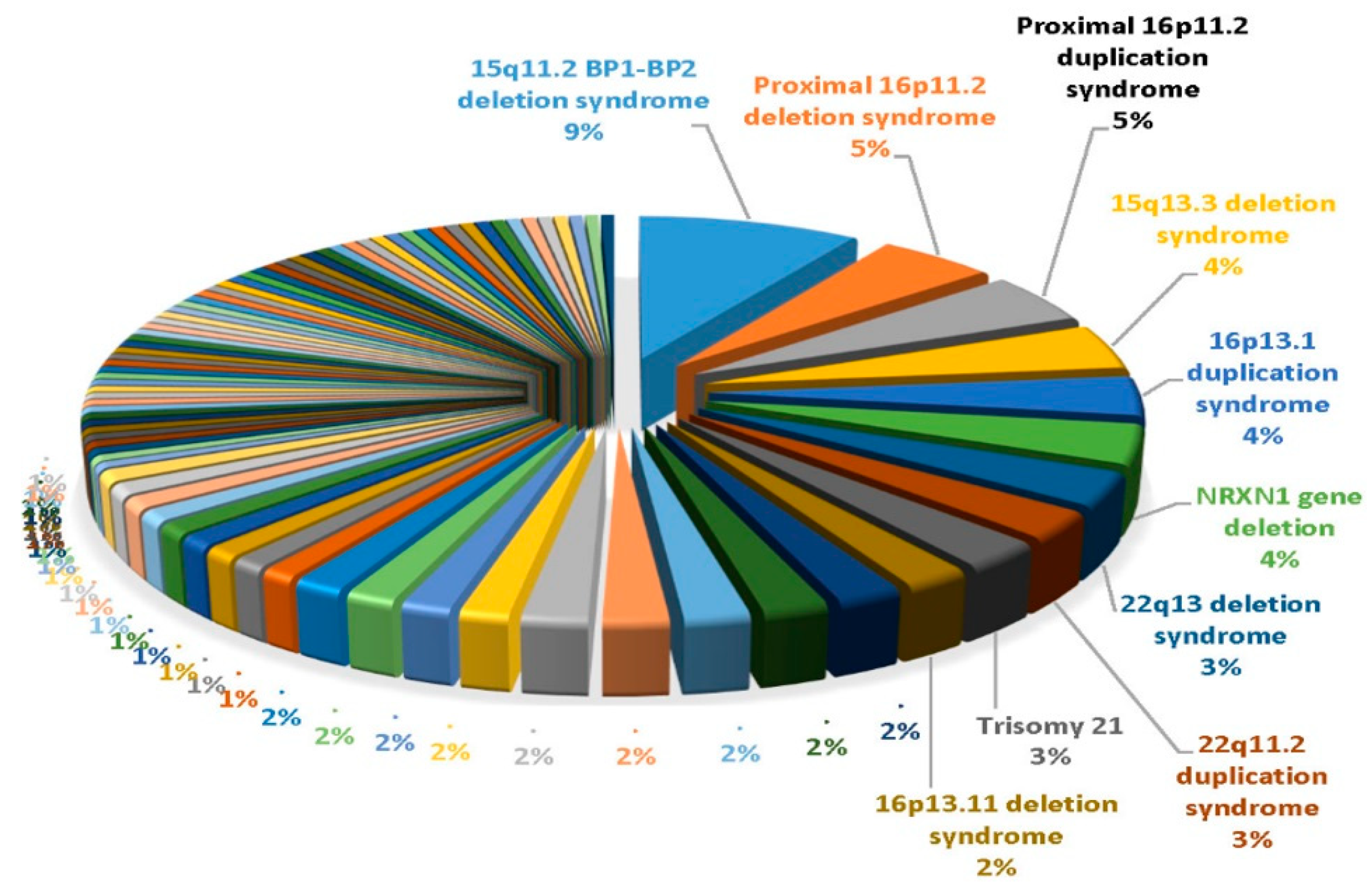
:
{kind=link}
{kind=link}
1. Introduction
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
| Modified from G.B. Schaefer and N.J. Mendelsohn, “Genetics evaluation for the etiologic diagnosis of autism spectrum disorders,” Genetics in Medicine, vol. 10, pp 4–12, 2008 [25] and from M.G. Butler and others, “Assessment and treatment in autism spectrum disorders: A focus on genetics and psychiatry”, Autism Research and Treatment, vol. 2012, 242537, 2012 [30]. | |
2. Diagnosis and Genetics of ASD
2.1. Genetic Factors Contributing to Autism
2.2. Metabolic Factors Contributing to Autism
3. Clinical Assessment and Testing
3.1. Initial Clinical Evaluation
3.2. High-Resolution Microarrays and ASD
3.3. Next-Generation Sequencing (NGS)
4. Treatment Approaches
4.1. Behavioral Interventions in ASD
4.1.1. For Children and Adolescents with ASD
4.1.2. For Adults with ASD
4.2. Medication Treatments in ASD
4.2.1. For the Treatment of ADHD Symptoms in ASD
4.2.2. For the Treatment of Irritability, Aggression, and Self-Injurious Behavior in ASD
4.2.3. For the Treatment of Repetitive Behaviors Including Stereotypies in ASD
4.2.4. For the Treatment of Persistent Insomnia in ASD
4.3. Pharmacogenetics and Role in Medication Selection and Management
5. Future Directions
6. Summary
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kanner, L. Autistic disturbances of affective contact. Nervous Child. 1943, 32, 217–253. [Google Scholar]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM-5), 5th ed.; American Psychiatric Association: Washington, DC, USA, 2013. [Google Scholar]
- World Health Organization. ICD-10: International Statistical Classification of Diseases and Related Health Problems: Tenth Revision, 2nd ed.; World Health Organization: Geneva, Switzerland, 2004. [Google Scholar]
- Hyman, S.L.; Levy, S.E.; Myers, S.M. AAP Council on Children with Disabilities, Section on Developmental and Behavioral Pediatrics. Identification, Evaluation, and Management of Children with Autism Spectrum Disorder. Pediatrics 2020, 145, e20193447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutter, M.; Le Couteur, A.; Lord, C. ADI-R: Autism Diagnostic Interview-Revised (ADI-R); Western Psychological Services: Los Angeles, CA, USA, 2003. [Google Scholar]
- Lord, C.; DiLavore, P.C.; Gotham, K.; Guthrie, W.; Luyster, R.J.; Risi, S.; Rutter, M. Autism Diagnostic Observation Schedule: ADOS-2; Western Psychological Services: Los Angeles, CA, USA, 2012. [Google Scholar]
- Tordjman, S.; Cohen, D.; Anderson, G.M.; Botbol, M.; Banitano, R.; Coulon, N.; Roubertous, P.L. Reprint of “Reframing autism as a behavioral syndrome and not a specific mental disorder: Implications of genetic and phenotypic heterogeneity”. Neurosci. Biobehav. Rev. 2018, 89, 132–150. [Google Scholar]
- Miles, J.H.; Takahashi, T.N.; Bagby, S.; Sahota, P.K.; Vaslow, D.F.; Wang, C.H.; Hillman, R.E.; Farmer, J.E. Essential versus complex autism: Definition of fundamental prognostic subtypes. Am. J. Med. Genet. 2005, 135, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Cohen, D.; Pichard, N.; Tordjman, S.; Baumann, C.; Burglen, L.; Excoffier, E.; Lazar, G.; Mazet, P.; Pinquier, C.; Verloes, A.; et al. Specific genetic disorders and autism: Clinical contribution towards their identification. J. Autism Dev. Disord. 2005, 35, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Rapin, I. Autistic regression and disintegrative disorder: How important the role of epilepsy? Semin. Pediatr. Neurol. 1995, 2, 278–285. [Google Scholar] [CrossRef]
- Miles, J.H.; Hillman, R.E. Value of a clinical morphology examination in autism. Am. J. Med. Genet. 2000, 10, 245–253. [Google Scholar] [CrossRef]
- Fombonne, E.; Roge, B.; Claverie, J.; Courty, S.; Frémolle, J. Microcephaly and macrocephaly in autism. J. Autism. Dev. Disord. 1999, 29, 113–119. [Google Scholar] [CrossRef]
- Miles, J.H. Autism spectrum disorders—A genetics review. Genet Med. 2011, 13, 278–294. [Google Scholar] [CrossRef] [Green Version]
- Butler, M.G.; Dasouki, M.J.; Zhou, X.P.; Talebizadeh, Z.; Brown, M.; Takahashi, T.N.; Miles, J.H.; Wang, C.H.; Stratton, R.; Pilarski, R.; et al. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J. Med. Genet. 2005, 42, 318–321. [Google Scholar] [CrossRef] [Green Version]
- Carper, R.A.; Courchesne, E. Inverse correlation between frontal lobe and cerebellum sizes in children with autism. Brain 2000, 123, 836–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carper, R.A.; Moses, P.; Tigue, Z.D.; Courchesne, E. Cerebral lobes in autism: Early hyperplasia and abnormal age effects. Neuroimage 2002, 16, 1038–1051. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.; Keeser, D.; Reiser, M.F.; Teipel, S.; Meindl, T. Functional and structural MR imaging in neuropsychiatric disorders, part 2: Application in schizophrenia and autism. AJNR Am. J. Neuroradiol. 2012, 33, 2033–2037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philip, R.C.; Daubermann, M.R.; Whalley, H.C.; Baynham, K.; Lawrie, S.M.; Stanfield, A.C. A systematic review and meta-analysis of the fMRI investigation of autism spectrum disorders. Neurosci. Biobehav. Rev. 2012, 36, 901–942. [Google Scholar] [CrossRef]
- Kobayashi, A.; Yokota, S.; Takeuchi, H.; Asano, K.; Asano, M.; Sassa, Y.; Taki, Y.; Kawashima, R. Increased grey matter volume of the right superior temporal gyrus in healthy children with autistic cognitive style: A VBM study. Brain Cogn. 2020, 139, 105514. [Google Scholar] [CrossRef]
- Gabrielli, A.P.; Manzardo, A.M.; Butler, M.G. GeneAnalytics pathways and profiling of shared autism and cancer genes. Int. J. Mol. Sci. 2019, 20, 1166. [Google Scholar] [CrossRef] [Green Version]
- Butler, M.G.; Rafi, S.K.; Manzardo, A.M. High-resolution chromosome ideogram representation of currently recognized genes for Autism spectrum disorders. Int. J. Mol Sci. 2015, 16, 6464–6495. [Google Scholar] [CrossRef] [Green Version]
- Tordjman, S.; Drapier, D.; Bonnot, O.; Graignic, R.; Fortes, S.; Cohen, D.; Millet, B.; Laurent, C.; Roubertoux, P.L. Animal models relevant to schizophrenia and autism: Validity and limitations. Behav. Genet. 2007, 37, 61–67. [Google Scholar] [CrossRef]
- Walsh, P.; Elsabbagh, M.; Bolton, P.; Singh, I. In search of biomarkers for autism: Scientific, social and ethical challenges. Nat. Rev. Neurosci. 2011, 2, 603–612. [Google Scholar] [CrossRef]
- Happé, F.; Ronald, A.; Plomin, R. Time to give up on a single explanation for Autism. Nat. Neurosci. 2006, 9, 1218–1220. [Google Scholar] [CrossRef]
- Schaefer, G.B.; Mendelsohn, N.J.; Professional Practice Guidelines Committee. Clinical genetics evaluation in identifying the etiology of autism spectrum disorders. Genet. Med. 2008, 10, 301–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Dies, K.A.; Holm, I.A.; Bridgemohan, C.; Sobeih, M.M.; Caronna, E.B.; Miller, K.J.; Frazier, J.A.; Silverstain, I.; Picker, J.; et al. Austism Consortium Clinical Genetics/DNA Diagnostics Collaboration. Clinical genetic testing for patients with autism spectrum disorders. Pediatrics 2010, 125, 727–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waye, M.M.Y.; Cheng, H.Y. Genetics and epigenetics of autism: A review. Psychiatry Clin. Neurosci. 2018, 72, 228–244. [Google Scholar] [CrossRef] [PubMed]
- Howes, O.D.; Rogdaki, M.; Findon, J.L.; Wichers, R.H.; Charman, T.; King, B.H.; Loth, E.; McAlonan, G.M.; McCracker, J.T.; Parr, J.R.; et al. Autism spectrum disorder: Consensus guidelines on assessment, treatment and research from the British Association for Psychopharmacology. J. Psychopharmacol. 2018, 32, 3–29. [Google Scholar] [CrossRef] [PubMed]
- Rosen, T.E.; Mazefsky, C.A.; Vasa, R.A.; Lerner, M.D. Co-occurring psychiatric conditions in autism spectrum disorder. Int Rev. Psychiatry. 2018, 30, 40–61. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.G.; Youngs, E.L.; Roberts, J.L.; Hellings, J.A. Assessment and treatment in autism spectrum disorders: A focus on genetics and psychiatry. Autism Res. Treat. 2012, 2012, 242537. [Google Scholar] [CrossRef]
- Rice, C. Prevalence of Autism spectrum disorders—Autism and developmental disabilities monitoring network. Morbid Mortal Wkly. Rep. 2006, 58, 1–20. [Google Scholar]
- Rose, S.; Niyazov, D.M.; Rossignol, D.A.; Golenthal, M.; Kahler, S.G.; Frye, R.E. Clinical and molecular characteristics of mitochondrial dysfunction in autism spectrum disorder. Mol. Diagn. Ther. 2018, 22, 571–593. [Google Scholar] [CrossRef] [Green Version]
- Abrahams, B.S.; Geschwind, D.H. Advances in autism genetics: On the threshold of a new neurobiology. Nat. Rev. Genet. 2008, 9, 341–355. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, G.B.; Mendelsohn, N.J.; Professional Practice and Guidelines Committee. Clinical genetics evaluation in identifying the etiology of autism spectrum disorders: 2013 guideline revisions. Genet. Med. 2013, 15, 399–407. [Google Scholar] [CrossRef] [Green Version]
- Sebat, J.; Lakshmi, B.; Malhotra, D.; Troge, J.; Lese-Martin, C.; Walsh, T.; Yamrom, B.; Yoon, S.; Krasnitz, A.; Kendall, J.; et al. Strong association of de novo copy number mutations with autism. Science 2007, 316, 445–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaefer, G.B.; Starr, L.; Pickering, D.; Skar, G.; Dehaai, K.; Sanger, W.G. Array comparative genomic hybridization findings in a cohort referred for an autism evaluation. J. Child. Neurol. 2010, 25, 1498–1503. [Google Scholar] [CrossRef] [PubMed]
- Wenger, T.L.; Kao, C.; McDonald-McGinn, D.M.; Zackair, E.H.; Bailey, A.; Schultz, R.T.; Morrow, B.E.; Emanuel, B.S.; Hakonarson, H. The role of mGluR copy number variation in genetic and environmental forms of syndromic autism spectrum disorder. Sci. Rep. 2016, 6, 19372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallmayer, J.; Cleveland, S.; Torres, A.; Phillips, J.; Cohen, B.; Torigoe, T.; Miller, J.; Fedele, A.; Collins, J.; Smith, K.; et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch. Gen. Psychiatry 2011, 68, 1095–1102. [Google Scholar] [CrossRef]
- Tick, B.; Bolton, P.; Happe, F.; Rutther, M.; Rijsdijk, F. Heritability of autism spectrum disorders: A meta-analysis of twin studies. J. Child Psychol. Psychiatry 2016, 57, 585–595. [Google Scholar] [CrossRef] [Green Version]
- Weinder, D.J.; Wigoder, E.M.; Riple, S.; Walters, R.K.; Kosmicki, J.A.; Grove, J.; Samocha, K.E.; Goldstein, J.I.; Okbay, A.; Bybjerg-Grauhom, J.; et al. Polygenic transmission disequlibirium confirms that common and rate vairation act additively to create risk for autism spectrum disorders. Nat. Genet. 2017, 49, 978–983. [Google Scholar] [CrossRef] [Green Version]
- Pizzo, L.; Jensen, M.; Polyak, A.; Rosenfeld, J.A.; Mannik, K.; Krishnan, A.; McCready, E.; Pichon, O.; Le Caignec, C.; Van Dijck, A.; et al. Rare variants in the genetic background modulate cognitive and developmental phenotypes in individuals carryng disease-associated varients. Genet Med. 2019, 21, 816–825. [Google Scholar] [CrossRef] [Green Version]
- Myers, S.M.; Challman, T.D.; Bernier, R.; Bourgeron, T.; Chung, W.K.; Constantinto, J.N.; Eichler, E.E.; Jacquemenot, S.; Miller, D.T.; Mitchell, K.J.; et al. Insufficient evidence for “Autism-Specific” genes. Am. J. Hum. Genet. 2020, 106, 587–595. [Google Scholar]
- Vorstman, J.A.; Parr, J.R.; Moreno-De-Luca, D.; Anney, R.J.L.; Nurnberger, J.I.; Hallmayer, J.F. Autism genetics: Opportunities and challenges for clinical translation. Nat. Rev. Genet. 2017, 18, 362–376. [Google Scholar] [CrossRef]
- Yenkoyan, K.; Griogryan, A.; Fereshetyan, K.; Ypremyan, D. Adances in understanding the pathophysiology of autism spectrum disorders. Behav. Brain Res. 2017, 331, 92–101. [Google Scholar] [CrossRef]
- Srivastava, S.; Love-Nichols, J.A.; Dies, K.A.; Ledbetter, D.H.; Martin, C.L.; Chung, W.K.; Firth, H.V.; Frazier, T.; Hansen, R.L.; Prock, L.; et al. NDD exome scoping review work group. Meta-analysis and multidisciplinary consensus statement: Exome sequencing is a first-tier clinical diagnostic test for individuals with neurodevelopmental disorders. Genet. Med. 2019, 27, 2413–2421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhillon, S.; Hellings, J.A.; Butler, M.G. Genetics and mitochondrial abnormalities in autism spectrum disorders: A review. Curr. Genom. 2011, 12, 322–332. [Google Scholar] [CrossRef] [Green Version]
- Ho, K.S.; Wassman, E.R.; Baxter, A.L.; Hensel, C.H.; Martin, M.M.; Prasad, A.; Twede, H.; Vanzo, R.J.; Butler, M.G. Chromosomal microarray analysis of consecutive individuals with autism spectrum disorders using an ultra-high resolution chromosomal microarray optimized for neurodevelopmental disorders. Int. J. Mol. Sci. 2016, 17, 2070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodbury-Smith, M.; Paterson, A.D.; O’Connor, I.; Zarrei, M.; Yuen, R.K.C.; Howe, J.L.; Thompson, A.; Parlier, M.; Fernandez, B.; Piven, J.; et al. A genome-wide linkage study of autism spectrum disorder and the broad autism phenotype in extended pedigrees. J. Neurodev. Disord. 2018, 10, 20. [Google Scholar] [CrossRef]
- Fernandez, B.A.; Roberts, W.; Chung, B.; Weksberg, R.; Meyn, S.; Szatmari, P.; Joseph-George, A.M.; Mackay, S.; Whitten, K.; Nble, B.; et al. Phenotypic spectrum associated with de novo and inherited deletions and duplications at 16p11.2 in individuals ascertained for diagnosis of autism spectrum disorder. J. Med. Genet. 2010, 47, 195–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, D.T.; Shen, Y.; Weiss, L.A.; Korn, J.; Anselm, I.; Bridgemohan, C.; Cox, G.F.; Dickinson, H.; Gentile, J.; Harris, D.J.; et al. Microdeletion/duplication at 15q13.2q13.3 among individuals with features of autism and other neuropsychiatric disorders. J. Med. Genet. 2009, 46, 242–248. [Google Scholar] [CrossRef] [Green Version]
- Rossi, M.; El-Khechen, D.; Black, M.H.; Hagmna, K.D.F.; Tang, S.; Powis, Z. Outcomes of diagnostic exome sequencing in patients with diagnosed or suspected autism spectrum disorders. Pediatr. Neurol. 2017, 70, 34–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tammimies, K.; Marshall, C.R.; Walker, S.; Kaur, G.; Thiruvahindrapuram, B.; Lionel, A.C.; Yuen, R.K.C.; Uddin, M.; Roberts, W.; Weksberg, R.; et al. Molecular diagnostic yield of chromosomal microarray analysis and whole-exome sequencing in children with autism spectrum disorder. JAMA 2017, 314, 895–903. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhang, H.; Ma, D.; Bucan, M.; Glessner, J.T.; Abrahams, B.S.; Salyakina, D.; Imielinski, M.; Bradfield, J.P.; Sleiman, P.M.A.; et al. Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature 2009, 459, 528–533. [Google Scholar] [CrossRef]
- Delvin, B.; Scherer, S.W. Genetic architecture in autism spectrum disorder. Curr. Opin. Genet. Dev. 2012, 22, 229–237. [Google Scholar]
- Geschwind, D.H.; State, M.W. Gene hunting in autism spectrum disorder: On the path to precision medicine. Lancet Neurol. 2015, 14, 1109–1120. [Google Scholar] [CrossRef] [Green Version]
- Pinto, D.; Delaby, E.; Merico, D.; Barbosa, M.; Merikangas, A.; Klei, L.; Thiruvahindrapuram, B.; Xu, X.; Ziman, R.; Wang, Z.; et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am. J. Hum. Genet. 2014, 94, 677–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuen, R.K.C.; Merico, D.; Bookman, M.; Howe, J.L.; Thiruvahindrapuram, B.; Patel, R.V.; Whitney, J.; Delaux, N.; Bingham, J.; Wang, Z.; et al. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat. Neurosci. 2017, 20, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Glessner, J.T.; Wang, K.; Cai, G.; Korvatska, O.; Kim, C.E.; Wood, S.; Zhang, H.; Estes, A.; Brune, C.W.; Bradfield, J.P.; et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 2009, 459, 569–573. [Google Scholar] [CrossRef]
- Holt, R.; Monaco, A.P. Links between genetics and pathophysiology in the autism spectrum disorders. EMBO Molecul. Med. 2011, 3, 438–450. [Google Scholar] [CrossRef] [PubMed]
- Khanzada, N.S.; Butler, M.G.; Manzardo, A.M. GeneAnalytics pathway analysis and genetic overlap among autism spectrum disorder, bipolar disorder and schizophrenia. Int. J. Mol Sci. 2017, 18, 527. [Google Scholar] [CrossRef]
- Chatterjee, M.; Schild, D.; Teunissen, C.E. Contactins in the central nervous system: Role in health and disease. Neural. Regen. Res. 2019, 14, 206–216. [Google Scholar]
- Varga, N.A.; Pentelenyi, K.; Balicza, P.; Gezsi, A.; Remenyi, V.; Harsfalvi, V.; Bencsik, R.; Iles, A.; Prekop, C.; Molnar, M.J. Mitochondrial dysfunction and autism: Comprehensive genetic analyses of children with autism and mtDNA deletion. Behav Brain Funct. 2018, 14, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, D.C. Mitochondrial genes and disease. Hosp. Pract. 1986, 21, 77–92. [Google Scholar]
- Wallace, D.C. Mitochondrial diseases in mand and mouse. Science 1999, 283, 1482–1488. [Google Scholar] [CrossRef] [Green Version]
- Schon, E.A.; Manfredi, G. Neuronal degeneration and mitochondrial dysfunction. J. Clin. Investig. 2003, 111, 303–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiMauro, S.; Schon, E.A. Mitochondrial respiratory-chain diseases. N. Engl. J. Med. 2003, 348, 2656–2668. [Google Scholar] [CrossRef] [PubMed]
- Pons, R.; Andreu, A.L.; Checcarelli, N.; Vila, M.R.; Engelstad, K.; Sue, C.M.; Shungu, D.; Haggerty, R.; de Vivo, D.C.; DiMauro, S. Mitochondrial DNA abnormalities and autistic spectrum disorders. J. Pediatr. 2004, 144, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Spelbrink, J.N. Functional organization of mammalian mitochondrial DNA in nucleotides: History, recent developments, and future challenges. IUBMB 2010, 62, 19–32. [Google Scholar]
- Boyle, C.A.; Boulet, S.; Schieve, L.A.; Cohen, R.A.; Blumberg, S.J.; Yeargin-Allsopp, M.; Visser, S.; Kogan, M.D. Trends in the prevalence of developmental disabilities in the US children, 1997–2008. Pediatrics 2011, 127, 1034–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heil, K.M.; Schaaf, C.P. The genetics of autism spectrum disorders—A guide for clinicians. Curr. Psychiatry Rep. 2013, 15, 334. [Google Scholar] [CrossRef]
- Roberts, J.L.; Jovanes, K.; Dasouki, M.; Manzardo, A.M.; Butler, M.G. Chromosomal microarray analysis of consecutive individuals with autism spectrum disorders or learning disability presenting for genetic services. Gene 2014, 535, 70–78. [Google Scholar] [CrossRef] [Green Version]
- Weitlauf, A.S.; McPheeters, M.L.; Peters, B.; Sathe, N.; Travis, R.; Aiello, R.; Williamson, E.; Veenstra-VanderWeele, J.; Krishnaswami, S.; Jerome, R.; et al. Therapies for Children with Autism Spectrum Disorder: Behavioral Interventions Update; Comparative Effectiveness Review No. 137. (Prepared by the Vanderbilt evidence-Based Practice Center Under Contract No. 290-2012-00009-I.) AHRQ Publication No. 14-EHC036-EF.; Agency for Healthcare Research and Quality: Rockville, MD, USA, 2014. [Google Scholar]
- Tiura, M.; Kim, J.; Detmers, D.; Baldi, H. Predictors of longitudinal ABA treatment outcomes for children with autism: A growth curve analysis. Res. Dev. Disabil. 2017, 70, 185–197. [Google Scholar] [CrossRef]
- Reichow, B.; Hume, K.; Barton, E.E.; Boyd, B.A. Early intensive behavioral intervention (EIBI) for young children with autism spectrum disorders (ASD). Cochrane Database Syst. Rev. 2018, 5, CD009260. [Google Scholar] [CrossRef]
- Frankel, F.; Myatt, R.; Sugar, C. A randomized controlled study of parent-assisted children’s friendship training with children having autism spectrum disorders. J. Autism Dev. Disord. 2010, 40, 827–842. [Google Scholar] [CrossRef] [Green Version]
- National Institute for Clinical Excellence. Autism: Recognition, referral, diagnosis and management of adults on the autism spectrum. Natl. Inst. Health Care Excell. 2012, 142, 18. [Google Scholar]
- Hillier, A.; Fish, T.; Cloppert, P. Outcomes of a social and vocational skills support group for adolescents and young adults on the autism spectrum. Focus Autism Other Dev. Disabl. 2007, 22, 107–115. [Google Scholar] [CrossRef]
- Lang, R.; Regester, A.; Lauderdale, S. Treatment of anxiety in autism spectrum disorders using cognitive behaviour therapy: A systematic review. Dev. Neurorehabil. 2010, 13, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Accordino, R.E.; Kidd, C.; Politte, L.C.; Henry, C.A.; McDougle, C.J. Psychopharmacological interventions in autism spectrum disorder. Expert Opin. Pharmacother. 2016, 17, 937–952. [Google Scholar]
- Madden, J.M.; Lakoma, M.D.; Lynch, F.L.; Rusinak, D.; Owen-Smith, A.A.; Coleman, K.J.; Quinn, V.P.; Yau, V.M.; Qian, Y.X.; Croen, L.A. Psychotropic medication use among insured children with autism spectrum disorder. J. Autism Dev. Disord. 2017, 47, 144–154. [Google Scholar] [CrossRef]
- Stepanova, E.; Dowling, S.; Phelps, M.; Findling, R.L. Pharmacotherapy of emotional and behavioral symptoms associated with autism spectrum disorder in children and adolescents. Dialogues Clin. Neurosci. 2017, 19, 395–402. [Google Scholar]
- Lamy, M.; Erickson, C.A. Pharmacological management of behavioral disturbances in children and adolescents with autism spectrum disorders. Curr. Prob. Ped. Adolesc Health Care 2012, 48, 250–264. [Google Scholar] [CrossRef] [PubMed]
- Research Units on Pediatric Psychopharmacology (RUPP). Randomized, controlled, crossover trial of methylphenidate in pervasive developmental disorders with hyperactivity. Arch. Gen. Psychiatry. 2005, 62, 1266–1275. [Google Scholar] [CrossRef]
- Reichow, B.; Volkmar, F.R.; Bloch, M.H. Systematic review and meta-analysis of pharmacological treatment of the symptoms of attention-deficit/hyperactivity disorder in children with pervasive developmental disorders. J. Autism Dev. Disord. 2013, 43, 2435–4241. [Google Scholar] [CrossRef] [Green Version]
- Harfterkamp, M.; van de Loo-Neus, G.; Minderaa, R.B.; van der Gaag, R.-J.; Escobar, R.; Schacht, A.; Pamulapati, S.; Buietelaar, J.K.; Hoekstra, P.J. A randomized double-blind study of atomoxetine versus placebo for attention-deficit/hyperactivity disorder symptoms in children with autism spectrum disorders. J. Am. Acad. Child Adol. Psychiatry 2012, 51, 733–741. [Google Scholar] [CrossRef]
- Guy, W. ECDEU Assessment Manual for Psychopharmacology, Revised.; Department of Health, Education, and Welfare Publication (ADM): National Institute of Mental Health: Rockville, MD, USA, 1976; pp. 76–338. [Google Scholar]
- Scahill, L.; McCracken, J.T.; King, B.H.; Rockhill, C.; Shah, B.; Politte, L.; Sanders, R.; Minjarez, M.; Cowen, J.; Mullett, J.; et al. Research Unites on Pediatric Psychopharmacology Autism Network. Extended-release Guanfacine for hyperactivity in children with autism spectrum disorder. Am. J. Psychiatry 2015, 172, 1197–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandina, G.J.; Bossie, C.A.; Youssef, E.; Zhu, Y.; Dunbar, F. Risperidone improves behavioral symptoms in children with autism in a randomized, double-blind, placebo-controlled trial. J. Autism Dev. Disord. 2007, 37, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Aman, M.G.; Singh, N.N. Aberrant Behavior Checklist Manual; Slosson Publications: East Aurora, NY, USA, 1986. [Google Scholar]
- Levine, S.Z.; Kodesh, A.; Goldberg, Y.; Reichenberg, A.; Furukawa, T.A.; Kolevzon, A.; Leucht, S. Initial severity and efficacy of risperidone in autism: Results from the RUPP trial. Eur. Psychiatry. 2016, 32, 16–20. [Google Scholar] [CrossRef]
- Wink, L.K.; Early, M.; Schaefer, T.; Pottenger, A.; Horn, P.; MDougle, C.J.; Erickson, C.A. Body mass index change in autism spectrum disorders: Comparison of treatment with risperidone and aripiprazole. J. Child Adolesc. Psychopharmacol. 2014, 24, 78–82. [Google Scholar] [CrossRef] [Green Version]
- Hollander, E.; Chaplin, W.; Soorya, L.; Wasserman, S.; Novotny, S.; Rusoff, J.; Feirsen, N.; Pepa, L.; Anagnostou, E. Divalproex sodium vs placebo for the treatment of irritability in children and adolescents with autism spectrum disorders. Neuropsychopharmacol 2010, 35, 990–998. [Google Scholar] [CrossRef] [Green Version]
- Hellings, J.A.; Weckbaugh, M.; Nickel, E.J.; Cain, S.E.; Zarcone, J.R.; Reese, R.M.; Hall, S.; Ermer, D.J.; Tsai, L.Y.; Schroeder, S.R.; et al. A double-blind, placebo-controlled study of valproate for aggression in youth with pervasive developmental disorders. J. Child Adolesc. Psychopharmacol. 2005, 15, 682–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzone, L.; Ruta, L. Topiramate in children with autistic spectrum disorders. Brain Dev. 2006, 28, 668. [Google Scholar] [CrossRef] [PubMed]
- Rezaei, V.; Mohammadi, M.R.; Ghanizadeh, A.; Sahraian, A.; Tabrizi, M.; Rezazadeh, S.-A.; Akhondzadeh, S. Double-blind, placebo-controlled trial of risperidone plus topiramate in children with autistic disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry 2010, 34, 1269–1272. [Google Scholar] [CrossRef] [PubMed]
- Swatzyna, R.J.; Boutros, N.N.; Genovese, A.C.; MacInerney, E.K.; Roark, A.J.; Kozlowski, G.P. Electroencephalogram (EEG) for children with autism spectrum disorder: Evidential considerations for routine screening. Eur. Child Adolesc. Psychiatry 2019, 28, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Hollander, E.; Soorya, L.; Chaplin, W.; Anagnostou, E.; Taylor, C.P.; Ferretti, C.J.; Wasserman, S.; Swanson, E.; Settipani, C. A double-blind placebo-controlled trial of fluoxetine for repetitive behaviors and global severity in adult autism spectrum disorders. Am. J. Psychiatry 2012, 169, 292–299. [Google Scholar] [CrossRef]
- Williams, K.; Brignell, A.; Randall, M.; Silove, N.; Hazell, P. Selective serotonin reuptake inhibitors (SSRIs) for autism spectrum disorders (ASD). Cochrane Database Syst. Rev. 2013, 8, CD004677. [Google Scholar] [CrossRef] [PubMed]
- Souders, M.C.; Zavodny, S.; Eriksen, W.; Sinko, R.; Connell, J.; Kerns, C.; Schaaf, R.; Pinto-Martin, J. Sleep in children with autism spectrum disorder. Curr. Psychiatry Rep. 2017, 19, 34. [Google Scholar] [CrossRef] [PubMed]
- Blackmer, A.B.; Feinstein, J.A. Management of sleep disorders in children with neurodevelopmental disorders: A review. Pharmacother 2016, 36, 84–98. [Google Scholar] [CrossRef] [PubMed]
- Ming, X.; Gordon, E.; Kang, N.; Wagner, G.C. Use of clonidine in children with autism spectrum disorders. Brain Dev. 2008, 30, 454–460. [Google Scholar] [CrossRef]
- Butler, M.G. Pharmacogenetics and psychiatric care: A review and commentary. J. Ment. Health Clin. Psychol. 2018, 2, 17–24. [Google Scholar] [CrossRef]
- Weinshilboum, R.M.; Wang, L. Pharmacogenetics and pharmacogenomics: Development, science and translation. Annu Rev. Genom. Hum. Genet. 2006, 7, 223–245. [Google Scholar] [CrossRef]
- Weng, L.; Zhang, L.; Peng, Y.; Huang, R.S. Pharamcogenetics and pharmacogenomics: A bridge to individualized cancer therapy. Pharmacogenomics 2013, 14, 315–324. [Google Scholar] [CrossRef] [Green Version]
- Kirchheiner, J.; Seeringer, A. Clinical implications of pharmacogenetics of cytochrome P450 drug metabolizing enzymes. Biochim. Biophys. Acta 2007, 1770, 489–494. [Google Scholar] [CrossRef]
- Reynolds, K.S. Achieving the promise of personalized medicine. Clin. Pharmacol. Ther. 2012, 92, 401–405. [Google Scholar] [CrossRef]
- Lee, J.W.; Aminken, F.; Bhavsar, A.P.; Shaw, K.; Carleton, B.C.; Hayden, M.R.; Ross, C.J.D. The emerging era of pharmacogenomics: Current successes, future potential, and challenges. Clin. Genet. 2014, 86, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Kalow, W. Human pharmacogenomics: The development of a science. Hum. Genom. 2004, 1, 375–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werck-Reichhart, D.; Feyereisen, R. Cytochromes P450: A success story. Genome Biol. 2000, 1, 3003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samer, C.F.; Lorenzini, K.I.; Rollason, V.; Daali, Y.; Desmeues, J.A. Applications of CYP450 testing in the clinical setting. Mol. Diagn. Ther. 2013, 17, 165–184. [Google Scholar] [CrossRef] [Green Version]
- Danielson, P.B. The cytochrome P450 superfamily: Biochemistry, evolution and drug metabolism in humans. Curr. Drug Metab. 2002, 3, 561–597. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, Y.; Guan, M.X. A peep into mitochondrial disorder: Multifaceted from mitochondrial DNA mutations to nuclear gene modulation. Protein Cell 2015, 6, 862–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaefer, G.B. Clinical genetic aspects of ASD spectrum disorders. Int. J. Mol. Sci. 2016, 17, 180. [Google Scholar] [CrossRef] [Green Version]
- Oikonomakis, V.; Kosma, K.; Mitrakos, A.; Sofocleous, C.; Pervanidou, P.; Syrmou, A.; Pampanos, A.; Psoni, S.; Fryssira, H.; Kanavakis, E.; et al. Recurrent copy number variations as risk factors for autism spectrum disorders: Analysis of the clinical implications. Clin. Genet. 2016, 89, 708–718. [Google Scholar] [CrossRef]
- Hall, L.; Kelley, E. The contribution of epigenetics to understanding genetic factors in autism. Autism 2014, 18, 872–881. [Google Scholar] [CrossRef]
- Hagerman, R.J.; Berry-Kravis, E.; Hazlett, H.C.; Bailey, D.B., Jr.; Moine, H.; Kooy, R.F.; Tassone, F.; Gantois, I.; Sonenberg, N.; Mandel, J.L.; et al. Fragile X Syndrome. Nat. Rev. Dis. Primers 2017, 29, 17065. [Google Scholar] [CrossRef]
- Rafi, S.K.; Butler, M.G. The 15q11.2 BP1-BP2 Microdeletion (Burnside–Butler) Syndrome: In Silico Analyses of the Four Coding Genes Reveal Functional Associations with Neurodevelopmental Phenotypes. Int. J. Med. Sci. 2020, 21, 3296. [Google Scholar] [CrossRef]
- Hartin, S.M.; Hossain, W.A.; Butler, M.G.; Francis, D.; Godler, D.E.; Barkataki, S. Analysis of the Prader-Willi syndrome imprinting center using droplet digitcal PCR and Next-Generation whole-exome sequencing. Mol. Genet. Genom. Med. 2019, 7, e00575. [Google Scholar] [CrossRef] [PubMed] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Genovese, A.; Butler, M.G. Clinical Assessment, Genetics, and Treatment Approaches in Autism Spectrum Disorder (ASD). Int. J. Mol. Sci. 2020, 21, 4726. https://doi.org/10.3390/ijms21134726
Genovese A, Butler MG. Clinical Assessment, Genetics, and Treatment Approaches in Autism Spectrum Disorder (ASD). International Journal of Molecular Sciences. 2020; 21(13):4726. https://doi.org/10.3390/ijms21134726
Chicago/Turabian StyleGenovese, Ann, and Merlin G. Butler. 2020. "Clinical Assessment, Genetics, and Treatment Approaches in Autism Spectrum Disorder (ASD)" International Journal of Molecular Sciences 21, no. 13: 4726. https://doi.org/10.3390/ijms21134726
APA StyleGenovese, A., & Butler, M. G. (2020). Clinical Assessment, Genetics, and Treatment Approaches in Autism Spectrum Disorder (ASD). International Journal of Molecular Sciences, 21(13), 4726. https://doi.org/10.3390/ijms21134726