Role of Tyr-39 for the Structural Features of α-Synuclein and for the Interaction with a Strong Modulator of Its Amyloid Assembly

 , and
, and
Abstract
:1. Introduction
2. Results
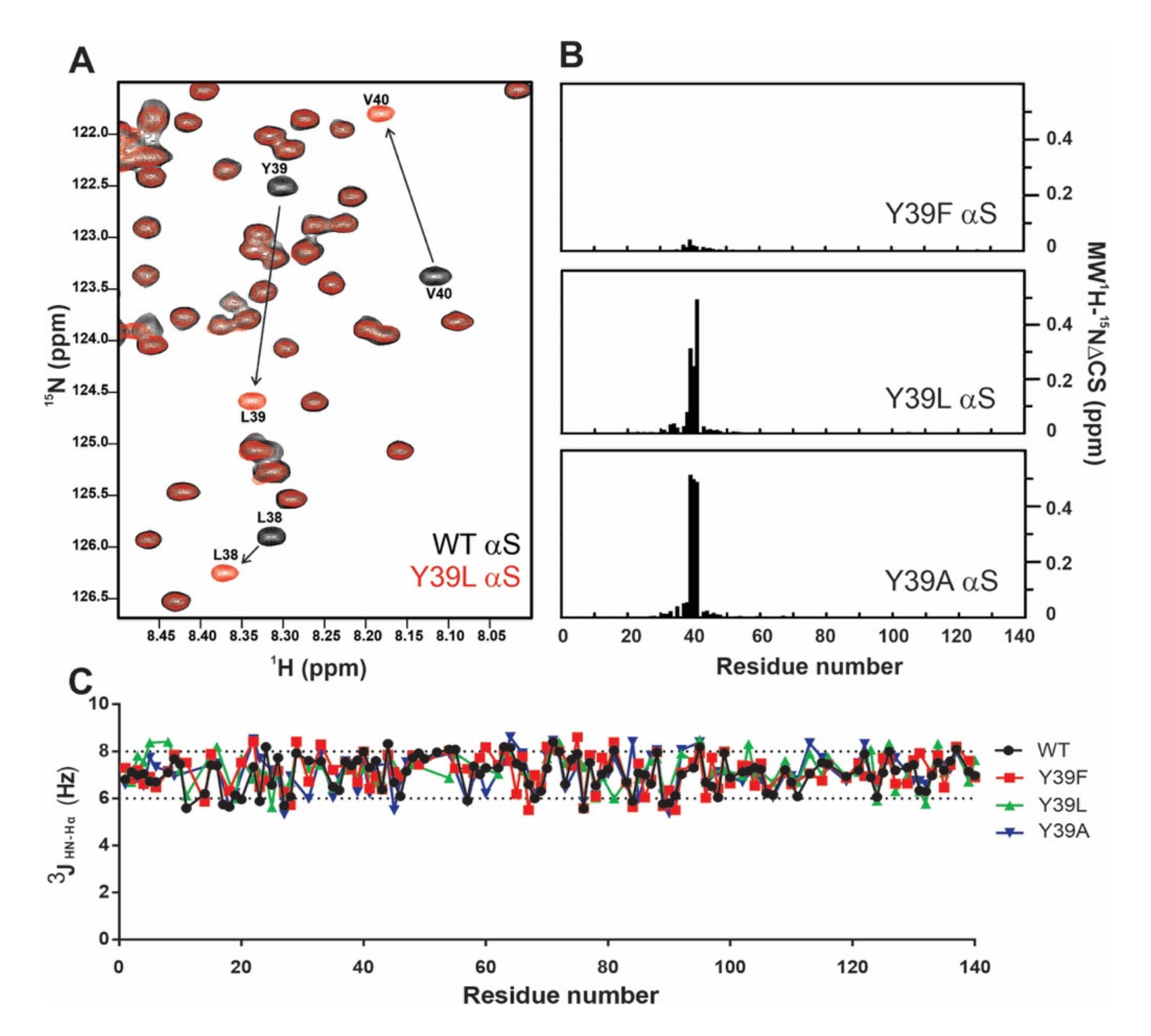
2.1. Monomeric αS Remains Disordered after Y39 Mutation
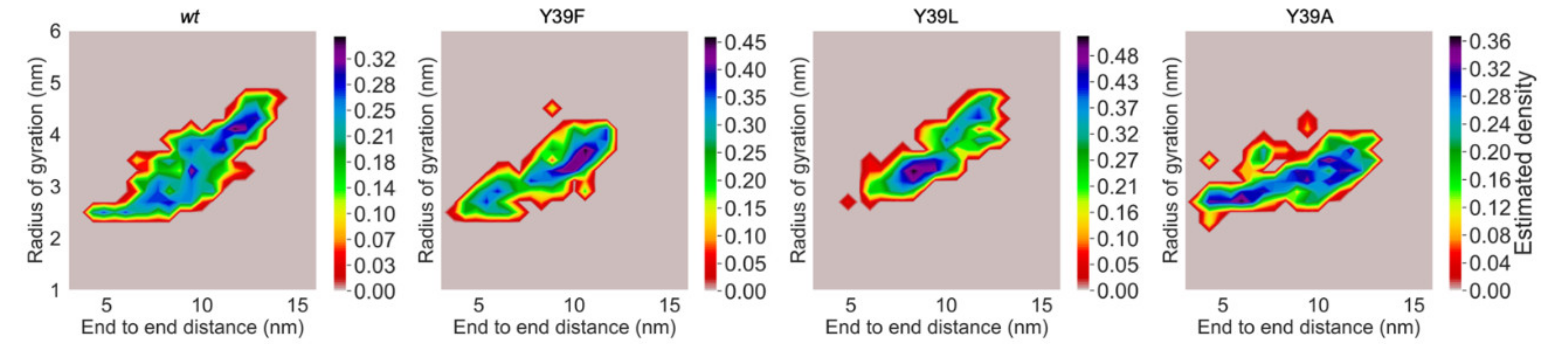
2.2. Structural Propensities of the Y39 αS Variants
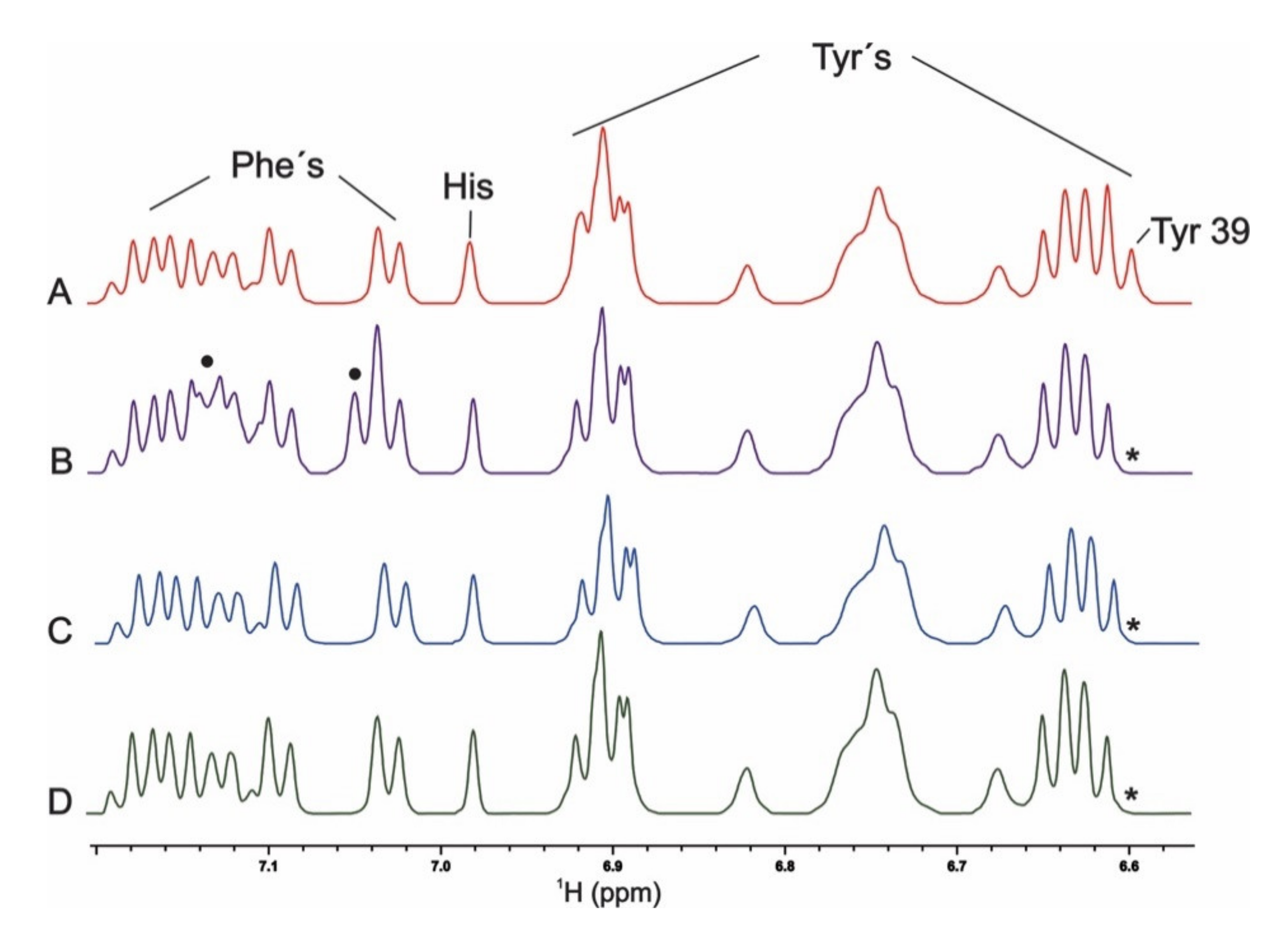
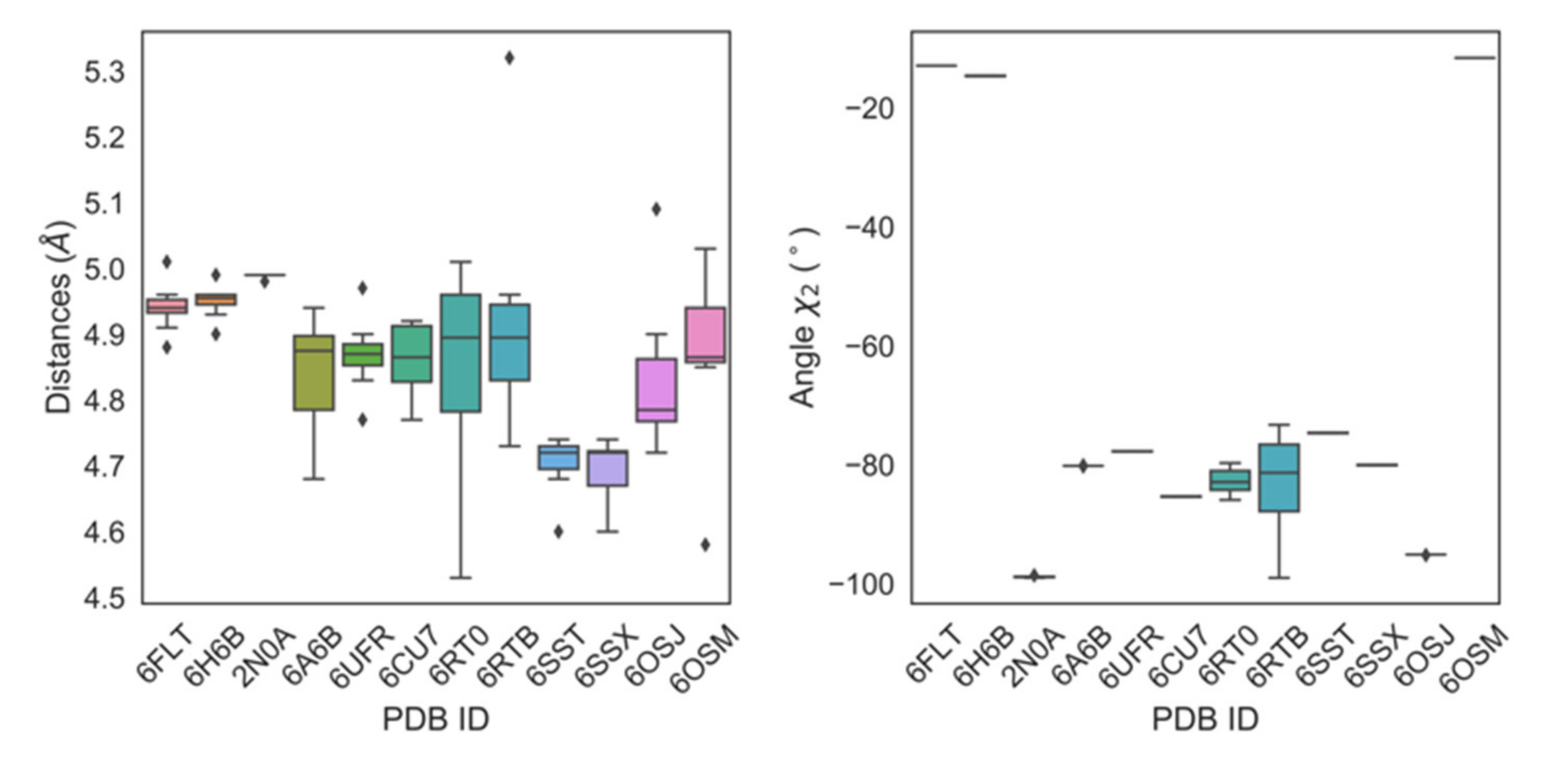
2.3. Aromaticity at Position 39 and PcTS Binding
3. Discussion
4. Materials and Methods
4.1. Proteins and Reagents
4.2. NMR Experiments
4.3. CD Spectroscopy
4.4. Molecular Simulations
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Soto, C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 2003, 4, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Bitan, G. Modulating self-assembly of amyloidogenic proteins as a therapeutic approach for neurodegenerative diseases: Strategies and mechanisms. ChemMedChem 2012, 7, 359–374. [Google Scholar] [CrossRef] [PubMed]
- Gazit, E. A possible role for pi-stacking in the self-assembly of amyloid fibrils. FASEB J. 2002, 16, 77–83. [Google Scholar] [CrossRef]
- Valiente-Gabioud, A.A.; Miotto, M.C.; Chesta, M.E.; Lombardo, V.; Binolfi, A.; Fernandez, C.O. Phthalocyanines as molecular scaffolds to block disease-associated protein aggregation. Acc. Chem. Res. 2016, 49, 801–808. [Google Scholar] [CrossRef] [Green Version]
- Lamberto, G.R.; Binolfi, A.; Orcellet, M.L.; Bertoncini, C.W.; Zweckstetter, M.; Griesinger, C.; Fernandez, C.O. Structural and mechanistic basis behind the inhibitory interaction of PcTS on alpha-synuclein amyloid fibril formation. Proc. Natl. Acad. Sci. USA 2009, 106, 21057–21062. [Google Scholar] [CrossRef] [Green Version]
- Bulic, B.; Pickhardt, M.; Khlistunova, I.; Biernat, J.; Mandelkow, E.M.; Mandelkow, E.; Waldmann, H. Rhodanine-based tau aggregation inhibitors in cell models of tauopathy. Angew. Chem. Int. Ed. 2007, 46, 9215–9219. [Google Scholar] [CrossRef]
- Schenk, D.; Basi, G.S.; Pangalos, M.N. Treatment strategies targeting amyloid β-protein. Cold Spring Harb. Perspect. Med. 2012, 2, a006387. [Google Scholar] [CrossRef] [Green Version]
- Masuda, M.; Suzuki, N.; Taniguchi, S.; Oikawa, T.; Nonaka, T.; Iwatsubo, T.; Hisanaga, S.-I.; Goedert, M.; Hasegawa, M. Small molecule inhibitors of alpha-synuclein filament assembly. Biochemistry 2006, 45, 6085–6094. [Google Scholar] [CrossRef]
- Caughey, B.; Caughey, W.S.; Kocisko, D.A.; Lee, K.S.; Silveira, J.R.; Morrey, J.D. Prions and transmissible spongiform encephalopathy (TSE) chemotherapeutics: A common mechanism for anti-TSE compounds? Acc. Chem. Res. 2006, 39, 646–653. [Google Scholar] [CrossRef]
- Ehrnhoefer, D.E.; Bieschke, J.; Boeddrich, A.; Herbst, M.; Masino, L.; Lurz, R.; Engemann, S.; Pastore, A.; Wanker, E.E.; Ehrnhoefer, D.E. EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat. Struct. Mol. Biol. 2008, 15, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Caughey, W.S.; Priola, S.A.; Kocisko, D.A.; Raymond, L.D.; Ward, A.; Caughey, B. Cyclic tetrapyrrrole sulfonation, metals, and oligomerization in antiprion activity. Antimicrob. Agents Chemother. 2007, 51, 3887–3894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, J.; Ryazanov, S.; Leonov, A.; Levin, J.; Shi, S.; Schmidt, F.; Prix, C.; Pan-Montojo, F.; Bertsch, U.; Mitteregger-Kretzschmar, G. Anle138b: A novel oligomer modulator for disease-modifying therapy of neurodegenerative diseases such as prion and Parkinson’s disease. Acta Neuropathol. 2013, 125, 795–813. [Google Scholar] [CrossRef] [Green Version]
- Levin, J.; Schmidt, F.; Boehm, C.; Prix, C.; Bötzel, K.; Ryazanov, S.; Leonov, A.; Griesinger, C.; Giese, A. The oligomer modulator anle138b inhibits disease progression in a Parkinson mouse model even with treatment started after disease onset. Acta Neuropathol. 2014, 127, 779–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherzer-Attali, R.; Shaltiel-Karyo, R.; Adalist, Y.H.; Segal, D.; Gazit, E. Generic inhibition of amyloidogenic proteins by two naphthoqui-none-tryptophan hybrid molecules. Proteins 2012, 80, 1962–1973. [Google Scholar] [CrossRef] [PubMed]
- Frydman-Marom, A.; Shaltiel-Karyo, R.; Moshe, S.; Gazit, E. The generic amyloid formation inhibition effect of a designed small aromatic β-breaking peptide. Amyloid 2011, 18, 119–127. [Google Scholar] [CrossRef]
- Chakraborty, R.; Sahoo, S.; Halder, N.; Rath, H.; Chattopadhyay, K. Conformational-Switch Based Strategy Triggered by [18] π Heteroannulenes toward Reduction of Alpha Synuclein Oligomer Toxicity. ACS Chem. Neurosci. 2019, 10, 573–587. [Google Scholar] [CrossRef]
- Lizarraga, F.G.; Socías, S.B.; Avila, C.L.; Torres-Bugeau, C.M.; Barbosa, L.R.; Binolfi, A.; Sepúlveda-Díaz, J.E.; Del Bel, E.; Fernández, C.O.; Papy-Garcia, D.; et al. Repurposing doxycycline for synucleinopathies: Remodelling of α-synuclein oligomers towards non-toxic parallel beta-sheet structured species. Sci. Rep. 2017, 7, 41755. [Google Scholar] [CrossRef] [Green Version]
- Pujols, J.; Díaz, S.P.; Lázaro, D.F.; Peccati, F.; Pinheiro, F.; González, D.; Carija, A.; Navarro, S.; Giménez, M.C.; García, J.; et al. Small molecule inhibits α-synuclein aggregation, disrupts amyloid fibrils, and prevents degeneration of dopaminergic neurons. Proc. Natl. Acad. Sci. USA 2018, 115, 10481–10486. [Google Scholar] [CrossRef] [Green Version]
- Valdinocci, D.; Grant, G.D.; Dickson, T.; Pountney, D. Epothilone D inhibits microglia-mediated spread of alpha-synuclein aggregates. Mol. Cell Neurosci. 2018, 89, 80–94. [Google Scholar] [CrossRef] [Green Version]
- Schwab, K.; Frahm, S.; Horsley, D.; Rickard, J.E.; Melis, V.; Goatman, E.A.; Magbagbeolu, M.; Douglas, M.; Leith, M.G.; Baddeley, T.C.; et al. A protein aggregation inhibitor, leuco-methylthioninium bis(hydromethanesulfonate), decreases α-synuclein inclusions in a transgenic mouse model of synucleinopathy. Front. Mol. Neurosci. 2017, 10, 447. [Google Scholar] [CrossRef] [PubMed]
- Palazzi, L.; Bruzzone, E.; Bisello, G.; Leri, M.; Stefani, M.; Bucciantini, M.; De Laureto, P.P. Oleuropein aglycone stabilizes the monomeric α-synuclein and favours the growth of non-toxic aggregates. Sci. Rep. 2018, 8, 8337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, N.N.; Kumar, R.; Panigrahi, R.; Navalkar, A.; Ghosh, D.; Sahay, S.; Mondal, M.; Kumar, A.; Maji, S.K. Comparison of α-synuclein fibril inhibition by four different amyloid inhibitors. ACS Chem. Neurosci. 2017, 8, 2722–2733. [Google Scholar] [CrossRef] [PubMed]
- Reiner, A.M.; Schmidt, F.; Ryazanov, S.; Leonov, A.; Weckbecker, D.; Deeg, A.A.; Griesinger, C.; Giese, A.; Zinth, W. Photophysics of diphenyl-pyrazole compounds in solutions and α-synuclein aggregates. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 800–807. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Freed, C.R. Tyrosine-to-cysteine modification of human α-synuclein enhances protein aggregation and cellular toxicity. J. Biol. Chem. 2004, 279, 10128–10135. [Google Scholar] [CrossRef] [Green Version]
- Ulrih, N.P.; Barry, C.H.; Fink, A.L. Impact of Tyr to Ala mutations on α-synuclein fibrillation and structural properties. Biochim. Biophys. Acta 2008, 1782, 581–585. [Google Scholar] [CrossRef] [Green Version]
- Kaylor, J.; Bodner, N.; Edridge, S.; Yamin, G.; Hong, D.P.; Fink, A.L. Characterization of oligomeric intermediates in alpha-synuclein fibrillation: FRET studies of Y125W/Y133F/Y136F alpha-synuclein. J. Mol. Biol. 2005, 353, 357–372. [Google Scholar] [CrossRef]
- Zhu, M.; Rajamani, S.; Kaylor, J.; Han, S.; Zhou, F.; Fink, A.L. The flavonoid baicalein inhibits fibrillation of alpha-synuclein and disaggregates existing fibrils. J. Biol. Chem. 2004, 279, 26846–26857. [Google Scholar] [CrossRef] [Green Version]
- Burai, R.; Ait-Bouziad, N.; Chiki, A.; Lashuel, H.A. Elucidating the Role of Site-Specific Nitration of α-Synuclein in the Pathogenesis of Parkinson’s Disease via Protein Semisynthesis and Mutagenesis. J. Am. Chem. Soc. 2015, 137, 5041–5052. [Google Scholar] [CrossRef]
- Wang, L.; Friesner, R.A.; Berne, B.J. Replica exchange with solute scaling: A more efficient version of replica exchange with solute tempering (REST2). J. Phys. Chem. B 2011, 115, 9431–9438. [Google Scholar] [CrossRef] [Green Version]
- Lamberto, G.R.; Torres-Monserrat, V.; Bertoncini, C.W.; Salvatella, X.; Zweckstetter, M.; Griesinger, C.; Fernandez, C.O. Towards the discovery of effective polycyclic inhibitors of alpha-Synuclein amyloid assembly. J. Biol. Chem. 2011, 286, 32036–32044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentile, I.; Garro, H.A.; Delgado Ocaña, S.; Gonzalez, N.; Strohäker, T.; Schibich, D.; Quintanar, L.; Sambrotta, L.; Zweckstetter, M.; Griesinger, C.; et al. Interaction of Cu(I) with the Met-X3-Met motif of alpha-synuclein: Binding ligands, affinity and structural features. Metallomics 2018, 10, 1383–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, J.A.; Wilkins, D.K.; Smith, L.J.; Dobson, C.M. Characterization of protein unfolding by NMR diffusion measurements. J. Biomol. NMR 1997, 10, 199–203. [Google Scholar] [CrossRef]
- Altieri, A.S.; Hinton, D.P.; Byrd, R.A. Association of Biomolecular Systems via Pulsed Field Gradient NMR Self-Diffusion Measurements. J. Am. Chem. Soc. 1995, 117, 7566–7567. [Google Scholar] [CrossRef]
- Binolfi, A.; Rasia, R.M.; Bertoncini, C.W.; Ceolin, M.; Zweckstetter, M.; Griesinger, C.; Jovin, T.M.; Fernández, C.O. Interaction of α-Synuclein with Divalent Metal Ions Reveals Key Differences: A Link between Structure, Binding Specificity and Fibrillation Enhancement. J. Am. Chem. Soc. 2006, 128, 9893–9901. [Google Scholar] [CrossRef]
- Bertoncini, C.W.; Rasia, R.M.; Lamberto, G.R.; Binolfi, A.; Zweckstetter, M.; Griesinger, C.; Fernandez, C.O. Structural characterization of the intrinsically unfolded protein beta-synuclein, a natural negative regulator of alpha-synuclein aggregation. J. Mol. Biol. 2007, 372, 708–722. [Google Scholar] [CrossRef]
- Morar, A.S.; Olteanu, A.; Young, G.B.; Pielak, G.J. Solvent-induced collapse of alpha-synuclein and acid-denatured cytochrome c. Protein Sci. 2001, 10, 2195–2199. [Google Scholar] [CrossRef]
- Davis-Searles, P.R.; Saunders, A.J.; Erie, D.A.; Winzor, D.J.; Pielak, G.J. Interpreting the effects of small uncharged solutes on protein-folding equilibria. Annu. Rev. Biophys. Biomol. Struct. 2001, 30, 271–306. [Google Scholar] [CrossRef] [Green Version]
- Wilkins, D.K.; Grimshaw, S.B.; Receveur, V.; Dobson, C.M.; Jones, J.A.; Smith, L.J. Hydrodynamic radii of native and denatured proteins measured by pulse field gradient NMR techniques. Biochemistry 1999, 38, 16424–16431. [Google Scholar] [CrossRef]
- Allison, J.R.; Rivers, R.C.; Christodoulou, J.C.; Vendruscolo, M.; Dobson, C.M. A relationship between the transient structure in the monomeric state and the aggregation propensities of α-synuclein and β-synuclein. Biochemistry 2014, 53, 7170–7183. [Google Scholar] [CrossRef]
- Maltsev, A.S.; Ying, J.; Bax, A. Impact of N-terminal acetylation of alpha-synuclein on its random coil and lipid binding properties. Biochemistry 2012, 51, 5004–5013. [Google Scholar] [CrossRef] [PubMed]
- Wise-Scira, O.; Dunn, A.; Aloglu, A.K.; Sakallioglu, I.T.; Coskuner, O. Structures of the E46K mutant-type alpha-synuclein protein and impact of E46K mutation on the structures of the wild-type alpha-synuclein protein. ACS Chem. Neurosci. 2013, 4, 498–508. [Google Scholar] [CrossRef] [Green Version]
- Hollingsworth, S.A.; Karplus, P.A. A fresh look at the Ramachandran plot and the occurrence of standard structures in proteins. Biomol. Concepts 2010, 1, 271–283. [Google Scholar] [CrossRef] [PubMed]
- Dusa, A.; Kaylor, J.; Edridge, S.; Bodner, N.; Hong, N.-P.; Fink, A.L. Characterization of oligomers during α-synuclein aggregation using intrinsic tryptophan fluorescence. Biochemistry 2006, 45, 2752–2760. [Google Scholar] [CrossRef]
- Guerrero-Ferreira, R.; Taylor, N.M.I.; Mona, D.; Ringler, P.; Lauer, M.E.; Riek, R.; Britschgi, M.; Stahlberg, H. Cryo-EM structure of alpha-synuclein fibrils. Elife 2018, 7, e36402. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhao, C.; Luo, F.; Liu, Z.; Gui, X.; Luo, Z.; Zhang, X.; Li, D.; Liu, C.; Li, X. Amyloid fibril structure of alpha-synuclein determined by cryo-electron microscopy. Cell Res. 2018, 28, 897–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meade, R.M.; Fairlie, D.P.; Mason, J.M. Alpha-synuclein structure and Parkinson’s disease—Lessons and emerging principles. Mol. Neurodegener 2019, 14, 29. [Google Scholar] [CrossRef] [Green Version]
- Der-Sarkissian, A.; Jao, C.C.; Chen, J.; Langen, R. Structural organization of alpha-synuclein fibrils studied by site-directed spin labeling. J. Biol. Chem. 2003, 278, 37530–37535. [Google Scholar] [CrossRef] [Green Version]
- Heise, H.; Hoyer, W.; Becker, S.; Andronesi, O.C.; Riedel, D.; Baldus, M. Molecular-level secondary structure, polymorphism, and dynamics of full-length alpha-synuclein fibrils studied by solid-state NMR. Proc. Natl. Acad. Sci. USA 2005, 102, 15871–15876. [Google Scholar] [CrossRef] [Green Version]
- Johnson, M.; Geeves, M.A.; Mulvihill, D.P. Production of amino-terminally acetylated recombinant proteins in E. coli. Methods Mol. Biol. 2013, 981, 193–200. [Google Scholar] [CrossRef] [Green Version]
- Hoyer, W.; Cherny, D.; Subramaniam, V.; Jovin, T.M. Impact of the acidic C-terminal region comprising amino acids 109-140 on alpha-synuclein aggregation in vitro. Biochemistry 2004, 43, 16233–16242. [Google Scholar] [CrossRef]
- Cavanagh, J.; Fairbrother, W.; Palmer, A., III; Skelton, N. Protein NMR Spectroscopy: Principles and Practice; Academic Press: Cambridge, MA, USA, 1995; ISBN 9780080515298. [Google Scholar]
- Vuister, G.W.; Bax, A. Quantitative J correlation: A new approach for measuring homonuclear three-bond J(HNHA) coupling constants in 15N-enriched proteins. J. Am. Chem. Soc. 1993, 115, 7772–7777. [Google Scholar] [CrossRef]
- Hill, R.B.; Flanagan, J.M.; Prestegard, J.H. 1H and 15N magnetic resonance assignments, secondary structure, and tertiary fold of Escherichia coli DnaJ(1-78). Biochemistry 1995, 34, 5587–5596. [Google Scholar] [CrossRef] [PubMed]
- Serrano, L. Comparison between the phi distribution of the amino acids in the protein database and NMR data indicates that amino acids have various phi propensities in the random coil conformation. J. Mol. Biol. 1995, 254, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, G.; Musiani, F.; Abad, E.; DiBenedetto, D.; Mouhib, H.; Fernández, C.O.; Carloni, P. Conformational ensemble of human alpha-synuclein physiological form predicted by molecular simulations. Phys. Chem. Chem. Phys. 2016, 18, 5702–5706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daura, X.; Gademann, K.; Jaun, B.; Seebach, D.; Van Gunsteren, W.F.; Mark, A.E. Peptide Folding: When Simulation Meets Experiment. Angew. Chem. Int. Ed. 1999, 38, 236–240. [Google Scholar] [CrossRef]
- Páll, S.; Abraham, M.; Kutzner, C.; Hess, B.; Lindahl, E. Tackling exascale software challenges in molecular dynamics simulations with GROMACS. In Solving Software Challenges for Exascale; Markidis, S., Laure, E., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 3–27. [Google Scholar]
- Schrödinger. Schrödinger Release 2019-3: Maestro. 2019: New York, NY. Available online: https://www.schrodinger.com (accessed on 15 July 2020).
- Tribello, G.A.; Bonomi, M.; Branduardi, D.; Camilloni, C.; Bussi, G. PLUMED 2: New feathers for an old bird. Comput. Phys. Commun. 2014, 185, 604–613. [Google Scholar] [CrossRef] [Green Version]
- Robustelli, P.; Piana, S.; Shaw, D.E. Developing a molecular dynamics force field for both folded and disordered protein states. Proc. Natl. Acad. Sci. USA 2018, 115, E4758–E4766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.; Fraaije, J.G. LINCS: A linear constraint solver for molecular simulations. J. Comp. Chem. 1998, 18, 9. [Google Scholar] [CrossRef]
- Nosé, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 2006, 52, 255–268. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A Gen. Phys. 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Chem. Phys. 1998, 52, 7182–7190. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Mavridis, L.; Janes, R.W. PDB2CD: A web-based application for the generation of circular dichroism spectra from protein atomic coordinates. Bioinformatics 2017, 33, 56–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, B.; Liu, Y.; Ginzinger, S.W.; Wishart, D.S. SHIFTX2: Significantly improved protein chemical shift prediction. J. Biomol. NMR 2011, 50, 43–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabsch, W.; Sander, C. Dictionary of Protein Secondary Structure: Pattern Recognition of Hydrogen-Bonded and Geometrical Features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Secondary Structure | wt | Y39F | Y39L | Y39A |
|---|---|---|---|---|
| Coil | 72 ± 6 | 67 ± 6 | 59 ± 6 | 68 ± 6 |
| Bend | 30 ± 5 | 31 ± 4 | 32 ± 5 | 25 ± 5 |
| Turn | 19 ± 5 | 23 ± 6 | 25 ± 5 | 24 ± 6 |
| α-Helix | 11 ± 4 | 8 ± 5 | 11 ± 4 | 11 ± 4 |
| 3-Helix | 5 ± 4 | 7 ± 4 | 7 ± 4 | 6 ± 4 |
| β-Sheet | 2 ± 4 | 1 ± 2 | 3 ± 3 | 2 ± 2 |
| β-Bridge | 1 ± 2 | 3 ± 2 | 4 ± 2 | 4 ± 2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palomino-Hernandez, O.; Buratti, F.A.; Sacco, P.S.; Rossetti, G.; Carloni, P.; Fernandez, C.O. Role of Tyr-39 for the Structural Features of α-Synuclein and for the Interaction with a Strong Modulator of Its Amyloid Assembly. Int. J. Mol. Sci. 2020, 21, 5061. https://doi.org/10.3390/ijms21145061
Palomino-Hernandez O, Buratti FA, Sacco PS, Rossetti G, Carloni P, Fernandez CO. Role of Tyr-39 for the Structural Features of α-Synuclein and for the Interaction with a Strong Modulator of Its Amyloid Assembly. International Journal of Molecular Sciences. 2020; 21(14):5061. https://doi.org/10.3390/ijms21145061
Chicago/Turabian StylePalomino-Hernandez, Oscar, Fiamma A. Buratti, Pamela S. Sacco, Giulia Rossetti, Paolo Carloni, and Claudio O. Fernandez. 2020. "Role of Tyr-39 for the Structural Features of α-Synuclein and for the Interaction with a Strong Modulator of Its Amyloid Assembly" International Journal of Molecular Sciences 21, no. 14: 5061. https://doi.org/10.3390/ijms21145061
APA StylePalomino-Hernandez, O., Buratti, F. A., Sacco, P. S., Rossetti, G., Carloni, P., & Fernandez, C. O. (2020). Role of Tyr-39 for the Structural Features of α-Synuclein and for the Interaction with a Strong Modulator of Its Amyloid Assembly. International Journal of Molecular Sciences, 21(14), 5061. https://doi.org/10.3390/ijms21145061