In Vivo Models for Cholangiocarcinoma—What Can We Learn for Human Disease?

 , ,
, ,  and
and 
Abstract
:1. Introduction
2. Current and Emerging Therapeutic Options for CCA
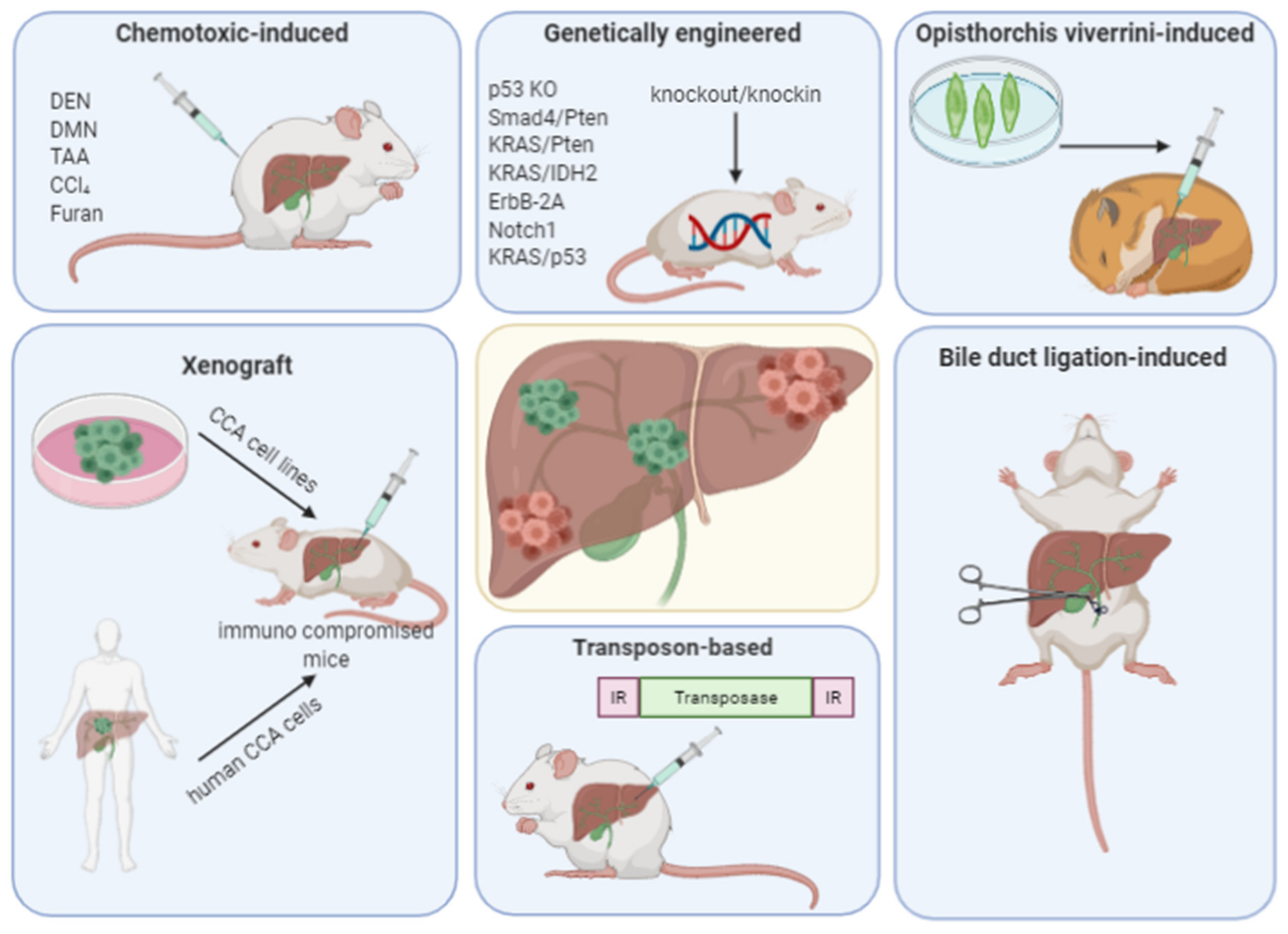
3. Animal Models
4. Chemotoxic-Induced Models
4.1. Furan Model
4.2. Thioacetamide Model
4.3. Diethylnitrosamine-Left Median Bile Duct Ligation Model
4.4. p53 Knockout-Carbon Tetrachloride (CCl4) Model
4.5. Opisthorchis viverrini Model
5. Genetically Engineered Models
5.1. Liver-Specific Deletion of Smad4 and Pten
5.2. Liver-Specific Activation of KRAS and Deletion of Pten
5.3. Liver-Specific Activation of KRAS and Deletion of Tp53
5.4. Liver-Specific Activation of KRAS and IDH2
5.5. ErbB-2A Overexpression
5.6. Notch1 Overexpression
5.7. Sleeping Beauty Transposons
6. Implantation Models
6.1. Xenograft Models
6.2. Syngeneic Models
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CCA | cholangiocarcinoma |
| CCl4 | carbon tetrachloride |
| CDA | cytidine deaminase |
| CI | confidence interval |
| COX | cyclooxygenase |
| DDC | 3,5-diethoxycarbonyl-1,4-dihydrocollidine |
| DEN | diethylnitrosamine |
| DMN | dimethylnitrosamine |
| DNA | deoxyribonucleotide acid |
| FGFR | fibroblast growth factor receptor |
| GABA | γ-aminobutyric acid |
| GEM | genetically engineered mice |
| GEMOX | gemcitabine/oxaliplatin |
| HCC | hepatocellular carcinoma |
| HR | hazard ratio |
| iCCA | intrahepatic cholangiocarcinoma |
| IDH | isocitrate dehydrogenase |
| IL | interleukin |
| LMBDL | left and median bile duct ligations |
| miRNA | micro ribonuleic acid |
| MPP | metalloproteinase |
| O. viverrini | Opisthorchis viverrini |
| OS | overall survival |
| OSBP | oxysterol binding protein |
| PDX | patient-derived xenograft |
| Pten | phosphatase and tensin homolog deleted chromosome 10 |
| ROS | reactive oxygen species |
| SB | sleeping beauty |
| TAA | thioacetamide |
| TGF | transforming growth factor |
| TKI | tyrosine-kinase inhibitors |
| VEGFR | vascular endothelial growth factor |
References
- Rizvi, S.; Gores, G.J. Pathogenesis, diagnosis, and management of cholangiocarcinoma. Gastroenterology 2013, 145, 1215–1229. [Google Scholar] [CrossRef] [Green Version]
- Marcano-Bonilla, L.; Mohamed, E.A.; Mounajjed, T.; Roberts, L.R. Biliary tract cancers: Epidemiology, molecular pathogenesis and genetic risk associations. Chin. Clin. Oncol. 2016, 5, 61. [Google Scholar] [CrossRef]
- Walter, D.; Ferstl, P.; Waidmann, O.; Trojan, J.; Hartmann, S.; Schnitzbauer, A.A.; Zeuzem, S.; Kraywinkel, K. Cholangiocarcinoma in Germany: Epidemiologic trends and impact of misclassification. Liver Int. 2019, 39, 316–323. [Google Scholar] [CrossRef]
- Von Hahn, T.; Ciesek, S.; Wegener, G.; Plentz, R.R.; Weismuller, T.J.; Wedemeyer, H.; Manns, M.P.; Greten, T.F.; Malek, N.P. Epidemiological trends in incidence and mortality of hepatobiliary cancers in Germany. Scand. J. Gastroenterol. 2011, 46, 1092–1098. [Google Scholar] [CrossRef] [PubMed]
- Mazzaferro, V.; Gorgen, A.; Roayaie, S.; Droz Dit Busset, M.; Sapisochin, G. Liver resection and transplantation for intrahepatic cholangiocarcinoma. J. Hepatol. 2020, 72, 364–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golub, D.; Iyengar, N.; Dogra, S.; Wong, T.; Bready, D.; Tang, K.; Modrek, A.S.; Placantonakis, D.G. Mutant Isocitrate Dehydrogenase Inhibitors as Targeted Cancer Therapeutics. Front. Oncol. 2019, 9, 417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, D.; Ma, J.; Wang, J.; Han, C.; Qian, Y.; Chen, G.; Li, X.; Zhang, J.; Cui, P.; Du, W.; et al. Anti-PD-1 therapy combined with chemotherapy in patients with advanced biliary tract cancer. Cancer Immunol. Immunother. 2019, 68, 1527–1535. [Google Scholar] [CrossRef] [Green Version]
- Abou-Alfa, G.K.; Macarulla, T.; Javle, M.M.; Kelley, R.K.; Lubner, S.J.; Adeva, J.; Cleary, J.M.; Catenacci, D.V.; Borad, M.J.; Bridgewater, J.; et al. Ivosidenib in IDH1-mutant, chemotherapy-refractory cholangiocarcinoma (ClarIDHy): A multicentre, randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2020, 21, 796–807. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Sahai, V.; Hollebecque, A.; Vaccaro, G.; Melisi, D.; Al-Rajabi, R.; Paulson, A.S.; Borad, M.J.; Gallinson, D.; Murphy, A.G.; et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: A multicentre, open-label, phase 2 study. Lancet Oncol. 2020, 21, 671–684. [Google Scholar] [CrossRef]
- Morton, J.J.; Bird, G.; Refaeli, Y.; Jimeno, A. Humanized Mouse Xenograft Models: Narrowing the Tumor-Microenvironment Gap. Cancer Res. 2016, 76, 6153–6158. [Google Scholar] [CrossRef] [Green Version]
- Hand, F.; Hoti, E. Contemporary role of liver transplantation for the treatment of cholangiocarcinoma. Expert Rev. Gastroenterol. Hepatol. 2020, 14, 1–7. [Google Scholar] [CrossRef]
- Doherty, B.; Nambudiri, V.E.; Palmer, W.C. Update on the Diagnosis and Treatment of Cholangiocarcinoma. Curr. Gastroenterol. Rep. 2017, 19, 2. [Google Scholar] [CrossRef] [PubMed]
- DeOliveira, M.L.; Cunningham, S.C.; Cameron, J.L.; Kamangar, F.; Winter, J.M.; Lillemoe, K.D.; Choti, M.A.; Yeo, C.J.; Schulick, R.D. Cholangiocarcinoma: Thirty-one-year experience with 564 patients at a single institution. Ann. Surg. 2007, 245, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Horgan, A.M.; Amir, E.; Walter, T.; Knox, J.J. Adjuvant therapy in the treatment of biliary tract cancer: A systematic review and meta-analysis. J. Clin. Oncol. 2012, 30, 1934–1940. [Google Scholar] [CrossRef] [PubMed]
- Primrose, J.N.; Fox, R.P.; Palmer, D.H.; Malik, H.Z.; Prasad, R.; Mirza, D.; Anthony, A.; Corrie, P.; Falk, S.; Finch-Jones, M.; et al. Capecitabine compared with observation in resected biliary tract cancer (BILCAP): A randomised, controlled, multicentre, phase 3 study. Lancet Oncol. 2019, 20, 663–673. [Google Scholar] [CrossRef] [Green Version]
- Edeline, J.; Benabdelghani, M.; Bertaut, A.; Watelet, J.; Hammel, P.; Joly, J.P.; Boudjema, K.; Fartoux, L.; Bouhier-Leporrier, K.; Jouve, J.-L.; et al. Gemcitabine and Oxaliplatin Chemotherapy or Surveillance in Resected Biliary Tract Cancer (PRODIGE 12-ACCORD 18-UNICANCER GI): A Randomized Phase III Study. J. Clin. Oncol. 2019, 37, 658–667. [Google Scholar] [CrossRef] [PubMed]
- Ebata, T.; Hirano, S.; Konishi, M.; Uesaka, K.; Tsuchiya, Y.; Ohtsuka, M.; Kaneoka, Y.; Yamamoto, M.; Ambo, Y.; Shimizu, Y.; et al. Randomized clinical trial of adjuvant gemcitabine chemotherapy versus observation in resected bile duct cancer. Br. J. Surg. 2018, 105, 192–202. [Google Scholar] [CrossRef]
- Shroff, R.T.; Kennedy, E.B.; Bachini, M.; Bekaii-Saab, T.; Crane, C.; Edeline, J.; El-Khoueiry, A.; Feng, M.; Katz, M.H.; Primrose, J.N.; et al. Adjuvant Therapy for Resected Biliary Tract Cancer: ASCO Clinical Practice Guideline. J. Clin. Oncol. 2019, 37, 1015–1027. [Google Scholar] [CrossRef] [Green Version]
- Valle, J.; Wasan, H.; Palmer, D.H.; Cunningham, D.; Anthoney, A.; Maraveyas, A.; Madhusudan, S.; Iveson, T.; Hughes, S.; Pereira, S.P.; et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N. Engl. J. Med. 2010, 362, 1273–1281. [Google Scholar] [CrossRef] [Green Version]
- Okusaka, T.; Nakachi, K.; Fukutomi, A.; Mizuno, N.; Ohkawa, S.; Funakoshi, A.; Nagino, M.; Kondo, S.; Nagaoka, S.; Funai, J.; et al. Gemcitabine alone or in combination with cisplatin in patients with biliary tract cancer: A comparative multicentre study in Japan. Br. J. Cancer 2010, 103, 469–474. [Google Scholar] [CrossRef] [Green Version]
- Valle, J.W.; Furuse, J.; Jitlal, M.; Beare, S.; Mizuno, N.; Wasan, H.; Bridgewater, J.; Okusaka, T. Cisplatin and gemcitabine for advanced biliary tract cancer: A meta-analysis of two randomised trials. Ann. Oncol. 2014, 25, 391–398. [Google Scholar] [CrossRef]
- Morizane, C.; Okusaka, T.; Mizusawa, J.; Katayama, H.; Ueno, M.; Ikeda, M.; Ozaka, M.; Okano, N.; Sugimori, K.; Fukutomi, A.; et al. Combination gemcitabine plus S-1 versus gemcitabine plus cisplatin for advanced/recurrent biliary tract cancer: The FUGA-BT (JCOG1113) randomized phase III clinical trial. Ann. Oncol. 2019, 30, 1950–1958. [Google Scholar] [CrossRef] [PubMed]
- Shroff, R.T.; Javle, M.M.; Xiao, L.; Kaseb, A.O.; Varadhachary, G.R.; Wolff, R.A.; Raghav, K.P.S.; Iwasaki, M.; Masci, P.; Ramanathan, R.K.; et al. Gemcitabine, Cisplatin, and nab-Paclitaxel for the Treatment of Advanced Biliary Tract Cancers: A Phase 2 Clinical Trial. JAMA Oncol. 2019, 5, 824–830. [Google Scholar] [CrossRef] [PubMed]
- McNamara, M.G.; Goyal, L.; Doherty, M.; Springfeld, C.; Cosgrove, D.; Sjoquist, K.M.; Park, J.O.; Verdaguer, H.; Braconi, C.; Ross, P.J.; et al. NUC-1031/cisplatin versus gemcitabine/cisplatin in untreated locally advanced/metastatic biliary tract cancer (NuTide:121). Future Oncol. 2020, 16, 1069–1081. [Google Scholar] [CrossRef] [PubMed]
- Arora, M.; Bogenberger, J.M.; Abdelrahman, A.; Leiting, J.L.; Chen, X.; Egan, J.B.; Kasimsetty, A.; Lenkiewicz, E.; Malasi, S.; Uson, P.L.S.; et al. Evaluation of NUC-1031: A first-in-class ProTide in biliary tract cancer. Cancer Chemother. Pharmacol. 2020, 6, 1063–1078. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Rim, H.J.; Sell, S. Heterogeneity of the “oval-cell” response in the hamster liver during cholangiocarcinogenesis following Clonorchis sinensis infection and dimethylnitrosamine treatment. J. Hepatol. 1997, 26, 1313–1323. [Google Scholar] [CrossRef]
- Loeuillard, E.; Fischbach, S.R.; Gores, G.J.; Rizvi, S. Animal models of cholangiocarcinoma. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1, 982–992. [Google Scholar] [CrossRef]
- Reznik, G.; Mohr, U.; Kmoch, N. Carcinogenic effects of different nitroso-compounds in Chinese hamsters. I. Dimethylnitrosamine and N-diethylnitrosamine. Br. J. Cancer 1976, 33, 411–418. [Google Scholar] [CrossRef] [Green Version]
- Praet, M.M.; Roels, H.J. Histogenesis of cholangiomas and cholangiocarcinomas in thioacetamide fed rats. Exp. Pathol. 1984, 26, 3–14. [Google Scholar] [CrossRef]
- Yeh, C.N.; Maitra, A.; Lee, K.F.; Jan, Y.Y.; Chen, M.F. Thioacetamide-induced intestinal-type cholangiocarcinoma in rat: An animal model recapitulating the multi-stage progression of human cholangiocarcinoma. Carcinogenesis 2004, 25, 631–636. [Google Scholar] [CrossRef]
- Maronpot, R.R.; Giles, H.D.; Dykes, D.J.; Irwin, R.D. Furan-induced hepatic cholangiocarcinomas in Fischer 344 rats. Toxicol. Pathol. 1991, 19 Pt 2, 561–570. [Google Scholar] [CrossRef] [Green Version]
- Hickling, K.C.; Hitchcock, J.M.; Oreffo, V.; Mally, A.; Hammond, T.G.; Evans, J.G.; Chipman, J.K. Evidence of oxidative stress and associated DNA damage, increased proliferative drive, and altered gene expression in rat liver produced by the cholangiocarcinogenic agent furan. Toxicol. Pathol. 2010, 38, 230–243. [Google Scholar] [CrossRef]
- Sirica, A.E. Biliary proliferation and adaptation in furan-induced rat liver injury and carcinogenesis. Toxicol. Pathol. 1996, 24, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Arora, D.S.; Ramsdale, J.; Lodge, J.P.; Wyatt, J.I. p53 but not bcl-2 is expressed by most cholangiocarcinomas: A study of 28 cases. Histopathology 1999, 34, 497–501. [Google Scholar] [CrossRef]
- Furubo, S.; Harada, K.; Shimonishi, T.; Katayanagi, K.; Tsui, W.; Nakanuma, Y. Protein expression and genetic alterations of p53 and ras in intrahepatic cholangiocarcinoma. Histopathology 1999, 35, 230–240. [Google Scholar] [CrossRef]
- Itoi, T.; Takei, K.; Shinohara, Y.; Takeda, K.; Nakamura, K.; Horibe, T.; Sanada, A.; Ohno, H.; Matsubayashi, H.; Saito, T.; et al. K-ras codon 12 and p53 mutations in biopsy specimens and bile from biliary tract cancers. Pathol. Int. 1999, 49, 30–37. [Google Scholar] [CrossRef]
- Kang, Y.K.; Kim, W.H.; Lee, H.W.; Lee, H.K.; Kim, Y.I. Mutation of p53 and K-ras, and loss of heterozygosity of APC in intrahepatic cholangiocarcinoma. Lab. Investig. 1999, 79, 477–483. [Google Scholar] [PubMed]
- Tannapfel, A.; Benicke, M.; Katalinic, A.; Uhlmann, D.; Kockerling, F.; Hauss, J.; Wittekind, C. Frequency of p16(INK4A) alterations and K-ras mutations in intrahepatic cholangiocarcinoma of the liver. Gut 2000, 47, 721–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirica, A.E.; Radaeva, S.; Caran, N. NEU overexpression in the furan rat model of cholangiocarcinogenesis compared with biliary ductal cell hyperplasia. Am. J. Pathol. 1997, 151, 1685–1694. [Google Scholar] [PubMed]
- Cadamuro, M.; Brivio, S.; Stecca, T.; Kaffe, E.; Mariotti, V.; Milani, C.; Fiorotto, R.; Spirli, C.; Strazzabosco, M.; Fabris, L. Animal models of cholangiocarcinoma: What they teach us about the human disease. Clin. Res. Hepatol. Gastroenterol. 2018, 42, 403–415. [Google Scholar] [CrossRef]
- Dashti, H.; Jeppsson, B.; Hägerstrand, I.; Hultberg, B.; Srinivas, U.; Abdulla, M.; Bengmark, S. Thioacetamide- and carbon tetrachloride-induced liver cirrhosis. Eur. Surg. Res. 1989, 21, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Hajovsky, H.; Hu, G.; Koen, Y.; Sarma, D.; Cui, W.; Moore, D.S.; Staudinger, J.L.; Hanzlik, R.P.; Hajovsky, L. Metabolism and toxicity of thioacetamide and thioacetamide S-oxide in rat hepatocytes. Chem. Res. Toxicol. 2012, 25, 1955–1963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Minicis, S.; Kisseleva, T.; Francis, H.; Baroni, G.S.; Benedetti, A.; Brenner, D.; Alvaro, D.; Alpini, G.; Marzioni, M. Liver carcinogenesis: Rodent models of hepatocarcinoma and cholangiocarcinoma. Dig. Liver Dis. 2013, 45, 450–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marzioni, M.; Torrice, A.; Saccomanno, S.; Rychlicki, C.; Agostinelli, L.; Pierantonelli, I.; Rhönnstad, P.; Trozzi, L.; Apelqvist, T.; Gentile, R.; et al. An oestrogen receptor beta-selective agonist exerts anti-neoplastic effects in experimental intrahepatic cholangiocarcinoma. Dig. Liver Dis. 2012, 44, 134–142. [Google Scholar] [CrossRef]
- Barker, E.A.; Smuckler, E.A. Altered microsome function during acute thioacetamide poisoning. Mol. Pharmacol. 1972, 8, 318–326. [Google Scholar] [PubMed]
- Thamavit, W.; Bhamarapravati, N.; Sahaphong, S.; Vajrasthira, S.; Angsubhakorn, S. Effects of dimethylnitrosamine on induction of cholangiocarcinoma in Opisthorchis viverrini-infected Syrian golden hamsters. Cancer Res. 1978, 38, 4634–4639. [Google Scholar] [PubMed]
- Yang, H.; Li, T.W.; Peng, J.; Tang, X.; Ko, K.S.; Xia, M.; Aller, M. A mouse model of cholestasis-associated cholangiocarcinoma and transcription factors involved in progression. Gastroenterology 2011, 141, 378–388. [Google Scholar] [CrossRef] [Green Version]
- Chong, D.Q.; Zhu, A.X. The landscape of targeted therapies for cholangiocarcinoma: Current status and emerging targets. Oncotarget 2016, 7, 46750–46767. [Google Scholar] [CrossRef] [Green Version]
- Eastmond, D.A. Evaluating genotoxicity data to identify a mode of action and its application in estimating cancer risk at low doses: A case study involving carbon tetrachloride. Environ. Mol. Mutagen. 2008, 49, 132–141. [Google Scholar] [CrossRef]
- Mekada, K.; Abe, K.; Murakami, A.; Nakamura, S.; Nakata, H.; Moriwaki, K.; Obata, Y.; Yoshiki, A. Genetic differences among C57BL/6 substrains. Exp. Anim. 2009, 58, 141–149. [Google Scholar] [CrossRef] [Green Version]
- Farazi, P.A.; Zeisberg, M.; Glickman, J.; Zhang, Y.; Kalluri, R.; DePinho, R.A. Chronic bile duct injury associated with fibrotic matrix microenvironment provokes cholangiocarcinoma in p53-deficient mice. Cancer Res. 2006, 66, 6622–6627. [Google Scholar] [CrossRef] [Green Version]
- Prueksapanich, P.; Piyachaturawat, P.; Aumpansub, P.; Ridtitid, W.; Chaiteerakij, R.; Rerknimitr, R. Liver Fluke-Associated Biliary Tract Cancer. Gut Liver 2018, 12, 236–245. [Google Scholar] [CrossRef] [Green Version]
- Sripa, B.; Mairiang, E.; Thinkhamrop, B.; Laha, T.; Kaewkes, S.; Sithithaworn, P.; Tessana, S.; Loukas, A.; Brindley, P.J.; Bethony, J.M. Advanced periductal fibrosis from infection with the carcinogenic human liver fluke Opisthorchis viverrini correlates with elevated levels of interleukin-6. Hepatology 2009, 50, 1273–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sripa, B.; Thinkhamrop, B.; Mairiang, E.; Laha, T.; Kaewkes, S.; Sithithaworn, P.; Periago, M.V.; Bhudhisawasdi, V.; Yonglitthipagon, P.; Mulvenna, J.; et al. Elevated plasma IL-6 associates with increased risk of advanced fibrosis and cholangiocarcinoma in individuals infected by Opisthorchis viverrini. PLoS Negl. Trop. Dis. 2012, 6, e1654. [Google Scholar] [CrossRef] [PubMed]
- Sawanyawisuth, K.; Silsirivanit, A.; Kunlabut, K.; Tantapotinan, N.; Vaeteewoottacharn, K.; Wongkham, S. A novel carbohydrate antigen expression during development of Opisthorchis viverrini-associated cholangiocarcinoma in golden hamster: A potential marker for early diagnosis. Parasitol. Int. 2012, 61, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Loilome, W.; Yongvanit, P.; Wongkham, C.; Tepsiri, N.; Sripa, B.; Sithithaworn, P.; Hanai, S.; Miwa, M. Altered gene expression in Opisthorchis viverrini-associated cholangiocarcinoma in hamster model. Mol. Carcinog. 2006, 45, 279–287. [Google Scholar] [CrossRef]
- Loilome, W.; Wechagama, P.; Namwat, N.; Jusakul, A.; Sripa, B.; Miwa, M.; Kuver, R.; Yongvanit, P. Expression of oxysterol binding protein isoforms in opisthorchiasis-associated cholangiocarcinoma: A potential molecular marker for tumor metastasis. Parasitol. Int. 2012, 61, 136–139. [Google Scholar] [CrossRef]
- Frese, K.K.; Tuveson, D.A. Maximizing mouse cancer models. Nat. Rev. Cancer 2007, 7, 645–658. [Google Scholar] [CrossRef]
- Adams, J.M.; Cory, S. Transgenic models of tumor development. Science 1991, 254, 1161–1167. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Razumilava, N.; Gores, G.J.; Walters, S.; Mizuochi, T.; Mourya, R.; Bessho, K.; Wang, Y.-H.; Glaser, S.S.; Shivakumar, P.; et al. Biliary repair and carcinogenesis are mediated by IL-33-dependent cholangiocyte proliferation. J. Clin. Investig. 2014, 124, 3241–3251. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.A.; Thomas, H.C.; Davidson, B.R.; Taylor-Robinson, S.D. Cholangiocarcinoma. Lancet 2005, 366, 1303–1314. [Google Scholar] [CrossRef]
- Kang, Y.K.; Kim, W.H.; Jang, J.J. Expression of G1-S modulators (p53, p16, p27, cyclin D1, Rb) and Smad4/Dpc4 in intrahepatic cholangiocarcinoma. Hum. Pathol. 2002, 33, 877–883. [Google Scholar] [CrossRef]
- Sansal, I.; Sellers, W.R. The biology and clinical relevance of the PTEN tumor suppressor pathway. J. Clin. Oncol. 2004, 22, 2954–2963. [Google Scholar] [CrossRef] [PubMed]
- Postic, C.; Magnuson, M.A. DNA excision in liver by an albumin-Cre transgene occurs progressively with age. Genesis 2000, 26, 149–150. [Google Scholar] [CrossRef]
- Andersen, J.B.; Spee, B.; Blechacz, B.R.; Avital, I.; Komuta, M.; Barbour, A.; Conner, E.A.; Gillen, M.C.; Roskams, T.; Roberts, L.R.; et al. Genomic and genetic characterization of cholangiocarcinoma identifies therapeutic targets for tyrosine kinase inhibitors. Gastroenterology 2012, 142, 1021–1031.e15. [Google Scholar] [CrossRef] [Green Version]
- Robertson, S.; Hyder, O.; Dodson, R.; Nayar, S.K.; Poling, J.; Beierl, K.; Eshleman, J.R.; Lin, M.-T.; Pawlik, T.M.; Anders, R.A. The frequency of KRAS and BRAF mutations in intrahepatic cholangiocarcinomas and their correlation with clinical outcome. Hum. Pathol. 2013, 44, 2768–2773. [Google Scholar] [CrossRef] [Green Version]
- Ikenoue, T.; Terakado, Y.; Nakagawa, H.; Hikiba, Y.; Fujii, T.; Matsubara, D.; Noguchi, R.; Zhu, C.; Yamamoto, K.; Kudo, Y.; et al. A novel mouse model of intrahepatic cholangiocarcinoma induced by liver-specific Kras activation and Pten deletion. Sci. Rep. 2016, 6, 23899. [Google Scholar] [CrossRef]
- Lin, Y.K.; Fang, Z.; Jiang, T.Y.; Wan, Z.H.; Pan, Y.F.; Ma, Y.H.; Shi, Y.-Y.; Tan, Y.-X.; Dong, L.; Zhang, Y.-J.; et al. Combination of Kras activation and PTEN deletion contributes to murine hepatopancreatic ductal malignancy. Cancer Lett. 2018, 421, 161–169. [Google Scholar] [CrossRef]
- O’Dell, M.R.; Huang, J.L.; Whitney-Miller, C.L.; Deshpande, V.; Rothberg, P.; Grose, V.; Rossi, R.M.; Zhu, A.X.; Land, H.; Bardeesy, N.; et al. Kras(G12D) and p53 mutation cause primary intrahepatic cholangiocarcinoma. Cancer Res. 2012, 72, 1557–1567. [Google Scholar] [CrossRef] [Green Version]
- Saha, S.K.; Parachoniak, C.A.; Ghanta, K.S.; Fitamant, J.; Ross, K.N.; Najem, M.S.; Gurumurthy, S.; Akbay, E.A.; Sia, D.; Cornella, H.; et al. Mutant IDH inhibits HNF-4alpha to block hepatocyte differentiation and promote biliary cancer. Nature 2014, 513, 110–114. [Google Scholar] [CrossRef] [Green Version]
- Lowery, M.A.; Burris, H.A.; Janku, F.; Shroff, R.T.; Cleary, J.M.; Azad, N.S.; Goyal, L.; Maher, E.A.; Gore, L.; Hollebecque, A.; et al. Safety and activity of ivosidenib in patients with IDH1-mutant advanced cholangiocarcinoma: A phase 1 study. Lancet Gastroenterol. Hepatol. 2019, 4, 711–720. [Google Scholar] [CrossRef]
- Yu, D.; Hung, M.C. Overexpression of ErbB2 in cancer and ErbB2-targeting strategies. Oncogene 2000, 19, 6115–6121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiguchi, K.; Carbajal, S.; Chan, K.; Beltran, L.; Ruffino, L.; Shen, J.; Matsumoto, T.; Yoshimi, N.; DiGiovanni, J. Constitutive expression of ErbB-2 in gallbladder epithelium results in development of adenocarcinoma. Cancer Res. 2001, 61, 6971–6976. [Google Scholar] [PubMed]
- Hofmann, J.J.; Zovein, A.C.; Koh, H.; Radtke, F.; Weinmaster, G.; Iruela-Arispe, M.L. Jagged1 in the portal vein mesenchyme regulates intrahepatic bile duct development: Insights into Alagille syndrome. Development 2010, 137, 4061–4072. [Google Scholar] [CrossRef] [Green Version]
- Zender, S.; Nickeleit, I.; Wuestefeld, T.; Sorensen, I.; Dauch, D.; Bozko, P.; El-Khatib, M.; Geffers, R.; Bektas, H.; Manns, M.P.; et al. A critical role for notch signaling in the formation of cholangiocellular carcinomas. Cancer Cell 2013, 23, 784–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, B.; Malato, Y.; Calvisi, D.F.; Naqvi, S.; Razumilava, N.; Ribback, S.; Gores, G.J.; Dombrowski, F.; Evert, M.; Chen, X.; et al. Cholangiocarcinomas can originate from hepatocytes in mice. J. Clin. Investig. 2012, 122, 2911–2915. [Google Scholar] [CrossRef] [Green Version]
- Yamada, D.; Rizvi, S.; Razumilava, N.; Bronk, S.F.; Davila, J.I.; Champion, M.D.; Borad, M.J.; Bezerra, J.A.; Chen, X.; Gores, G.J. IL-33 facilitates oncogene-induced cholangiocarcinoma in mice by an interleukin-6-sensitive mechanism. Hepatology 2015, 61, 1627–1642. [Google Scholar] [CrossRef] [PubMed]
- Gurlevik, E.; Fleischmann-Mundt, B.; Armbrecht, N.; Longerich, T.; Woller, N.; Kloos, A.; Hoffmann, D.; Schambach, A.; Wirth, T.C.; Manns, M.P.; et al. Adjuvant gemcitabine therapy improves survival in a locally induced, R0-resectable model of metastatic intrahepatic cholangiocarcinoma. Hepatology 2013, 58, 1031–1041. [Google Scholar] [CrossRef]
- Hudd, C.; LaRegina, M.C.; Devine, J.E.; Palmer, D.C.; Herbold, D.R.; Beinfeld, M.C.; Gelder, F.B.; Johnson, F.E. Response to exogenous cholecystokinin of six human gastrointestinal cancers xenografted in nude mice. Am. J. Surg. 1989, 157, 386–394. [Google Scholar] [CrossRef]
- Fava, G.; Marucci, L.; Glaser, S.; Francis, H.; De Morrow, S.; Benedetti, A.; Alvaro, D.; Venter, J.; Meininger, C.; Patel, T.; et al. gamma-Aminobutyric acid inhibits cholangiocarcinoma growth by cyclic AMP-dependent regulation of the protein kinase A/extracellular signal-regulated kinase 1/2 pathway. Cancer Res. 2005, 65, 11437–11446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Zhang, C.; Zhou, H.; Xiao, B.; Cheng, Y.; Wang, J.; Yao, F.; Duan, C.; Chen, R.; Liu, Y.; et al. Synergistic antitumor activity of the combination of salubrinal and rapamycin against human cholangiocarcinoma cells. Oncotarget 2016, 7, 85492–85501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, M.H.; Chen, L.J.; Chen, Y.L.; Tsai, M.S.; Shiau, C.W.; Chao, T.I.; Kao, J.; Chen, K. Targeting SHP-1-STAT3 signaling: A promising therapeutic approach for the treatment of cholangiocarcinoma. Oncotarget 2017, 8, 65077–65089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, P.L.; Miller, A.L.; Gamblin, T.L.; Council, L.N.; Christein, J.D.; Arnoletti, J.P.; Heslin, M.J.; Reddy, S.; Richardson, J.H.; Cui, X.; et al. JQ1 Induces DNA Damage and Apoptosis, and Inhibits Tumor Growth in a Patient-Derived Xenograft Model of Cholangiocarcinoma. Mol. Cancer Ther. 2018, 17, 107–118. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.J.; Dong, L.W.; Tan, Y.X.; Yang, G.Z.; Pan, Y.F.; Li, Z.; Tang, L.; Wang, M.; Wang, Q.; Wang, H.-Y. Inhibition of active autophagy induces apoptosis and increases chemosensitivity in cholangiocarcinoma. Lab. Investig. 2011, 91, 1146–1157. [Google Scholar] [CrossRef] [PubMed]
- Jo, H.J.; Shim, H.E.; Han, M.E.; Kim, H.J.; Kim, K.S.; Baek, S.; Choi, K.-U.; Hur, G.-Y.; Oh, S.-O. WTAP regulates migration and invasion of cholangiocarcinoma cells. J. Gastroenterol. 2013, 48, 1271–1282. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Yamagiwa, Y.; Ueno, Y.; Patel, T. Over-expression of interleukin-6 enhances cell survival and transformed cell growth in human malignant cholangiocytes. J. Hepatol. 2006, 44, 1055–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merino-Azpitarte, M.; Lozano, E.; Perugorria, M.J.; Esparza-Baquer, A.; Erice, O.; Santos-Laso, Á.; O’Rourke, C.J.; Andersen, J.B.; Jiménez-Agüero, R.; La Casta, A.; et al. SOX17 regulates cholangiocyte differentiation and acts as a tumor suppressor in cholangiocarcinoma. J. Hepatol. 2017, 67, 72–83. [Google Scholar] [CrossRef]
- Tang, J.; Liao, Y.; He, S.; Shi, J.; Peng, L.; Xu, X.; Xie, F.; Diao, N.; Huang, J.; Xie, Q.; et al. Autocrine parathyroid hormone-like hormone promotes intrahepatic cholangiocarcinoma cell proliferation via increased ERK/JNK-ATF2-cyclinD1 signaling. J. Transl. Med. 2017, 15, 17–1342. [Google Scholar] [CrossRef] [Green Version]
- Olaru, A.V.; Ghiaur, G.; Yamanaka, S.; Luvsanjav, D.; An, F.; Popescu, I.; Alexandrescu, S.; Allen, S.; Pawlik, T.M.; Torbenson, M.; et al. MicroRNA down-regulated in human cholangiocarcinoma control cell cycle through multiple targets involved in the G1/S checkpoint. Hepatology 2011, 54, 2089–2098. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Han, C.; Wu, T. MicroRNA-26a promotes cholangiocarcinoma growth by activating β-catenin. Gastroenterology 2012, 143, 246–256. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Han, C.; Lu, D.; Wu, T. miR-17–92 cluster promotes cholangiocarcinoma growth: Evidence for PTEN as downstream target and IL-6/Stat3 as upstream activator. Am. J. Pathol. 2014, 184, 2828–2839. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Jiang, X.; Zhou, X.; Dong, X.; Xie, K.; Yang, C.; Jiang, H.; Sun, X.; Lu, J. Neuropilin-1 regulated by miR-320 contributes to the growth and metastasis of cholangiocarcinoma cells. Liver Int. 2018, 38, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Leiting, J.L.; Murphy, S.J.; Bergquist, J.R.; Hernandez, M.C.; Ivanics, T.; Abdelrahman, A.M.; Yang, L.; Lynch, I.; Smadbeck, J.B.; Cleary, S.P.; et al. Biliary tract cancer patient-derived xenografts: Surgeon impact on individualized medicine. JHEP Rep. 2020, 2, 100068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirica, A.E.; Zhang, Z.; Lai, G.H.; Asano, T.; Shen, X.N.; Ward, D.J.; Mahatme, A.; DeWitt, J.L. A novel “patient-like” model of cholangiocarcinoma progression based on bile duct inoculation of tumorigenic rat cholangiocyte cell lines. Hepatology 2008, 47, 1178–1190. [Google Scholar] [CrossRef]
- Fingas, C.D.; Blechacz, B.R.; Smoot, R.L.; Guicciardi, M.E.; Mott, J.; Bronk, S.F.; Werneburg, N.W.; Sirica, A.E.; Gores, G.J. A smac mimetic reduces TNF related apoptosis inducing ligand (TRAIL)-induced invasion and metastasis of cholangiocarcinoma cells. Hepatology 2010, 52, 550–561. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.; Fischbach, S.R.; Bronk, S.F.; Hirsova, P.; Krishnan, A.; Dhanasekaran, R.; Smadbeck, J.B.; Smoot, R.L.; Vasmatzis, G.; Gores, G.J. YAP-associated chromosomal instability and cholangiocarcinoma in mice. Oncotarget 2018, 9, 5892–5905. [Google Scholar] [CrossRef] [Green Version]
- Banales, J.M.; Marin, J.J.G.; Lamarca, A.; Rodrigues, P.M.; Khan, S.A.; Roberts, L.R.; Cardinale, V.; Carpino, G.; Andersen, J.B.; Braconi, C.; et al. Cholangiocarcinoma 2020: The next horizon in mechanisms and management. Nat. Rev. Gastroenterol. Hepatol. 2020. [Google Scholar] [CrossRef]


{kind=link}
| Model | Strengths | Weaknesses |
|---|---|---|
| Toxic and Surgery | Early stage assessment of carcinogenesis inflammatory background | slow tumor development procedures mainly adapted to rats |
| Genetically Engineered Mice | recapitulation of most common genetic alterations early stage assessment of carcinogenesis immune-competent animals allow the study stroma–immune–tumor interactions | expensive/technically challenging no inflammatory background |
| Implantation | ||
| Syngeneic | fully functional immune system preservation of tumor microenvironment | incomplete mimicry of genetic heterogeneity of human CCA |
| PDX | preservation of histopathologic, transcriptomic, and genomic characteristics of a patient’s CCA testing of chemotherapeutic drug response/personalized precision medicine | absence of tumor microenvironment immunodeficient host |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohr, R.; Özdirik, B.; Knorr, J.; Wree, A.; Demir, M.; Tacke, F.; Roderburg, C. In Vivo Models for Cholangiocarcinoma—What Can We Learn for Human Disease? Int. J. Mol. Sci. 2020, 21, 4993. https://doi.org/10.3390/ijms21144993
Mohr R, Özdirik B, Knorr J, Wree A, Demir M, Tacke F, Roderburg C. In Vivo Models for Cholangiocarcinoma—What Can We Learn for Human Disease? International Journal of Molecular Sciences. 2020; 21(14):4993. https://doi.org/10.3390/ijms21144993
Chicago/Turabian StyleMohr, Raphael, Burcin Özdirik, Jana Knorr, Alexander Wree, Münevver Demir, Frank Tacke, and Christoph Roderburg. 2020. "In Vivo Models for Cholangiocarcinoma—What Can We Learn for Human Disease?" International Journal of Molecular Sciences 21, no. 14: 4993. https://doi.org/10.3390/ijms21144993
APA StyleMohr, R., Özdirik, B., Knorr, J., Wree, A., Demir, M., Tacke, F., & Roderburg, C. (2020). In Vivo Models for Cholangiocarcinoma—What Can We Learn for Human Disease? International Journal of Molecular Sciences, 21(14), 4993. https://doi.org/10.3390/ijms21144993