Transglutaminase 2-Mediated p53 Depletion Promotes Angiogenesis by Increasing HIF-1α-p300 Binding in Renal Cell Carcinoma

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
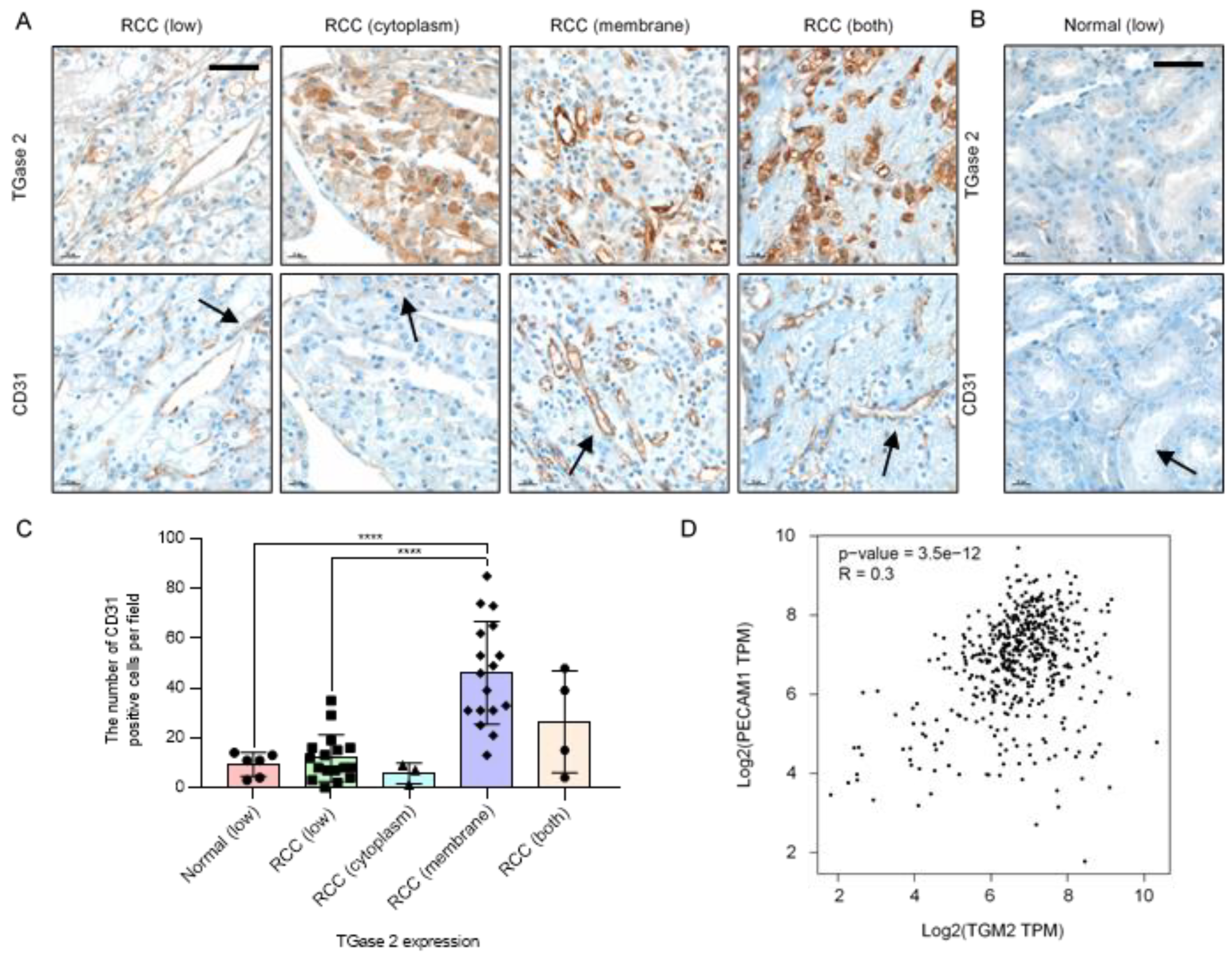
2.1. TGase 2 Expression Is Correlated with the Expression Level of CD31
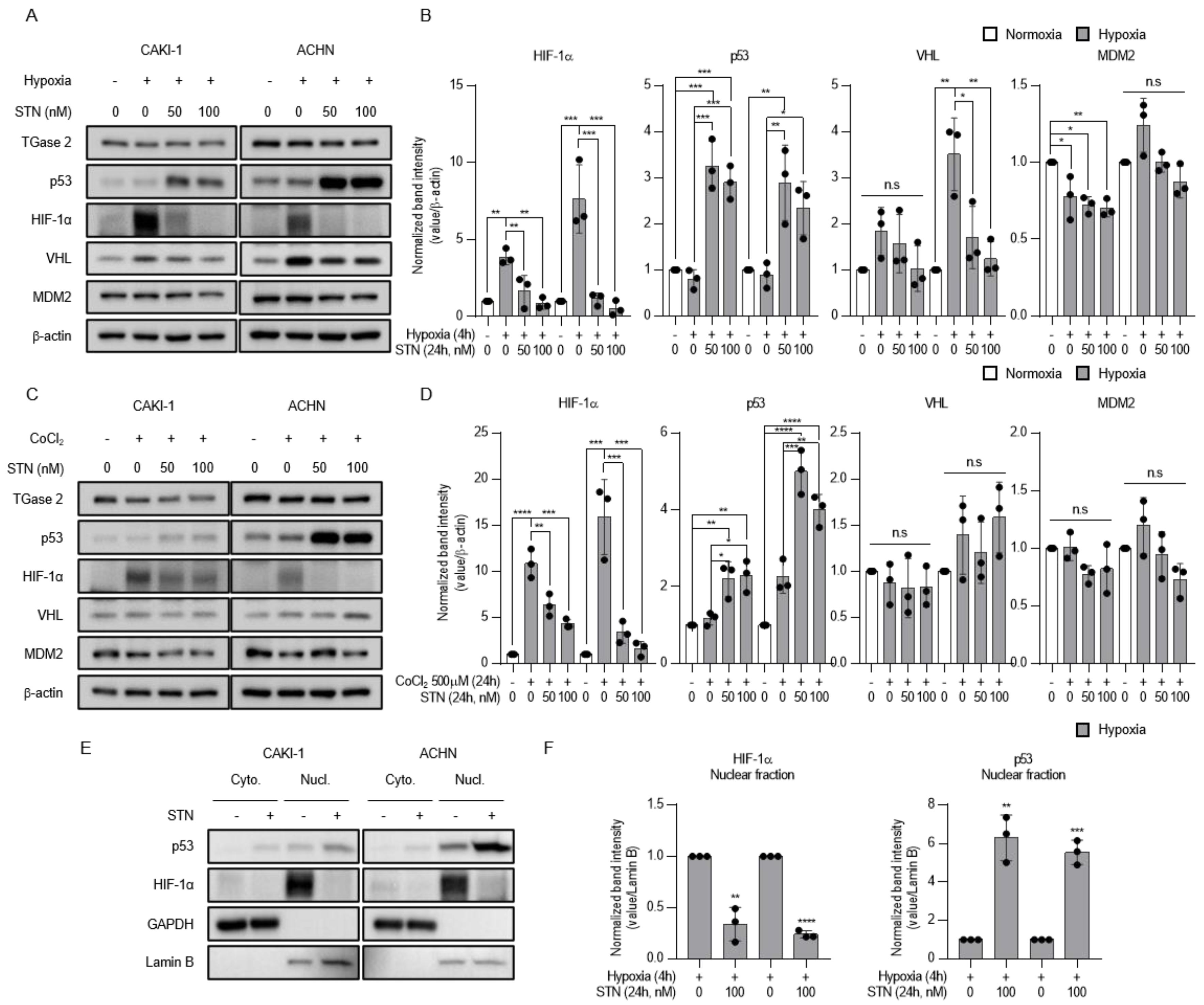
2.2. TGase 2 Inhibition Induces p53-Dependent Downregulation of Hypoxia-Inducible Factor (HIF)-1α
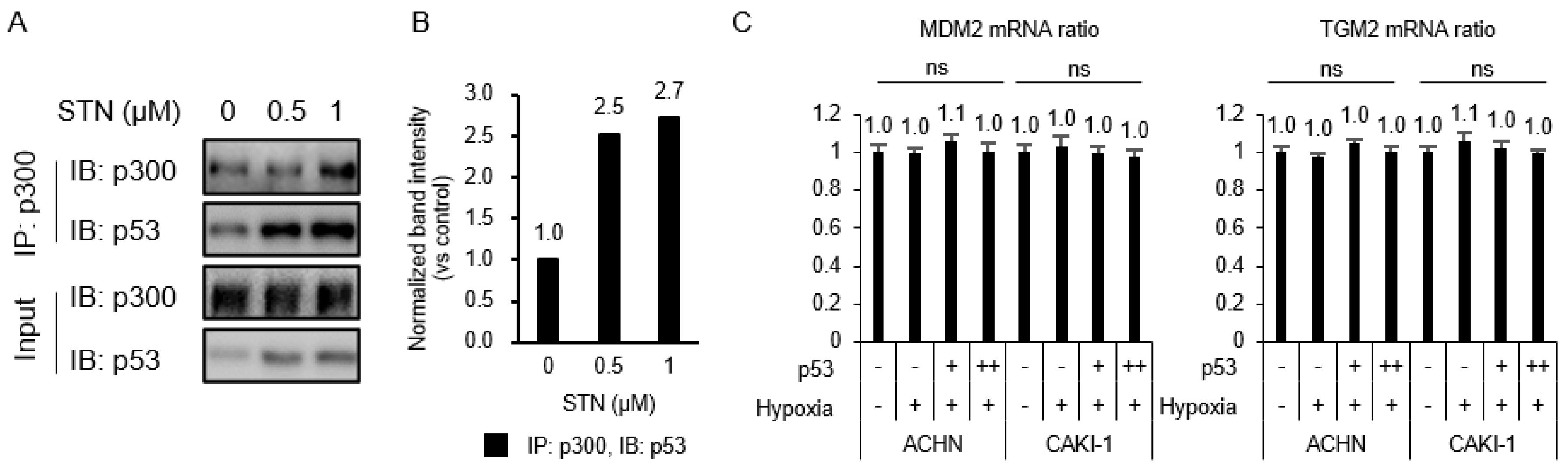
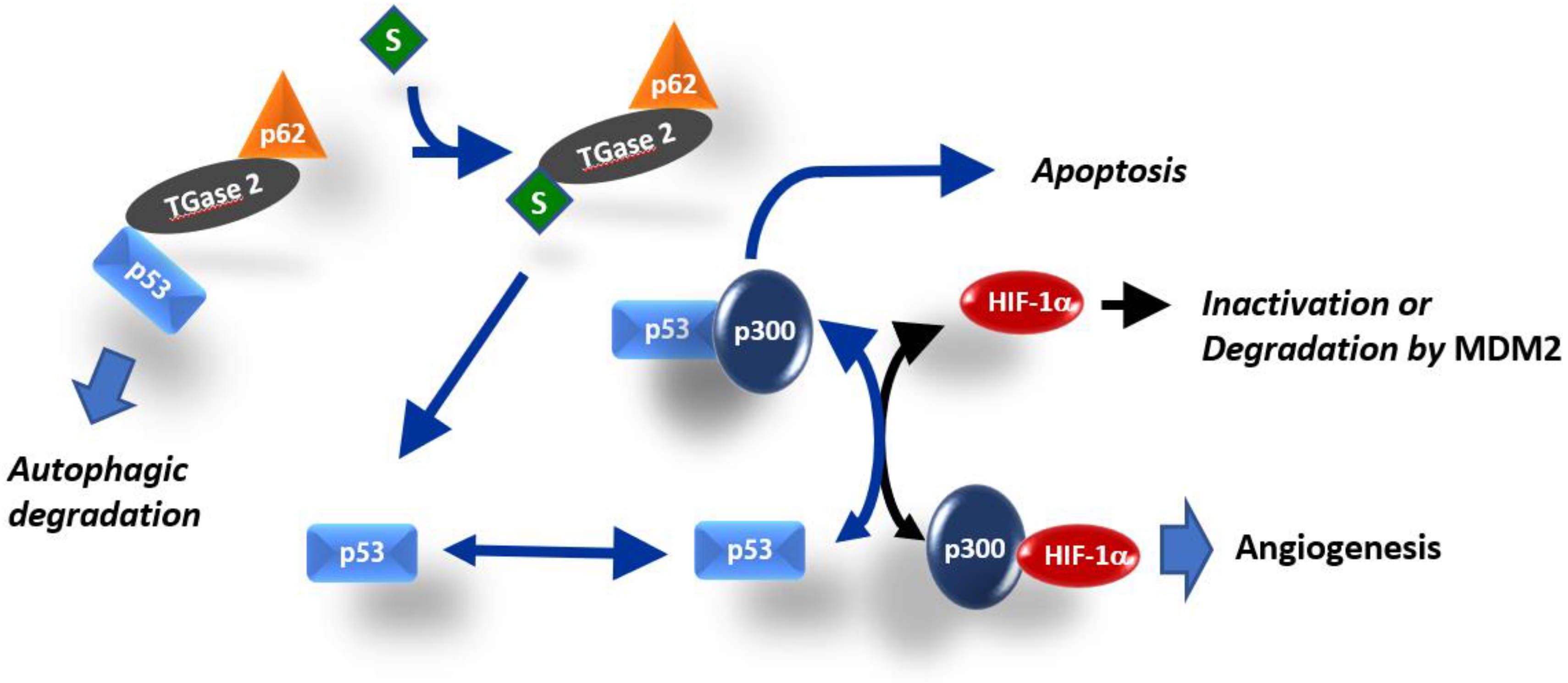
2.3. Competition between p53 and HIF-1α for Binding to p300
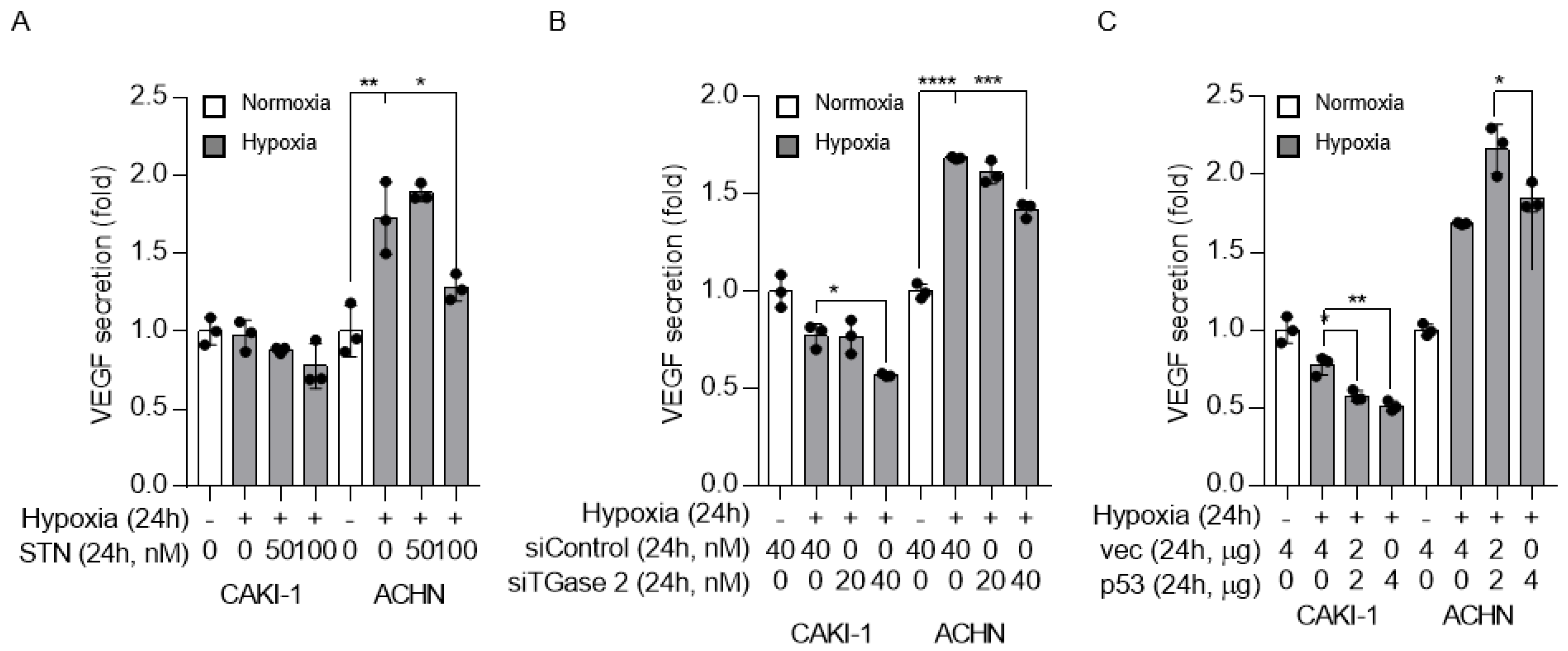
2.4. TGase 2 and p53 Modulate VEGF Secretion under Hypoxia
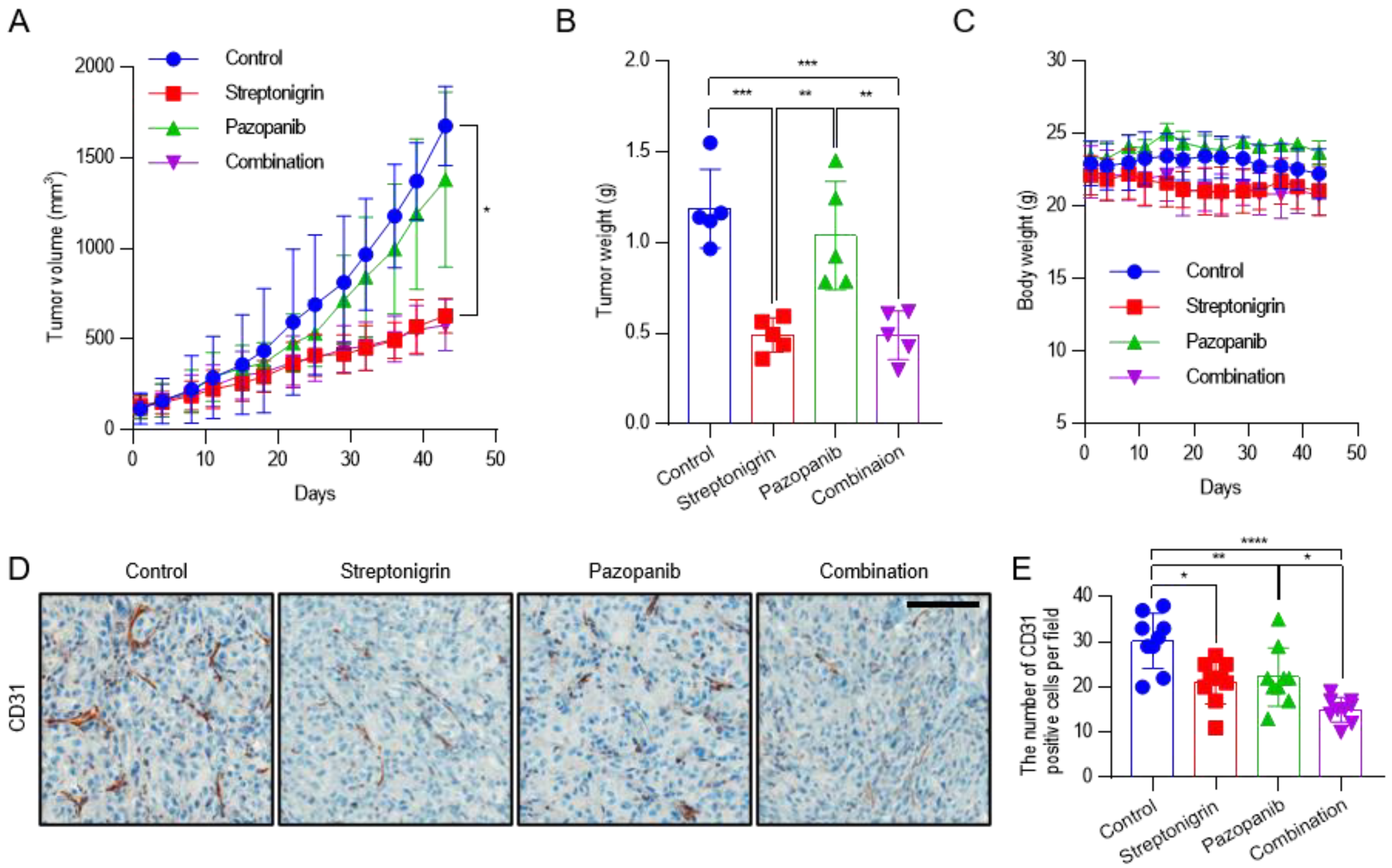
2.5. Pazopanib Combined with Streptonigrin Reduces Tumor Vascular Density
3. Discussion
4. Materials and Methods
4.1. Antibodies and Reagents
4.2. Cell Culture
4.3. Western Blotting
4.4. Real-Time PCR
4.5. VEGF ELISA
4.6. Preclinical Xenograft Tumor Models
4.7. Automated Immunohistochemistry
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mancilla-Jimenez, R.; Stanley, R.J.; Blath, R.A. Papillary renal cell carcinoma: A clinical, radiologic, and pathologic study of 34 cases. Cancer 1976, 38, 2469–2480. [Google Scholar] [CrossRef]
- Folkman, J.; Klagsbrun, M. Angiogenic factors. Science 1987, 235, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Sasaki, H.; Kim, S.J.; Tobisu, K.; Kakizoe, T.; Tsukamoto, T.; Kumamoto, Y.; Sugimura, T.; Terada, M. Markedly increased amounts of messenger RNAs for vascular endothelial growth factor and placenta growth factor in renal cell carcinoma associated with angiogenesis. Cancer Res. 1994, 54, 4233–4237. [Google Scholar]
- Nicol, D.; Hii, S.I.; Walsh, M.; Teh, B.; Thompson, L.; Kennett, C.; Gotley, D. Vascular endothelial growth factor expression is increased in renal cell carcinoma. J. Urol. 1997, 157, 1482–1486. [Google Scholar] [CrossRef]
- Park, M.J.; Baek, H.W.; Rhee, Y.Y.; Lee, C.; Park, J.W.; Kim, H.W.; Moon, K.C. Transglutaminase 2 expression and its prognostic significance in clear cell renal cell carcinoma. J. Pathol. Transl. Med. 2015, 49, 37–43. [Google Scholar] [CrossRef]
- Lee, S.H.; Lee, W.K.; Kim, N.; Kang, J.H.; Kim, K.H.; Kim, S.G.; Lee, J.S.; Lee, S.; Lee, J.; Joo, J.; et al. Renal cell carcinoma is abrogated by p53 stabilization through transglutaminase 2 inhibition. Cancers (Basel) 2018, 10, 455. [Google Scholar] [CrossRef] [Green Version]
- Folk, J.E. Mechanism and basis for specificity of transglutaminase-catalyzed epsilon-(gamma-glutamyl) lysine bond formation. Adv. Enzymol. Relat. Areas Mol. Biol. 1983, 54, 1–56. [Google Scholar]
- Kim, S.Y.; Keillor, J.W. A precision strategy to cure renal cell carcinoma by targeting transglutaminase 2. Int. J. Mol. Sci. 2020, 21, 2493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdem, S.; Yegen, G.; Telci, D.; Yildiz, I.; Tefik, T.; Issever, H.; Kilicaslan, I.; Sanli, O. The increased transglutaminase 2 expression levels during initial tumorigenesis predict increased risk of metastasis and decreased disease-free and cancer-specific survivals in renal cell carcinoma. World J. Urol. 2015, 33, 1553–1560. [Google Scholar] [CrossRef]
- Haroon, Z.A.; Hettasch, J.M.; Lai, T.S.; Dewhirst, M.W.; Greenberg, C.S. Tissue transglutaminase is expressed, active, and directly involved in rat dermal wound healing and angiogenesis. FASEB J. 1999, 13, 1787–1795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dardik, R.; Inbal, A. Complex formation between tissue transglutaminase II (tTG) and vascular endothelial growth factor receptor 2 (VEGFR-2): Proposed mechanism for modulation of endothelial cell response to VEGF. Exp. Cell Res. 2006, 312, 2973–2982. [Google Scholar] [CrossRef] [PubMed]
- Faye, C.; Chautard, E.; Olsen, B.R.; Ricard-Blum, S. The first draft of the endostatin interaction network. J. Biol. Chem. 2009, 284, 22041–22047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faye, C.; Inforzato, A.; Bignon, M.; Hartmann, D.J.; Muller, L.; Ballut, L.; Olsen, B.R.; Day, A.J.; Ricard-Blum, S. Transglutaminase-2: A new endostatin partner in the extracellular matrix of endothelial cells. Biochem. J. 2010, 427, 467–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soluri, M.F.; Boccafoschi, F.; Cotella, D.; Moro, L.; Forestieri, G.; Autiero, I.; Cavallo, L.; Oliva, R.; Griffin, M.; Wang, Z.; et al. Mapping the minimum domain of the fibronectin binding site on transglutaminase 2 (TG2) and its importance in mediating signaling, adhesion, and migration in TG2-expressing cells. FASEB J. 2019, 33, 2327–2342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hang, J.; Zemskov, E.A.; Lorand, L.; Belkin, A.M. Identification of a novel recognition sequence for fibronectin within the NH2-terminal beta-sandwich domain of tissue transglutaminase. J. Biol. Chem. 2005, 280, 23675–23683. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, I.; Osterlund, E.C.; Stamnaes, J.; Iversen, R.; Andersen, J.T.; Jorgensen, T.J.; Sollid, L.M. Dissecting the interaction between transglutaminase 2 and fibronectin. Amino Acids 2017, 49, 489–500. [Google Scholar] [CrossRef] [Green Version]
- Amelio, I.; Melino, G. The p53 family and the hypoxia-inducible factors (HIFs): Determinants of cancer progression. Trends Biochem. Sci. 2015, 40, 425–434. [Google Scholar] [CrossRef]
- Arany, Z.; Huang, L.E.; Eckner, R.; Bhattacharya, S.; Jiang, C.; Goldberg, M.A.; Bunn, H.F.; Livingston, D.M. An essential role for p300/CBP in the cellular response to hypoxia. Proc. Natl. Acad. Sci. USA 1996, 93, 12969–12973. [Google Scholar] [CrossRef] [Green Version]
- Schmid, T.; Zhou, J.; Kohl, R.; Brune, B. p300 relieves p53-evoked transcriptional repression of hypoxia-inducible factor-1 (HIF-1). Biochem. J. 2004, 380, 289–295. [Google Scholar] [CrossRef] [Green Version]
- Ravi, R.; Mookerjee, B.; Bhujwalla, Z.M.; Sutter, C.H.; Artemov, D.; Zeng, Q.; Dillehay, L.E.; Madan, A.; Semenza, G.L.; Bedi, A. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1alpha. Genes Dev. 2000, 14, 34–44. [Google Scholar]
- Blagosklonny, M.V.; An, W.G.; Romanova, L.Y.; Trepel, J.; Fojo, T.; Neckers, L. p53 inhibits hypoxia-inducible factor-stimulated transcription. J. Biol. Chem. 1998, 273, 11995–11998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, W.; Shi, X.L.; Roeder, R.G. Synergistic activation of transcription by CBP and p53. Nature 1997, 387, 819–823. [Google Scholar] [CrossRef]
- Lill, N.L.; Grossman, S.R.; Ginsberg, D.; DeCaprio, J.; Livingston, D.M. Binding and modulation of p53 by p300/CBP coactivators. Nature 1997, 387, 823–827. [Google Scholar] [CrossRef]
- Haupt, Y.; Rowan, S.; Shaulian, E.; Vousden, K.H.; Oren, M. Induction of apoptosis in HeLa cells by trans-activation-deficient p53. Genes Dev. 1995, 9, 2170–2183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, A.J. p53, the cellular gatekeeper for growth and division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.H.; Lee, J.S.; Hong, D.; Lee, S.H.; Kim, N.; Lee, W.K.; Sung, T.W.; Gong, Y.D.; Kim, S.Y. Renal cell carcinoma escapes death by p53 depletion through transglutaminase 2-chaperoned autophagy. Cell Death Dis. 2016, 7, e2163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ku, B.M.; Kim, D.S.; Kim, K.H.; Yoo, B.C.; Kim, S.H.; Gong, Y.D.; Kim, S.Y. Transglutaminase 2 inhibition found to induce p53 mediated apoptosis in renal cell carcinoma. FASEB J. 2013, 27, 3487–3495. [Google Scholar] [CrossRef] [PubMed]
- Ku, B.M.; Kim, S.J.; Kim, N.; Hong, D.; Choi, Y.B.; Lee, S.H.; Gong, Y.D.; Kim, S.Y. Transglutaminase 2 inhibitor abrogates renal cell carcinoma in xenograft models. J. Cancer Res. Clin. Oncol. 2014, 140, 757–767. [Google Scholar] [CrossRef]
- Kang, J.H.; Lee, S.H.; Cheong, H.; Lee, C.H.; Kim, S.Y. Transglutaminase 2 promotes autophagy by LC3 induction through p53 depletion in cancer cell. Biomol. Ther. Seoul 2019, 27, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.H.; Lee, S.H.; Kim, S.Y. Discovery of a novel target for renal cell carcinoma: Transglutaminase 2. Cell Death Dis. 2016, 7, e2200. [Google Scholar] [CrossRef] [PubMed]
- Nyberg, P.; Xie, L.; Kalluri, R. Endogenous inhibitors of angiogenesis. Cancer Res. 2005, 65, 3967–3979. [Google Scholar] [CrossRef] [Green Version]
- Karumanchi, S.A.; Jha, V.; Ramchandran, R.; Karihaloo, A.; Tsiokas, L.; Chan, B.; Dhanabal, M.; Hanai, J.I.; Venkataraman, G.; Shriver, Z.; et al. Cell surface glypicans are low-affinity endostatin receptors. Mol. Cell 2001, 7, 811–822. [Google Scholar] [CrossRef] [Green Version]
- Feng, Q.; Zhang, C.; Lum, D.; Druso, J.E.; Blank, B.; Wilson, K.F.; Welm, A.; Antonyak, M.A.; Cerione, R.A. A class of extracellular vesicles from breast cancer cells activates VEGF receptors and tumour angiogenesis. Nat. Commun. 2017, 8, 14450. [Google Scholar] [CrossRef] [PubMed]
- Di Simone, N.; De Spirito, M.; Di Nicuolo, F.; Tersigni, C.; Castellani, R.; Silano, M.; Maulucci, G.; Papi, M.; Marana, R.; Scambia, G.; et al. Potential new mechanisms of placental damage in celiac disease: Anti-transglutaminase antibodies impair human endometrial angiogenesis. Biol. Reprod. 2013, 88, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Dieterich, W.; Ehnis, T.; Bauer, M.; Donner, P.; Volta, U.; Riecken, E.O.; Schuppan, D. Identification of tissue transglutaminase as the autoantigen of celiac disease. Nat. Med. 1997, 3, 797–801. [Google Scholar] [CrossRef]
- Alstead, E.M.; Nelson-Piercy, C. Inflammatory bowel disease in pregnancy. Gut 2003, 52, 159–161. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Hnida, K.; Graewert, M.A.; Andersen, J.T.; Iversen, R.; Tuukkanen, A.; Svergun, D.; Sollid, L.M. Structural basis for antigen recognition by transglutaminase 2-specific autoantibodies in celiac disease. J. Biol. Chem. 2015, 290, 21365–21375. [Google Scholar] [CrossRef] [PubMed] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.-H.; Kang, J.H.; Ha, J.S.; Lee, J.-S.; Oh, S.-J.; Choi, H.-J.; Song, J.; Kim, S.-Y. Transglutaminase 2-Mediated p53 Depletion Promotes Angiogenesis by Increasing HIF-1α-p300 Binding in Renal Cell Carcinoma. Int. J. Mol. Sci. 2020, 21, 5042. https://doi.org/10.3390/ijms21145042
Lee S-H, Kang JH, Ha JS, Lee J-S, Oh S-J, Choi H-J, Song J, Kim S-Y. Transglutaminase 2-Mediated p53 Depletion Promotes Angiogenesis by Increasing HIF-1α-p300 Binding in Renal Cell Carcinoma. International Journal of Molecular Sciences. 2020; 21(14):5042. https://doi.org/10.3390/ijms21145042
Chicago/Turabian StyleLee, Seon-Hyeong, Joon Hee Kang, Ji Sun Ha, Jae-Seon Lee, Su-Jin Oh, Hyun-Jung Choi, Jaewhan Song, and Soo-Youl Kim. 2020. "Transglutaminase 2-Mediated p53 Depletion Promotes Angiogenesis by Increasing HIF-1α-p300 Binding in Renal Cell Carcinoma" International Journal of Molecular Sciences 21, no. 14: 5042. https://doi.org/10.3390/ijms21145042
APA StyleLee, S. -H., Kang, J. H., Ha, J. S., Lee, J. -S., Oh, S. -J., Choi, H. -J., Song, J., & Kim, S. -Y. (2020). Transglutaminase 2-Mediated p53 Depletion Promotes Angiogenesis by Increasing HIF-1α-p300 Binding in Renal Cell Carcinoma. International Journal of Molecular Sciences, 21(14), 5042. https://doi.org/10.3390/ijms21145042