Tumors Resistant to Checkpoint Inhibitors Can Become Sensitive after Treatment with Vascular Disrupting Agents


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
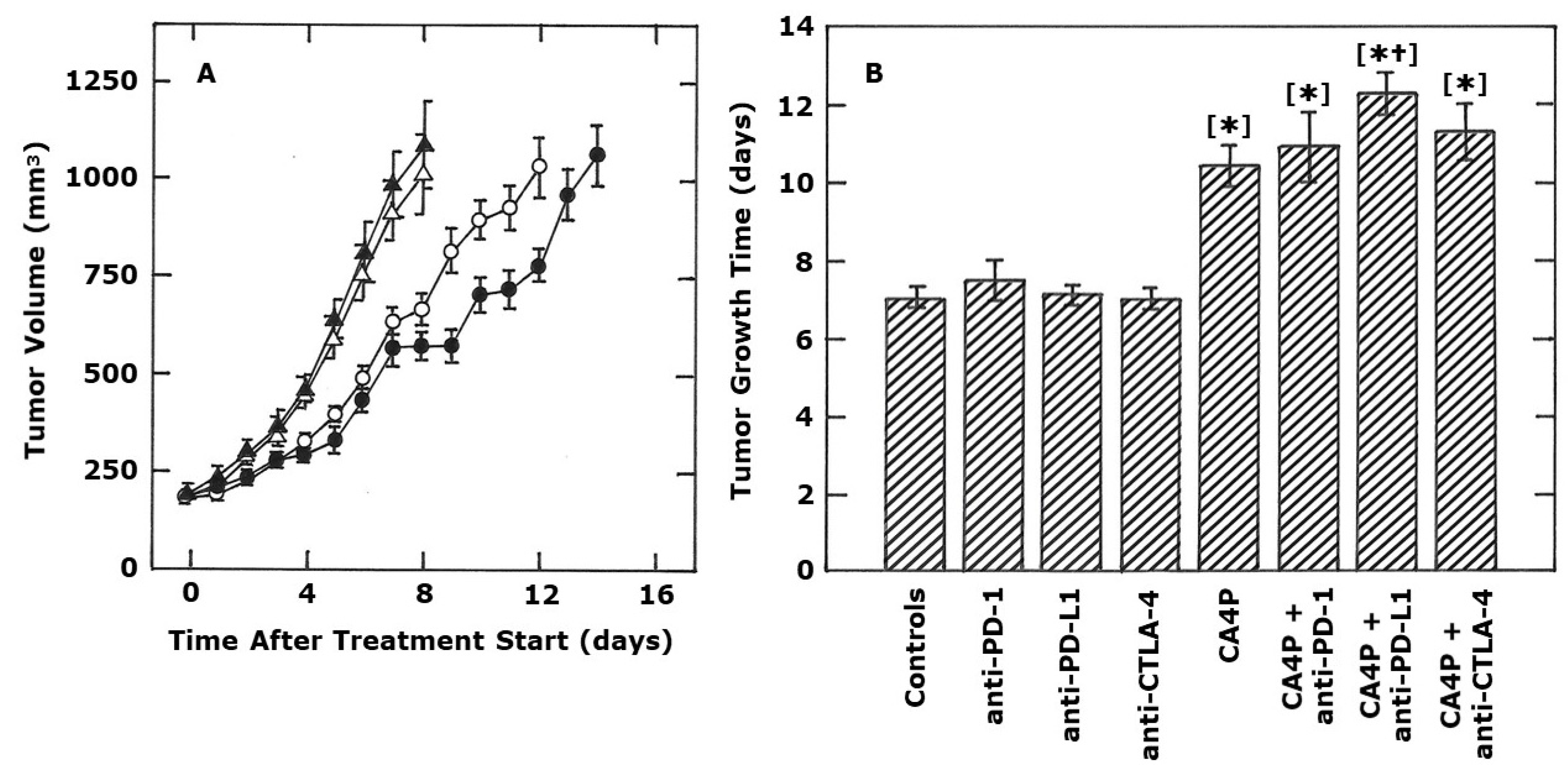
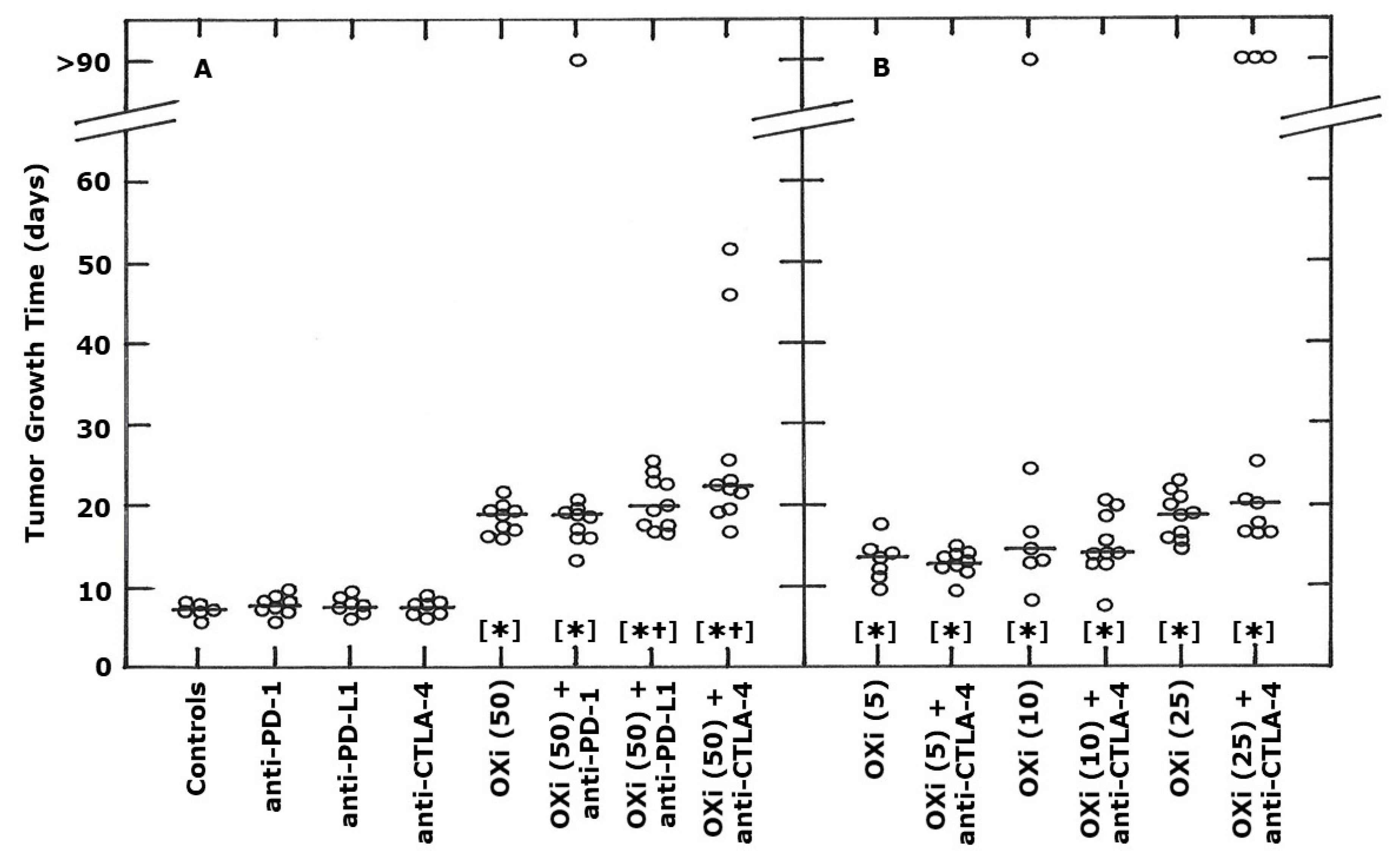
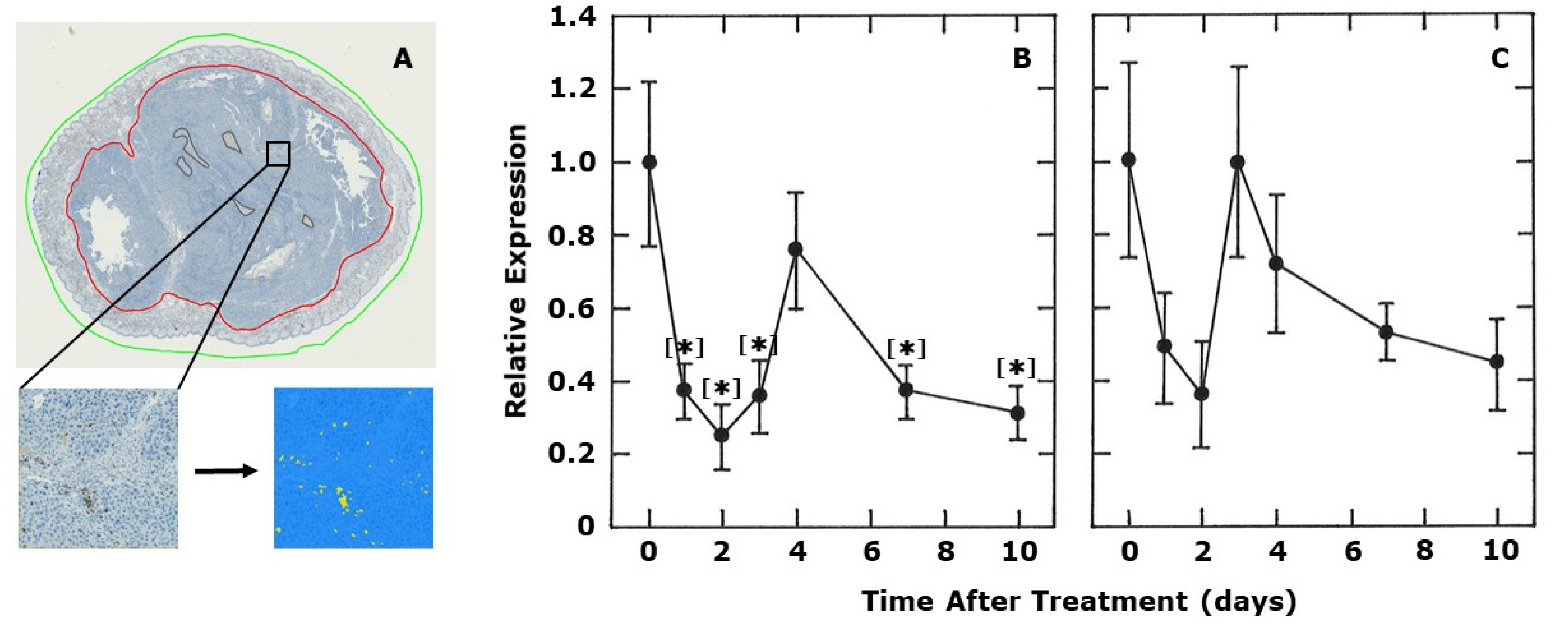
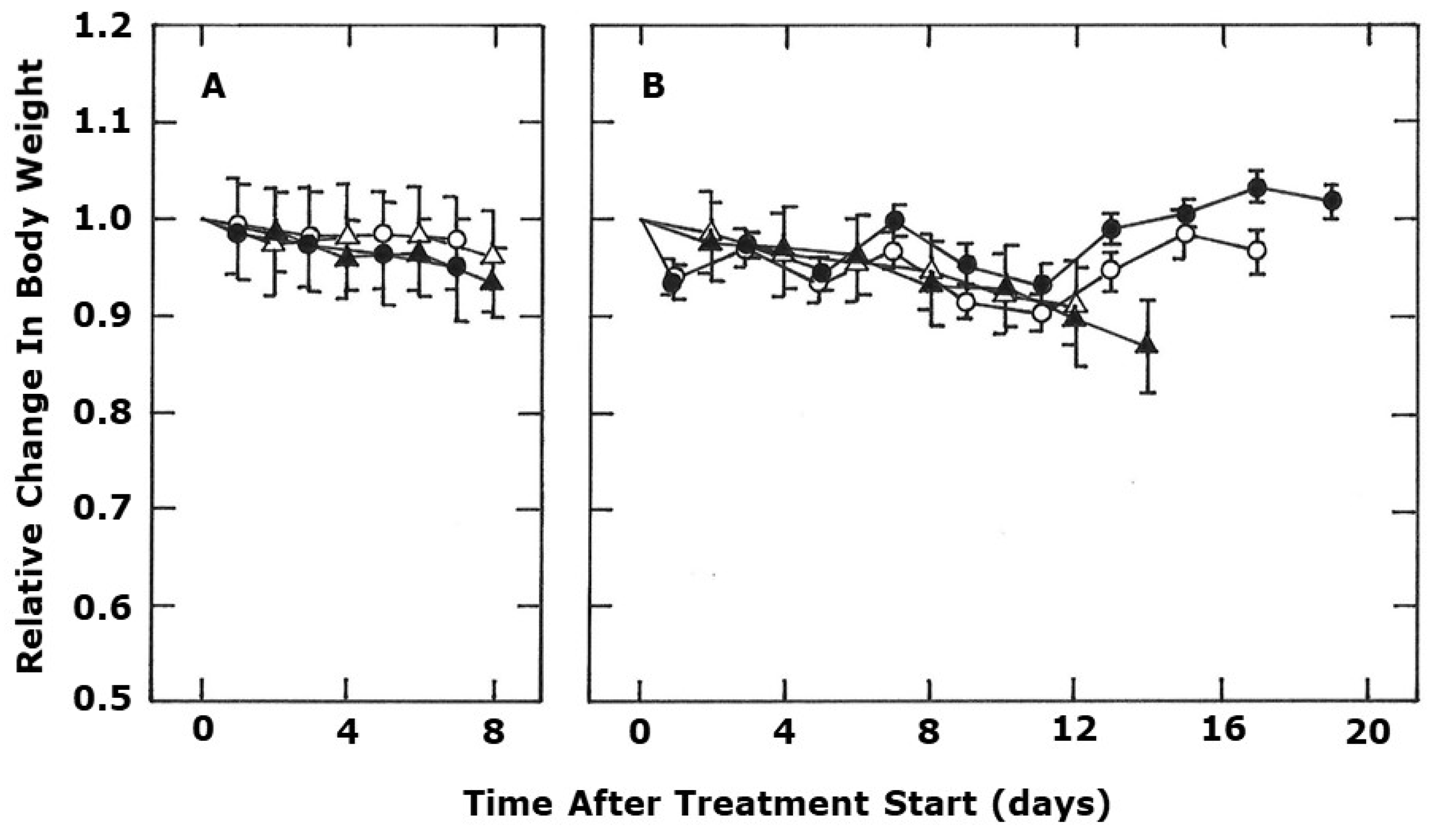
2. Results
3. Discussion
4. Materials and Methods
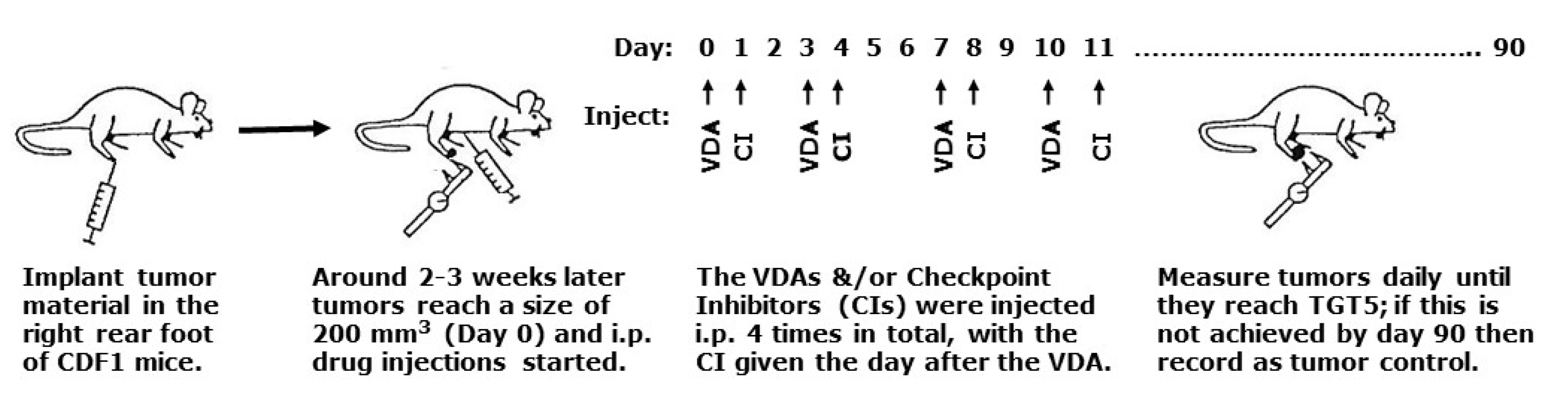
4.1. Animal and Tumor Model
4.2. Drug Preparation and Treatment
4.3. Tumor Response Assessment
4.4. CD4 and CD8 Immunohistochemistry
4.5. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| VDAs | Vascular disrupting agents |
| CA4P | Combretastatin A-4 phosphate |
| OXi | OXi4503 |
| Anti-PD-1 | Anti-programmed death 1 |
| Anti-PD-L1 | Anti-programmed death 1 ligand |
| Anti-CTLA-4 | Anti-cytotoxic T-lymphocyte-associated protein 4 |
| APCs | Antigen presenting cells |
| ICPs | Immune checkpoint proteins |
| DAMPs | Damage associated molecular patterns |
| HIF | Hypoxia inducible factor |
| i.p. | Intraperitoneal |
| TGT5 | Tumor growth time 5; days taken to grow to 5 times treatment volume |
| CIs | Checkpoint Inhibitors |
| SEM | Standard Error of the Mean |
References
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The immunobiology of cancer immunosurveillance and immunoediting. Immunity 2004, 21, 137–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brem, S.; Brem, H.; Folkman, J.; Finkelstein, D.; Patz, A. Prolonged tumor dormancy by prevention of neo-vascularization in the vitreous. Cancer Res. 1976, 36, 2807–2812. [Google Scholar] [PubMed]
- Folkman, J. How is blood vessel growth regulated in normal and neoplastic tissue? Cancer Res. 1986, 46, 467–473. [Google Scholar] [PubMed]
- Horsman, M.R.; Vaupel, P. Pathophysiological basis for the formation of the tumor microenvironment. Front. Oncol. Res. Top. 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Salama, A.K.S.; Moschos, S.J. Next steps in immuno-oncology: Enhancing antitumor effects through appropriate patient selection and rationally designed combination strategies. Ann. Oncol. 2017, 28, 57–74. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef]
- Drake, C.G.; Lipson, E.J.; Brahmer, J.R. Breathing new life into immunotherapy: Review of melanoma, lung and kidney cancer. Nat. Rev. Clin. Oncol. 2014, 11, 24–37. [Google Scholar] [CrossRef]
- Callahan, M.K.; Postow, M.A.; Wolchok, J.P. CTLA-4 and PD-1 pathway blockade: Combinations in the clinic. Front. Oncol. 2015, 4, 385. [Google Scholar] [CrossRef] [Green Version]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Yokoi, T.; Chiappori, A.; Lee, K.H.; de Wit, M.; et al. Durvalumab after chemoradiotherapy in stage III non-small-cell lung cancer. NEJM 2017, 377, 1919–1929. [Google Scholar] [CrossRef]
- Syn, N.L.; Teng, M.W.L.; Mok, T.S.K.; Soo, R.A. De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol. 2017, 18, e731–e741. [Google Scholar] [CrossRef]
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune Checkpoint Blockade in Cancer Therapy. J. Clin. Oncol. 2015, 33, 1974–1982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauber, K.; Brix, N.; Ernst, A.; Hennel, R.; Krombach, J.; Anders, H.; Belka, C. Targeting the heat shock response in combination with radiotherapy: Sensitizing cancer cells to irradiation-induced cell death and heating up their immunogenicity. Cancer Letts. 2015, 368, 209–229. [Google Scholar] [CrossRef] [PubMed]
- Stone, H.B.; Peters, L.J.; Milas, L. Effect of host immune capability on radiocurability and subsequent transplantability of a murine fibrosarcoma. J. Natl. Cancer Inst. 1979, 63, 1229–1235. [Google Scholar] [PubMed]
- Abuodeh, Y.; Venkat, P.; Kim, S. Systematic review of case reports on the abscopal effect. Curr. Probl. Cancer 2016, 40, 25–37. [Google Scholar] [CrossRef]
- Demaria, S.; Kawashima, N.; Yang, A.M.; Devitt, M.L.; Babb, J.S.; Allison, J.P.; Formenti, S.C. Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clin. Cancer Res. 2005, 11, 728–734. [Google Scholar]
- Dewan, M.Z.; Galloway, A.E.; Kawashima, N.; Dewyngaert, J.K.; Babb, J.S.; Formenti, S.C.; Demaria, S. Fractionated but Not Single-Dose Radiotherapy Induces an Immune-Mediated Abscopal Effect when Combined with Anti–CTLA-4 Antibody. Clin. Cancer Res. 2009, 15, 5379–5388. [Google Scholar] [CrossRef]
- Dovedi, S.J.; Adlard, A.L.; Lipowska-Bhalla, G.; McKenna, C.; Jones, S.; Cheadle, E.J.; Stratford, I.J.; Poon, E.; Morrow, M.; Stewart, R.; et al. Acquired Resistance to Fractionated Radiotherapy Can Be Overcome by Concurrent PD-L1 Blockade. Cancer Res. 2014, 74, 5458–5468. [Google Scholar] [CrossRef] [Green Version]
- Durante, M.; Reppingen, N.; Held, K.D. Immunologically augmented cancer treatment using modern radiotherapy. Trends Mol. Med. 2013, 19, 565–582. [Google Scholar] [CrossRef]
- Bernier, J. Immuno-oncology: Allying forces of radio- and immuno-therapy to enhance cancer cell killing. Crit. Rev. Oncol. Hematol. 2016, 108, 97–108. [Google Scholar] [CrossRef]
- Van Limbergen, E.J.; De Ruysscher, D.K.; Olivo Pimentel, V.; Marcus, D.; Berbee, M.; Hoeben, A.; Rekers, N.; Theys, J.; Yaromina, A.; Dubois, L.J.; et al. Combining radiotherapy with immunotherapy: The past, the present and the future. Br. J. Radiol. 2017, 90, 20170157. [Google Scholar] [CrossRef] [Green Version]
- Moulder, J.E.; Rockwell, S. Hypoxic fractions of solid tumors: Experimental techniques, methods of analysis, and a survey of existing data. Int. J. Radiat. Oncol. Biol. Phys. 1984, 10, 695–712. [Google Scholar] [CrossRef]
- Vaupel, P.; Kallinowski, F.; Okunieff, P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: A review. Cancer Res. 1989, 49, 6449–6465. [Google Scholar]
- Horsman, M.R.; Mortensen, L.S.; Petersen, J.B.; Busk, M.; Overgaard, J. Imaging hypoxia to improve radiotherapy outcome. Nat. Rev. Clin. Oncol. 2012, 9, 674–687. [Google Scholar] [CrossRef]
- Siemann, D.W.; Horsman, M.R. Modulation of the tumor vasculature and oxygenation to improve therapy. Pharmacol. Ther. 2015, 153, 107–124. [Google Scholar] [CrossRef] [Green Version]
- Horsman, M.R.; Overgaard, J. The impact of hypoxia and its modification on the outcome of radiotherapy. J. Radiat. Res. 2016, 57, i90–i98. [Google Scholar] [CrossRef] [Green Version]
- Vaupel, P.; Multhoff, G. Accomplices of the hypoxic tumor microenvironment compromising antitumor activity: Adenosine, lactate, acidosis, vascular endothelial growth factor, potassium ions, and phosphatidylserine. Front. Immunol. 2017, 8, 1887. [Google Scholar] [CrossRef] [Green Version]
- Multhoff, G.; Vaupel, P. Hypoxia compromises anti-cancer immune responses. Adv. Exp. Med. Biol. 2020, 1232, 131–143. [Google Scholar] [CrossRef]
- Siemann, D.W.; Bibby, M.C.; Dark, G.G.; Dicker, A.P.; Eskens, F.A.; Horsman, M.R.; Marmé, D.; LoRusso, P.M. Differentiation and definition of vascular-targeted therapies. Clin. Cancer Res. 2005, 11, 416–420. [Google Scholar] [PubMed]
- Horsman, M.R.; Siemann, D.W. Pathophysiological effects of vascular targeting agents and the implications for combination with conventional therapies. Cancer Res. 2006, 66, 11520–11539. [Google Scholar] [CrossRef] [Green Version]
- National Cancer Institute Web Site. Available online: http://www.cancer.gov/clinicaltrials (accessed on 1 March 2020).
- Siemann, D.W.; Chaplin, D.J.; Horsman, M.R. Realizing the potential of vascular targeted therapy: The rationale for combining vascular disrupting agents and anti-angiogenic agents to treat cancer. Cancer Invest. 2017, 35, 519–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overgaard, K.; Overgaard, J. Investigations on the possibility of a thermic tumour therapy—1. Short-wave treatment of a transplanted isologous mouse mammary carcinoma. Eur. J. Cancer 1972, 8, 65–78. [Google Scholar] [CrossRef]
- Overgaard, J. Simultaneous and sequential hyperthermia and radiation treatment of an experimental tumor and its surrounding normal tissue in vivo. Int. J. Radiat. Oncol. Biol. Phys. 1980, 6, 1507–1517. [Google Scholar] [CrossRef]
- Tozer, G.M.; Kanthou, C.; Baguley, B.C. Disrupting tumour blood vessels. Nat. Rev. Cancer 2005, 5, 423–435. [Google Scholar] [CrossRef]
- Siemann, D.W.; Chaplin, D.J.; Walicke, P.A. A review and update of the current status of the vasculature-disabling agent combretastatin-A4 phosphate (CA4P). Expert Opin. Invest. Drugs 2009, 18, 189–197. [Google Scholar] [CrossRef]
- Horsman, M.R.; Murata, R.; Breidahl, T.; Nielsen, F.U.; Maxwell, R.J.; Stødkiled-Jørgensen, H.; Overgaard, J. Combretastatins: Novel vascular targeting drugs for improving anti-cancer therapy. Adv. Exp. Med. Biol. 2000, 476, 311–323. [Google Scholar]
- Murata, R.; Overgaard, J.; Horsman, M.R. Comparative effects of combretastatin A-4 disodium phosphate and 5,6-dimethylxanthenone-4-acetic acid on blood perfusion in a murine tumour and normal tissues. Int. J. Radiat. Biol. 2001, 77, 195–204. [Google Scholar] [CrossRef]
- Murata, R.; Siemann, D.W.; Overgaard, J.; Horsman, M.R. Interaction between combretastatin A-4 disodium phosphate and radiation in murine tumours. Radiother Oncol. 2001, 60, 155–161. [Google Scholar] [CrossRef]
- Iversen, A.B.; Busk, M.; Bertelsen, L.B.; Laustsen, C.; Munk, O.L.; Nielsen, T.; Wittenborn, T.R.; Bussink, J.; Lok, J.; Stødkilde-Jørgensen, H.; et al. The potential of hyperpolarized 13C magnetic resonance spectroscopy to monitor the effect of combretastatin based vascular disrupting agents. Acta Oncol. 2017, 56, 1626–1633. [Google Scholar] [CrossRef] [Green Version]
- Murata, R.; Overgaard, J.; Horsman, M.R. Combretastatin A-4 disodium phosphate: A vascular targeting agent that improves the anti-tumor effects of hyperthermia, radiation and mild thermoradiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2001, 51, 1018–1024. [Google Scholar] [CrossRef]
- Salmon, H.W.; Siemann, D.W. Effect of the second-generation vascular disrupting agent OXi4503 on tumor vascularity. Clin. Cancer Res. 2006, 12, 4090–4094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folkes, L.K.; Christlieb, M.; Madej, E.; Stratford, M.R.L.; Wardman, P. Oxidative metabolism of combretastatin A-1 produces quinine intermediates with the potential to bind to nucleophiles and to enhance oxidative stress via free radicals. Chem. Res. Toxicol. 2007, 20, 1885–1894. [Google Scholar] [CrossRef] [PubMed]
- Rankin, E.B.; Giaccia, A.J. Hypoxic control of metastasis. Science 2016, 352, 175–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Triner, D.; Shah, Y.M. Hypoxia-inducible factors: A central link between inflammation and cancer. J. Clin. Invest. 2016, 126, 3689–3698. [Google Scholar] [CrossRef]
- Sørensen, B.S.; Horsman, M.R. Tumor hypoxia: Impact on radiation therapy and molecular pathways. Front. Oncol. 2020, 10, 562. [Google Scholar] [CrossRef]
- Barsoum, I.B.; Koti, M.; Siemens, D.R.; Graham, C.H. Mechanisms of hypoxia-mediated immune escape in cancer. Cancer Res. 2014, 74, 7185–7190. [Google Scholar] [CrossRef] [Green Version]
- Hatfield, S.M.; Kjaergaard, J.; Lukashev, D.; Schreiber, T.H.; Belikoff, B.; Abbott, R.; Sethumadhavan, S.; Philbrook, P.; Ko, M.; Cannici, R.; et al. Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci. Transl. Med. 2015, 7, 277. [Google Scholar] [CrossRef] [Green Version]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef]
- Petrova, V.; Annicchiarico-Petruzzelli, M.; Melino, G.; Amelio, I. The hypoxic tumour microenvironment. Oncogenesis 2018, 7, 10. [Google Scholar] [CrossRef]
- Vaupel, P.; Multhoff, G. Fatal alliance of hypoxia-/HIF-1α-driven microenvironmental traits promoting cancer progression. Adv. Exp. Med. Biol. 2020, 1232, 169–176. [Google Scholar] [CrossRef]
- Horsman, M.R.; Khalil, A.A.; Nordsmark, M.; Grau, C.; Overgaard, J. Relationship between radiobiological hypoxia and direct estimates of tumour oxygenation in a mouse tumour model. Radiother Oncol. 1993, 28, 69–71. [Google Scholar] [CrossRef]
- Horsman, M.R.; Khalil, A.A.; Siemann, D.W.; Grau, C.; Hill, S.A.; Lynch, E.M.; Chaplin, D.J.; Overgaard, J. Relationship between radiobiological hypoxia in tumors and electrode measurements of tumor oxygenation. Int. J. Radiat. Oncol. Biol. Phys. 1994, 29, 439–442. [Google Scholar] [CrossRef]
- Bentzen, L.; Keiding, S.; Horsman, M.R.; Gronroos, T.; Hansen, S.B.; Overgaard, J. Assessment of hypoxia in experimental mice tumours by [18F]fluoromisonidazole PET and pO2 electrode measurements: Influence of tumour volumes and carbogen breathing. Acta Oncol. 2002, 41, 304–312. [Google Scholar] [CrossRef]
- Bentzen, L.; Keiding, S.; Horsman, M.R.; Falborg, L.; Hansen, S.B.; Overgaard, J. Feasibility of detecting hypoxia in experimental mouse tumours with 18F-fluorinated tracers and positron emission tomography: A study evaluat ing [18F]fluoromisonidazole and [18F]fluoro-2-deoxy-d-glucose. Acta Oncol. 2000, 39, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Siemann, D.W.; Warrington, K.H.; Horsman, M.R. Vascular targeting agents: Adjuvants to radiation therapy. Radiother Oncol. 2000, 57, 5–12. [Google Scholar] [CrossRef]
- Siemann, D.W.; Horsman, M.R. Targeting the tumor vasculature: A strategy to improve radiation therapy. Expert Rev. Anticancer Ther. 2004, 4, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Wittenborn, T.R.; Horsman, M.R. Targeting tumour hypoxia to improve outcome of stereotactic radiotherapy. Acta Oncol. 2015, 54, 1385–1392. [Google Scholar] [CrossRef] [Green Version]
- Iversen, A.B.; Busk, M.; Horsman, M.R. Induction of hypoxia by vascular disrupting agents and the significance for their combination with radiation therapy. Acta Oncol. 2013, 52, 1320–1326. [Google Scholar] [CrossRef] [Green Version]
- Horsman, M.R. The therapeutic potential of using the vascular disrupting agent OXi4503 to enhance mild temperature thermoradiation. Int. J. Hyperth. 2015, 31, 453–459. [Google Scholar] [CrossRef]
- Horsman, M.R. Enhancing the radiation response of tumours but not early or late responding normal tissues using a vascular disrupting agent. Acta Oncol. 2017, 56, 1634–1638. [Google Scholar] [CrossRef] [Green Version]
- Verbrugge, I.; Hagekyriakou, J.; Sharp, L.L.; Galli, M.; West, A.; McLaughlin, N.M.; Duret, H.; Yagita, H.; Johnstone, R.W.; Smyth, M.J.; et al. Radiotherapy Increases the Permissiveness of Established Mammary Tumors to Rejection by Immunomodulatory Antibodies. Cancer Res. 2012, 72, 3163–3174. [Google Scholar] [CrossRef] [PubMed] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Horsman, M.R.; Wittenborn, T.R.; Nielsen, P.S.; Elming, P.B. Tumors Resistant to Checkpoint Inhibitors Can Become Sensitive after Treatment with Vascular Disrupting Agents. Int. J. Mol. Sci. 2020, 21, 4778. https://doi.org/10.3390/ijms21134778
Horsman MR, Wittenborn TR, Nielsen PS, Elming PB. Tumors Resistant to Checkpoint Inhibitors Can Become Sensitive after Treatment with Vascular Disrupting Agents. International Journal of Molecular Sciences. 2020; 21(13):4778. https://doi.org/10.3390/ijms21134778
Chicago/Turabian StyleHorsman, Michael R., Thomas R. Wittenborn, Patricia S. Nielsen, and Pernille B. Elming. 2020. "Tumors Resistant to Checkpoint Inhibitors Can Become Sensitive after Treatment with Vascular Disrupting Agents" International Journal of Molecular Sciences 21, no. 13: 4778. https://doi.org/10.3390/ijms21134778
APA StyleHorsman, M. R., Wittenborn, T. R., Nielsen, P. S., & Elming, P. B. (2020). Tumors Resistant to Checkpoint Inhibitors Can Become Sensitive after Treatment with Vascular Disrupting Agents. International Journal of Molecular Sciences, 21(13), 4778. https://doi.org/10.3390/ijms21134778