Integrative Analyses of Widely Targeted Metabolic Profiling and Transcriptome Data Reveals Molecular Insight into Metabolomic Variations during Apple (Malus domestica) Fruit Development and Ripening



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
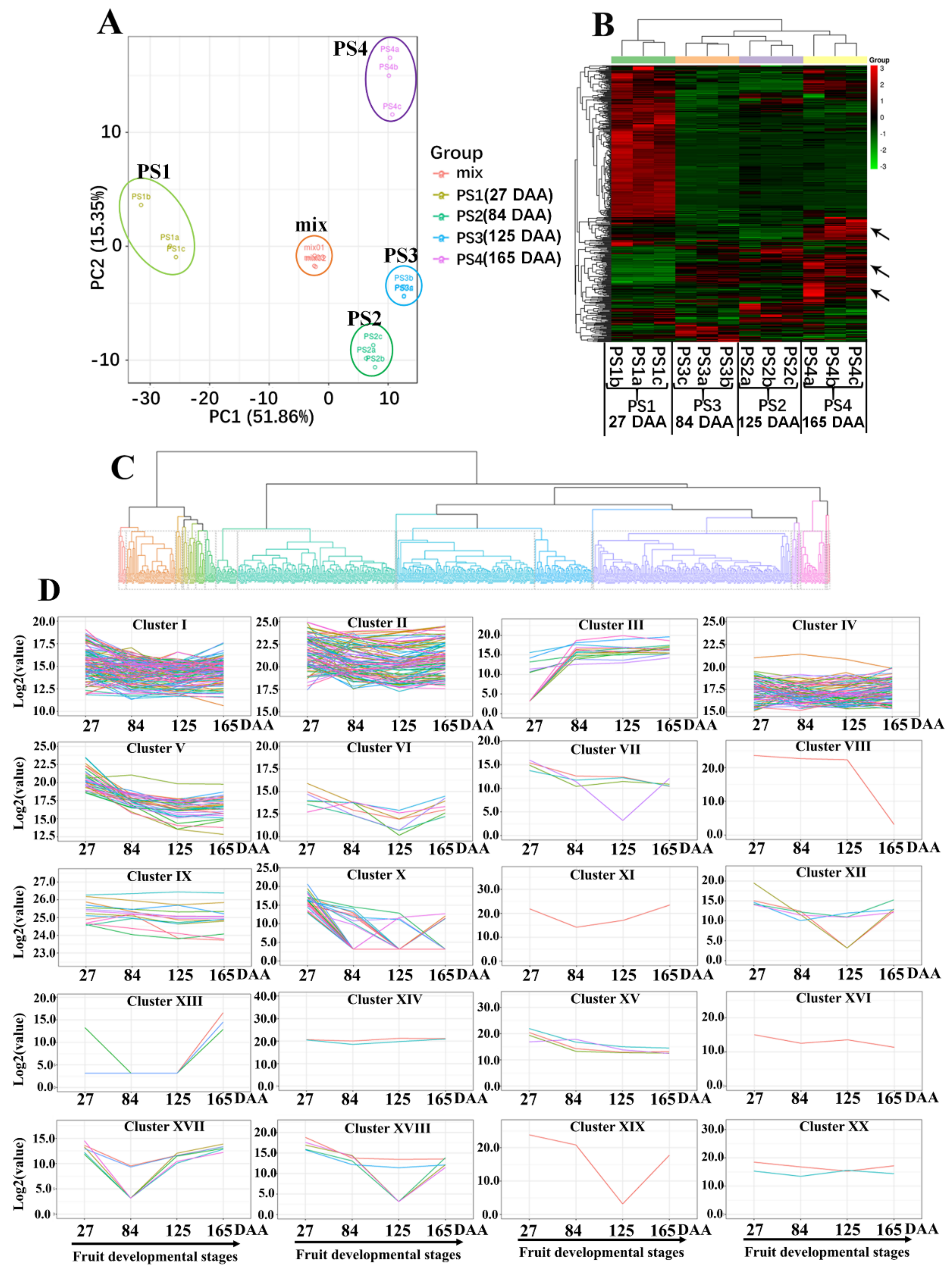
2.1. UPLC-MS Analyses of Apple Reveals Dynamic Metabolic Profiles During the Four Developmental Stages
2.2. Primary Metabolism Profiles of Apple Development and Ripening Process
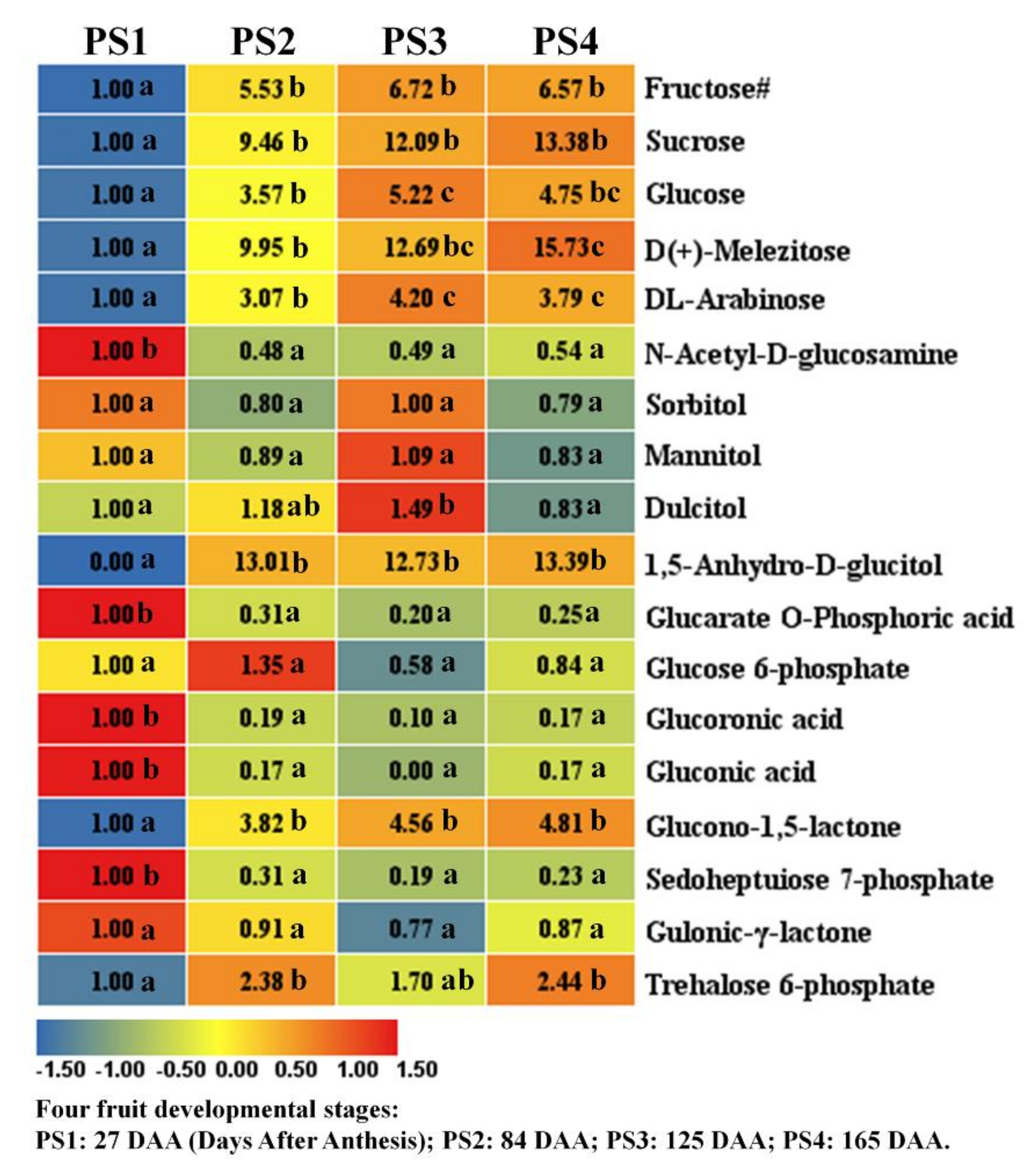
2.2.1. Metabolism of Sugar, Sugar Alcohols and Their Phosphates
2.2.2. Organic Acids Metabolism During Apple Development
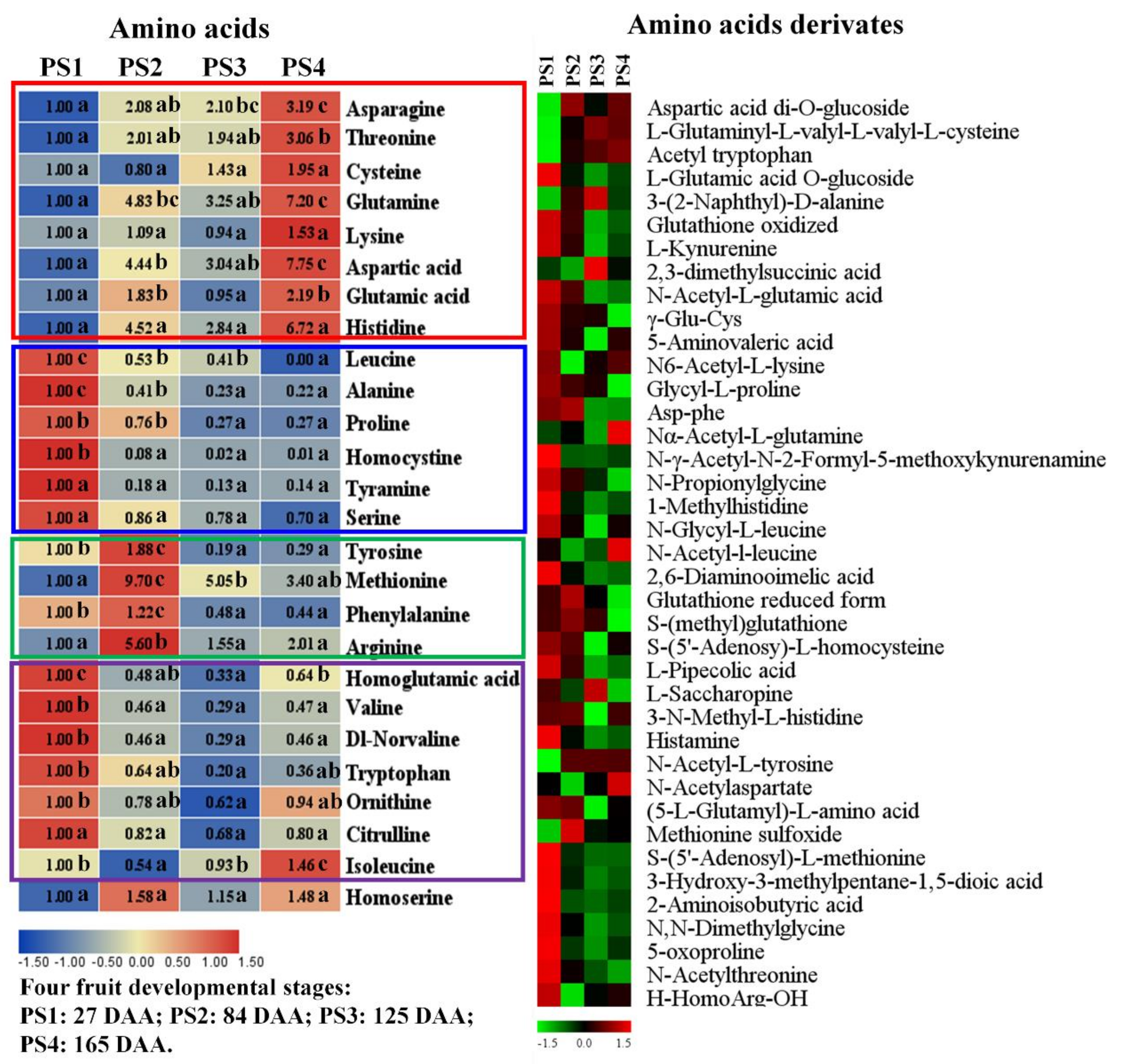
2.2.3. Accumulation Profiles of Amino Acids and Their Derivates
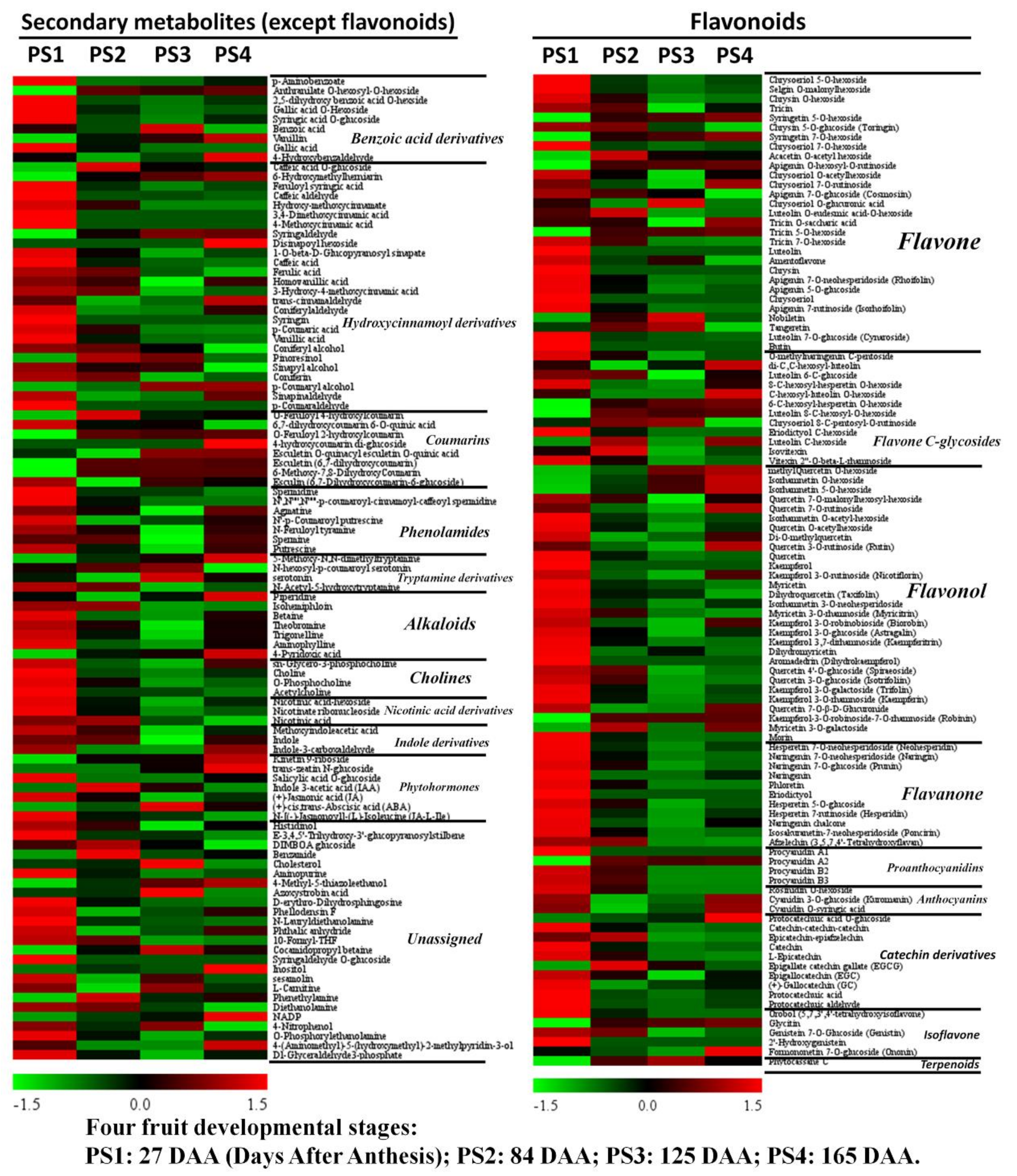
2.3. Parallel Accumulation of Secondary Metabolites During Apple Development and Ripening
2.3.1. Accumulation Profiles of Secondary Metabolites (Except Flavonoids)
Benzoic Acids
Hydroxycinnamoyl Derivatives
Polyamines
Phytohormones
2.3.2. Metabolism of Flavonoids
Flavone and Flavone C-glycosides
Flavonol
Flavanone
Proanthocyanidins and Anthocyanins
Catechin Derivatives and Isoflavones
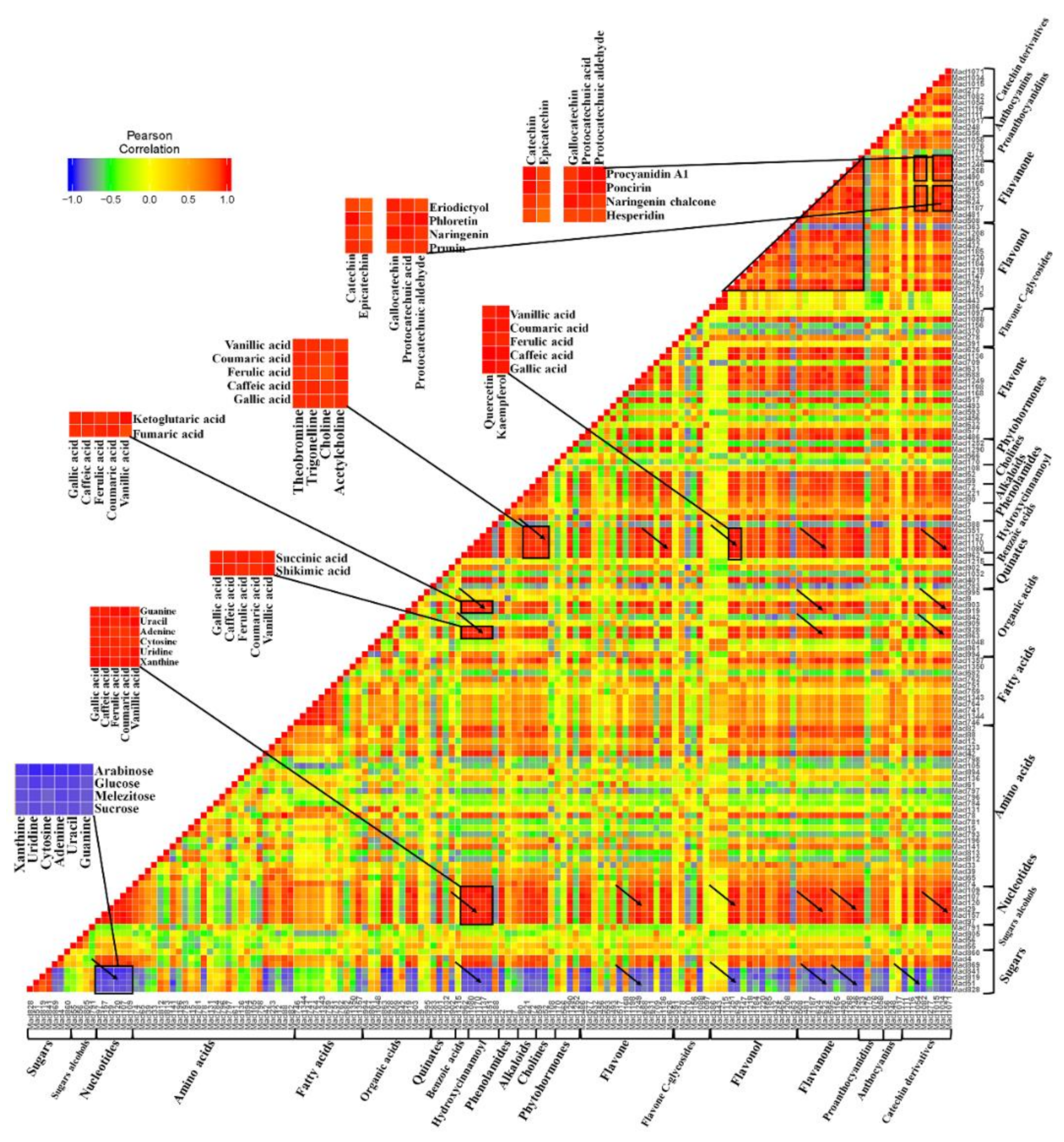
2.4. Metabolite–Metabolite Correlation During Apple Development and Ripening
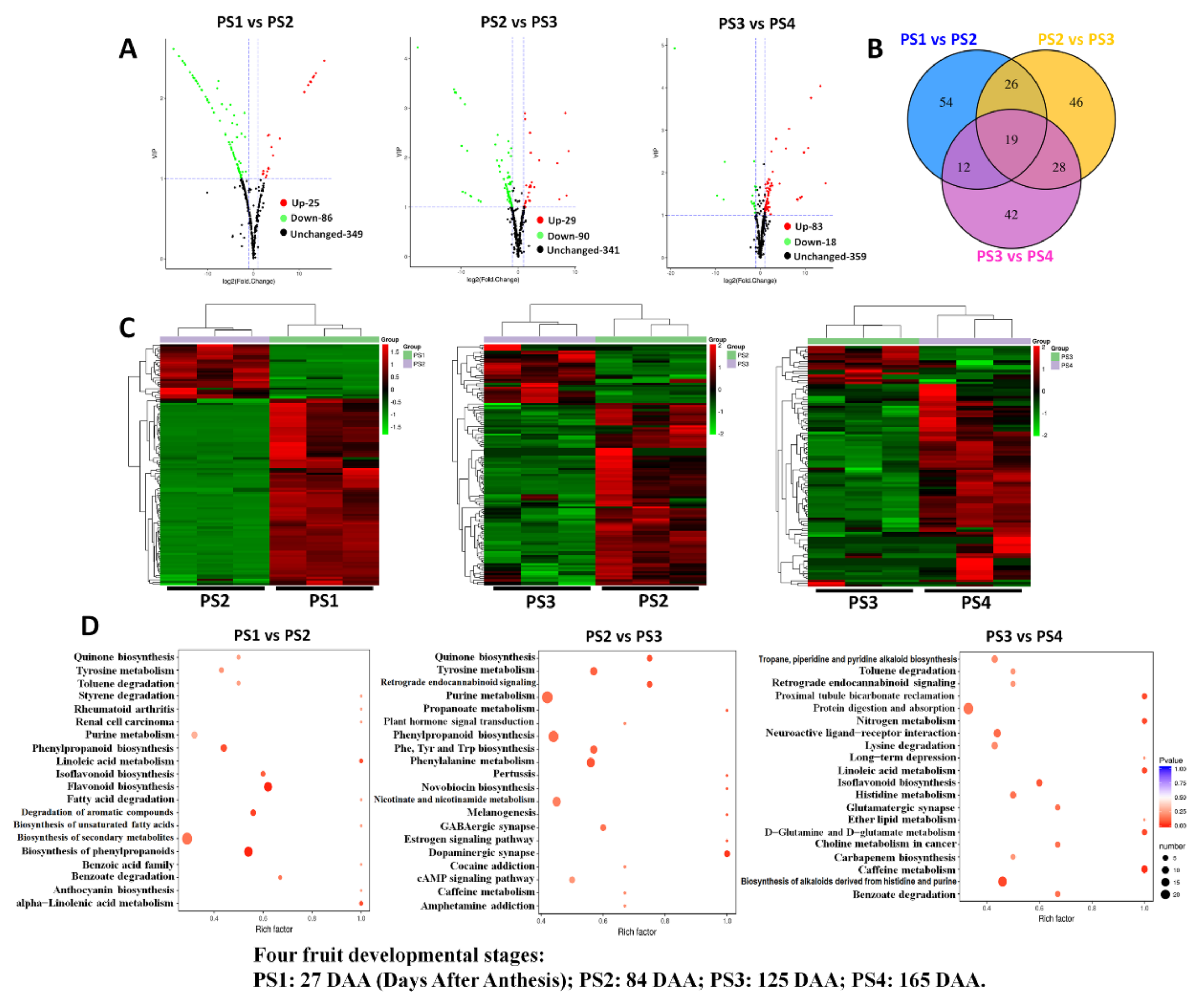
2.5. Differentially Accumulated Metabolites Among Different Fruit-Development Stages
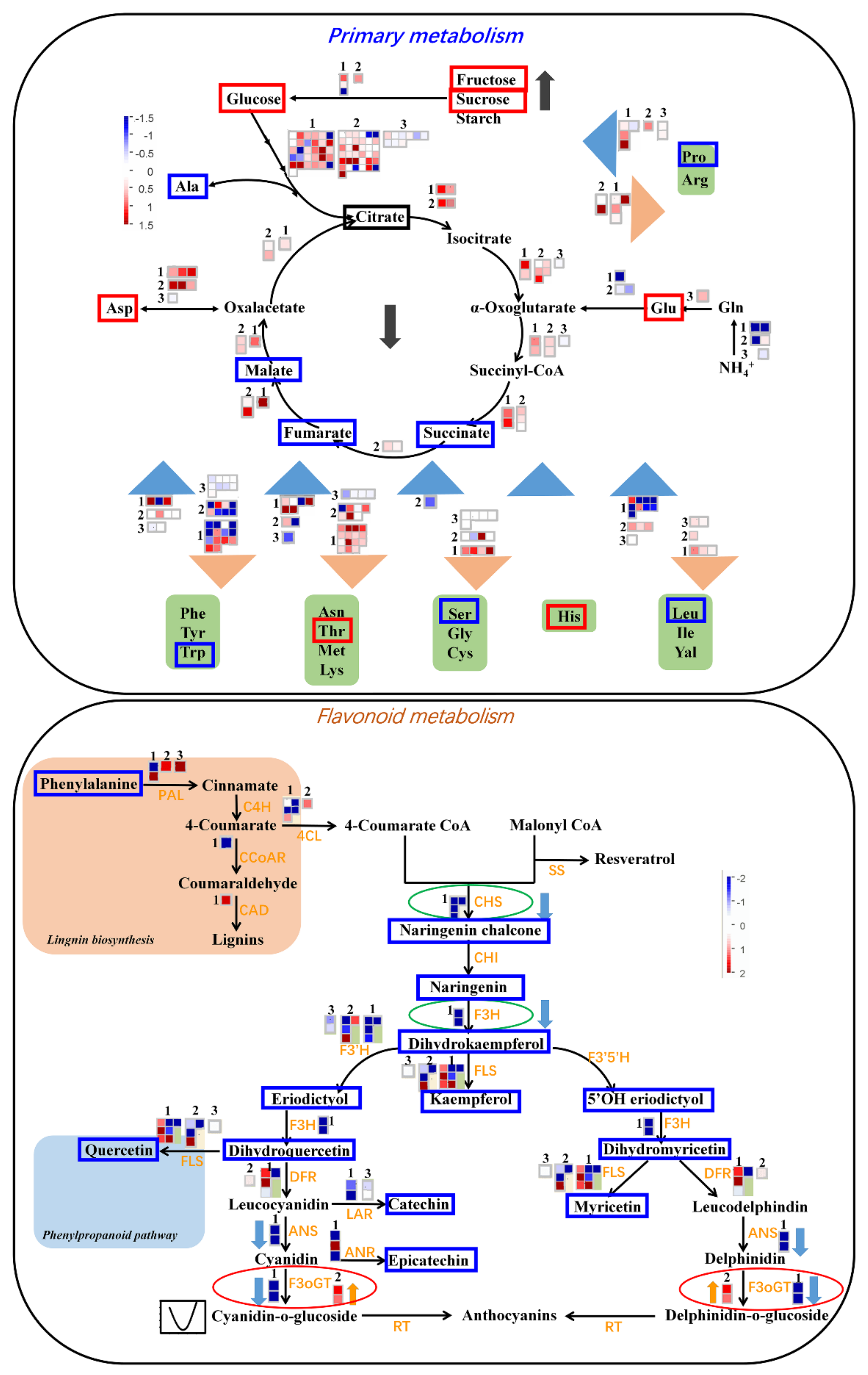
2.6. Expression Patterns Associated with Primary and Secondary Metabolisms Provide the Molecular Basis of Metabolic Changes During Apple Development and Ripening
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Sample Collection
4.2. Sample Preparation and Extraction
4.3. LC-ESI-MS/MS System-Based Widely Targeted Metabolomics Analysis
4.4. Metabolomics Data Processing and Statistical Analysis
4.5. RNA Extraction and Transcriptome Analysis
4.6. Availability of Data and Materials
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Giovannoni, J.J. Genetic regulation of fruit development and ripening. Plant Cell 2004, 16, S170–S180. [Google Scholar] [CrossRef] [Green Version]
- Holb, I.J.; Dremak, P.; Bitskey, K.; Gonda, I. Yield response, pest damage and fruit quality parameters of scab-resistant and scab-susceptible apple cultivars in integrated and organic production systems. Sci. Hortic. 2012, 145, 109–117. [Google Scholar] [CrossRef]
- Janssen, B.J.; Thodey, K.; Schaffer, R.J.; Alba, R.; Balakrishnan, L.; Bishop, R.; Bowen, J.H.; Crowhurst, R.N.; Gleave, A.P.; Ledger, S.; et al. Global gene expression analysis of apple fruit development from the floral bud to ripe fruit. BMC Plant Biol. 2008, 8, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patricia Denne, M. Fruit development and some tree factors affecting it. N. Z. J. Bot. 1963, 1, 265–294. [Google Scholar] [CrossRef]
- Denne, M.P. The growth of apple fruitlets, and the effect of early thinning on fruit development. Ann. Bot. 1960, 24, 397–406. [Google Scholar] [CrossRef]
- Brookfield, P.; Murphy, P.; Harker, R.; MacRae, E. Starch degradation and starch pattern indices; Interpretation and relationship to maturity. Postharvest Biol. Technol. 1997, 11, 23–30. [Google Scholar] [CrossRef]
- Li, M.J.; Feng, F.J.; Cheng, L.L. Expression patterns of genes involved in sugar metabolism and accumulation during apple fruit development. PLoS ONE 2012, 7, e33055. [Google Scholar] [CrossRef] [Green Version]
- Ackermann, J.; Fischer, M.; Amado, R. Changes in sugars, acids, and amino-acids during ripening and storage of apples (Cv Glockenapfel). J. Agric. Food Chem. 1992, 40, 1131–1134. [Google Scholar] [CrossRef]
- Etienne, A.; Genard, M.; Lobit, P.; Mbeguie-A-Mbeguie, D.; Bugaud, C. What controls fleshy fruit acidity? A review of malate and citrate accumulation in fruit cells. J. Exp. Bot. 2013, 64, 1451–1469. [Google Scholar] [CrossRef] [Green Version]
- Vrhovsek, U.; Rigo, A.; Tonon, D.; Mattivi, F. Quantitation of polyphenols in different apple varieties. J. Agric. Food Chem. 2004, 52, 6532–6538. [Google Scholar] [CrossRef]
- Kahle, K.; Kempf, M.; Schreier, P.; Scheppach, W.; Schrenk, D.; Kautenburger, T.; Hecker, D.; Huemmer, W.; Ackermann, M.; Richling, E. Intestinal transit and systemic metabolism of apple polyphenols. Eur. J. Nutr. 2011, 50, 507–522. [Google Scholar] [CrossRef]
- Tsao, R.; Yang, R.; Christopher, J.; Zhu, Y.; Zhu, H.H. Polyphenolic profiles in eight apple cultivars using high-performance liquid chromatography (HPLC). J. Agric. Food Chem. 2003, 51, 6347–6353. [Google Scholar] [CrossRef] [PubMed]
- Cuthbertson, D.; Andrews, P.K.; Reganold, J.P.; Davies, N.M.; Lange, B.M. Utility of metabolomics toward assessing the metabolic basis of quality traits in apple fruit with an emphasis on antioxidants. J. Agric. Food Chem. 2012, 60, 8552–8560. [Google Scholar] [CrossRef] [Green Version]
- Ernst, M.; Silva, D.B.; Silva, R.R.; Vencio, R.Z.N.; Lopes, N.P. Mass spectrometry in plant metabolomics strategies: From analytical platforms to data acquisition and processing. Nat. Prod. Rep. 2014, 31, 784–806. [Google Scholar] [CrossRef] [PubMed]
- Lei, Z.T.; Huhman, D.V.; Sumner, L.W. Mass spectrometry strategies in metabolomics. J. Biol. Chem. 2011, 286, 25435–25442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudell, D.R.; Mattheis, J.P.; Curry, E.A. Prestorage ultraviolet-white light irradiation alters apple peel metabolome. J. Agric. Food Chem. 2008, 56, 1138–1147. [Google Scholar] [CrossRef]
- Rudell, D.R.; Mattheis, J.P.; Hertog, M.L.A.T.M. Metabolomic change precedes apple superficial scald symptoms. J. Agric. Food Chem. 2009, 57, 8459–8466. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Mattheis, J.P.; Rudell, D.R. Antioxidant treatment alters metabolism associated with internal browning in “Braeburn” apples during controlled atmosphere storage. Postharvest Biol. Technol. 2012, 68, 32–42. [Google Scholar] [CrossRef]
- Vaclavik, L.; Schreiber, A.; Lacina, O.; Cajka, T.; Hajslova, J. Liquid chromatography–mass spectrometry-based metabolomics for authenticity assessment of fruit juices. Metabolomics 2011, 8, 793–803. [Google Scholar] [CrossRef]
- Fait, A.; Hanhineva, K.; Beleggia, R.; Dai, N.; Rogachev, I.; Nikiforova, V.J.; Fernie, A.R.; Aharoni, A. Reconfiguration of the achene and receptacle metabolic networks during strawberry fruit development. Plant Physiol. 2008, 148, 730–750. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wang, X.; Yu, O.; Tang, J.; Gu, X.; Wan, X.; Fang, C. Metabolic profiling of strawberry (Fragaria x ananassa Duch.) during fruit development and maturation. J. Exp. Bot. 2011, 62, 1103–1118. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Tohge, T.; Cuadros-Inostroza, A.; Tong, H.; Tenenboim, H.; Kooke, R.; Meret, M.; Keurentjes, J.B.; Nikoloski, Z.; Fernie, A.R.; et al. Mapping the Arabidopsis metabolic landscape by untargeted metabolomics at different environmental conditions. Mol. Plant 2018, 11, 118–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, G.T.; Wang, S.C.; Huang, Z.J.; Zhang, S.B.; Liao, Q.G.; Zhang, C.Z.; Lin, T.; Qin, M.; Peng, M.; Yang, C.K.; et al. Rewiring of the fruit metabolome in tomato breeding. Cell 2018, 172, 249. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Gao, Y.Q.; Xie, W.B.; Gong, L.; Lu, K.; Wang, W.S.; Li, Y.; Liu, X.Q.; Zhang, H.Y.; Dong, H.X.; et al. Genome-wide association analyses provide genetic and biochemical insights into natural variation in rice metabolism. Nat. Genet. 2014, 46, 714–721. [Google Scholar] [CrossRef]
- Chen, W.; Gong, L.; Guo, Z.L.; Wang, W.S.; Zhang, H.Y.; Liu, X.Q.; Yu, S.B.; Xiong, L.Z.; Luo, J. A novel integrated method for large-scale detection, identification, and quantification of widely targeted metabolites: Application in the study of rice metabolomics. Mol. Plant 2013, 6, 1769–1780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, N.; Bano, A.; Rahman, M.A.; Rathinasabapathi, B.; Babar, M.A. UPLC-HRMS based untargeted metabolic profiling reveals changes in chickpea (Cicer arietinum) metabolome following long-term drought stress. Plant Cell Environ. 2019, 42, 115–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Zhou, S.; Gong, X.; Song, Y.; van Nocker, S.; Ma, F.; Guan, Q. Single-base methylome analysis reveals dynamic epigenomic differences associated with water deficit in apple. Plant Biotechnol. J. 2018, 16, 672–687. [Google Scholar] [CrossRef] [PubMed]
- Oksman-Caldentey, K.M.; Saito, K. Integrating genomics and metabolomics for engineering plant metabolic pathways. Curr. Opin. Biotechnol. 2005, 16, 174–179. [Google Scholar] [CrossRef]
- Riedelsheimer, C.; Lisec, J.; Czedik-Eysenberg, A.; Sulpice, R.; Flis, A.; Grieder, C.; Altmann, T.; Stitt, M.; Willmitzer, L.; Melchinger, A.E. Genome-wide association mapping of leaf metabolic profiles for dissecting complex traits in maize. Proc. Natl. Acad. Sci. USA 2012, 109, 8872–8877. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Rudell, D.R.; Davies, P.J.; Watkins, C.B. Metabolic changes in 1-methylcyclopropene (1-MCP)-treated ‘Empire’apple fruit during storage. Metabolomics 2012, 8, 742–753. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, P.; Cheng, L. Developmental changes of carbohydrates, organic acids, amino acids, and phenolic compounds in ‘Honeycrisp’ apple flesh. Food Chem. 2010, 123, 1013–1018. [Google Scholar] [CrossRef]
- Carrari, F.; Baxter, C.; Usadel, B.; Urbanczyk-Wochniak, E.; Zanor, M.-I.; Nunes-Nesi, A.; Nikiforova, V.; Centero, D.; Ratzka, A.; Pauly, M. Integrated analysis of metabolite and transcript levels reveals the metabolic shifts that underlie tom06ato fruit development and highlight regulatory aspects of metabolic network behavior. Plant Physiol. 2006, 142, 1380–1396. [Google Scholar] [CrossRef] [Green Version]
- Maeda, H.; Dudareva, N. The shikimate pathway and aromatic amino acid biosynthesis in plants. Annu. Rev. Plant Biol. 2012, 63, 73–105. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.H.; Gao, H.Y.; Zhao, L.; Liao, X.J.; Chen, F.; Wang, Z.F.; Hu, X.S. Chemical compositional characterization of some apple cultivars. Food Chem. 2007, 103, 88–93. [Google Scholar] [CrossRef]
- Hecke, K.; Herbinger, K.; Veberič, R.; Trobec, M.; Toplak, H.; Štampar, F.; Keppel, H.; Grill, D. Sugar-, acid-and phenol contents in apple cultivars from organic and integrated fruit cultivation. Eur. J. Clin. Nutr. 2006, 60, 1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Abrosca, B.; Pacifico, S.; Cefarelli, G.; Mastellone, C.; Fiorentino, A. ‘Limoncella’ apple, an Italian apple cultivar: Phenolic and flavonoid contents and antioxidant activity. Food Chem. 2007, 104, 1333–1337. [Google Scholar] [CrossRef]
- Hagen, S.F.; Borge, G.I.A.; Bengtsson, G.B.; Bilger, W.; Berge, A.; Haffner, K.; Solhaug, K.A. Phenolic contents and other health and sensory related properties of apple fruit (Malus domestica Borkh., cv. Aroma): Effect of postharvest UV-B irradiation. Postharvest Biol. Technol. 2007, 45, 1–10. [Google Scholar] [CrossRef]
- Xie, D.Y.; Sharma, S.B.; Paiva, N.L.; Ferreira, D.; Dixon, R.A. Role of anthocyanidin reductase, encoded by BANYULS in plant flavonoid biosynthesis. Science 2003, 299, 396–399. [Google Scholar] [CrossRef] [Green Version]
- Pang, Y.Z.; Abeysinghe, I.S.B.; He, J.; He, X.Z.; Huhman, D.; Mewan, K.M.; Sumner, L.W.; Yun, J.F.; Dixon, R.A. Functional characterization of proanthocyanidin pathway enzymes from tea and their application for metabolic engineering. Plant Physiol. 2013, 161, 1103–1116. [Google Scholar] [CrossRef] [Green Version]
- Ashihara, H.; Deng, W.W.; Mullen, W.; Crozier, A. Distribution and biosynthesis of flavan-3-ols in Camellia sinensis seedlings and expression of genes encoding biosynthetic enzymes. Phytochemistry 2010, 71, 559–566. [Google Scholar] [CrossRef]
- Yun, Z.; Gao, H.J.; Liu, P.; Liu, S.Z.; Luo, T.; Jin, S.; Xu, Q.; Xu, J.; Cheng, Y.J.; Deng, X.X. Comparative proteomic and metabolomic profiling of citrus fruit with enhancement of disease resistance by postharvest heat treatment. BMC Plant Biol. 2013, 13, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef] [PubMed]
- Daccord, N.; Celton, J.M.; Linsmith, G.; Becker, C.; Choisne, N.; Schijlen, E.; van de Geest, H.; Bianco, L.; Micheletti, D.; Velasco, R.; et al. High-quality de novo assembly of the apple genome and methylome dynamics of early fruit development. Nat. Genet. 2017, 49, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thimm, O.; Blasing, O.; Gibon, Y.; Nagel, A.; Meyer, S.; Kruger, P.; Selbig, J.; Muller, L.A.; Rhee, S.Y.; Stitt, M. MAPMAN: A user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 2004, 37, 914–939. [Google Scholar] [CrossRef]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, J.; Yan, J.; Li, W.; Wang, Q.; Wang, C.; Guo, J.; Geng, D.; Guan, Q.; Ma, F. Integrative Analyses of Widely Targeted Metabolic Profiling and Transcriptome Data Reveals Molecular Insight into Metabolomic Variations during Apple (Malus domestica) Fruit Development and Ripening. Int. J. Mol. Sci. 2020, 21, 4797. https://doi.org/10.3390/ijms21134797
Xu J, Yan J, Li W, Wang Q, Wang C, Guo J, Geng D, Guan Q, Ma F. Integrative Analyses of Widely Targeted Metabolic Profiling and Transcriptome Data Reveals Molecular Insight into Metabolomic Variations during Apple (Malus domestica) Fruit Development and Ripening. International Journal of Molecular Sciences. 2020; 21(13):4797. https://doi.org/10.3390/ijms21134797
Chicago/Turabian StyleXu, Jidi, Jinjiao Yan, Wenjie Li, Qianying Wang, Caixia Wang, Junxing Guo, Dali Geng, Qingmei Guan, and Fengwang Ma. 2020. "Integrative Analyses of Widely Targeted Metabolic Profiling and Transcriptome Data Reveals Molecular Insight into Metabolomic Variations during Apple (Malus domestica) Fruit Development and Ripening" International Journal of Molecular Sciences 21, no. 13: 4797. https://doi.org/10.3390/ijms21134797
APA StyleXu, J., Yan, J., Li, W., Wang, Q., Wang, C., Guo, J., Geng, D., Guan, Q., & Ma, F. (2020). Integrative Analyses of Widely Targeted Metabolic Profiling and Transcriptome Data Reveals Molecular Insight into Metabolomic Variations during Apple (Malus domestica) Fruit Development and Ripening. International Journal of Molecular Sciences, 21(13), 4797. https://doi.org/10.3390/ijms21134797