Downregulation of GLI3 Expression Mediates Chemotherapy Resistance in Acute Myeloid Leukemia

 , ,
, , 

Abstract
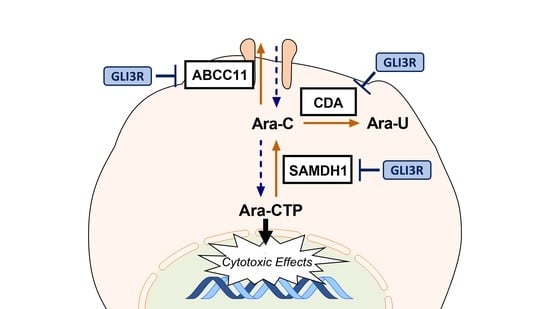
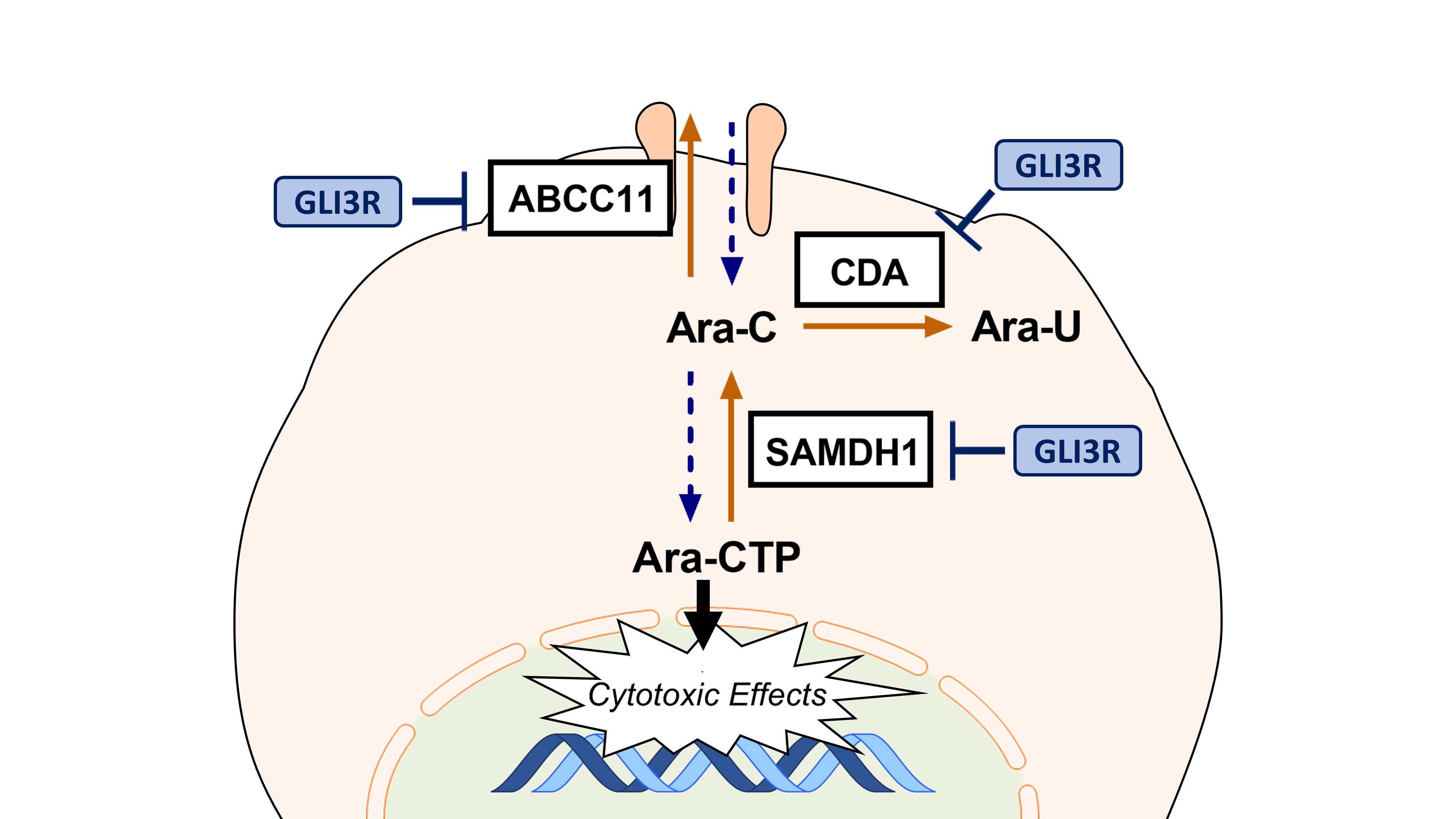
:
1. Introduction
2. Results
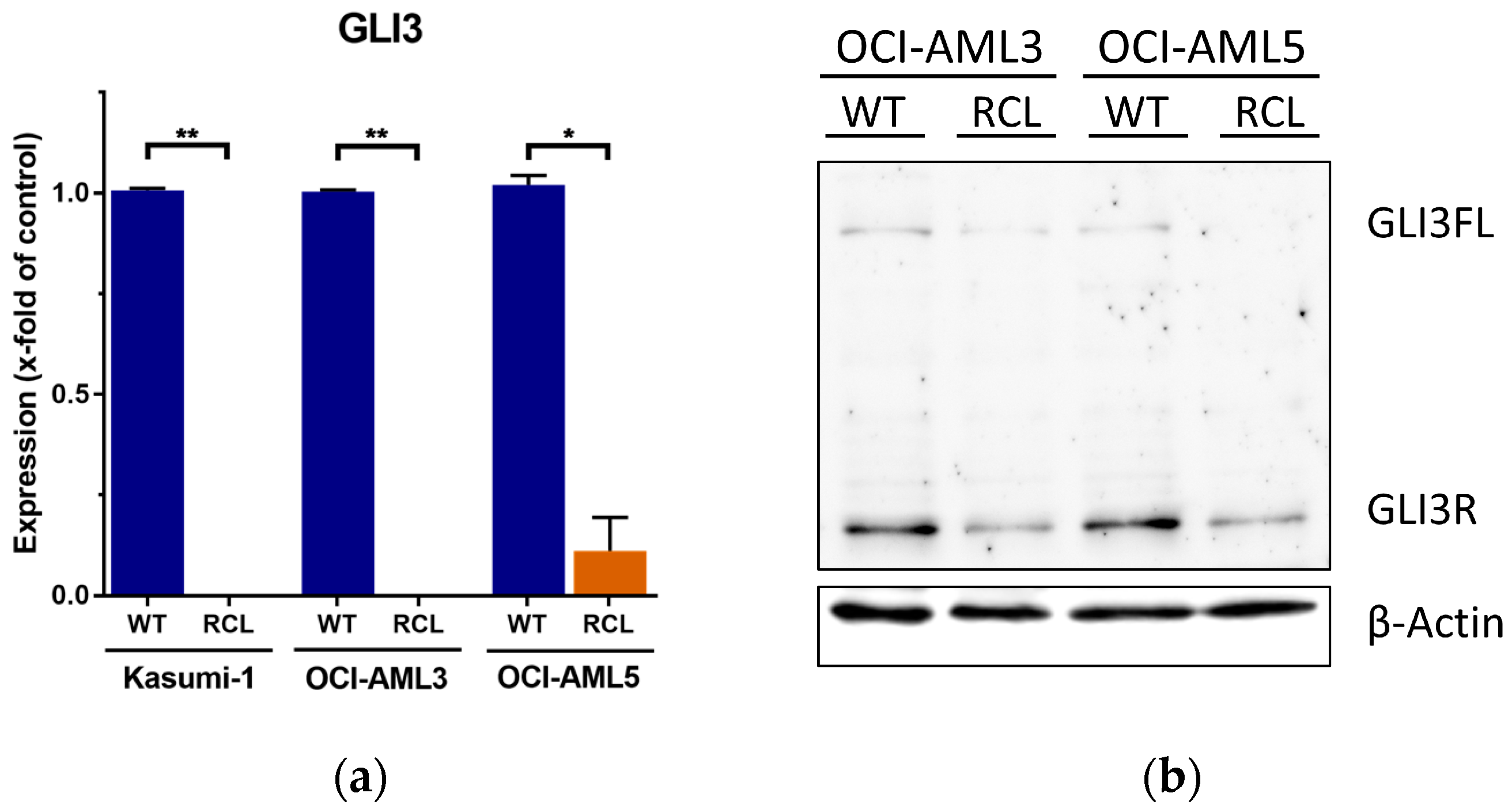
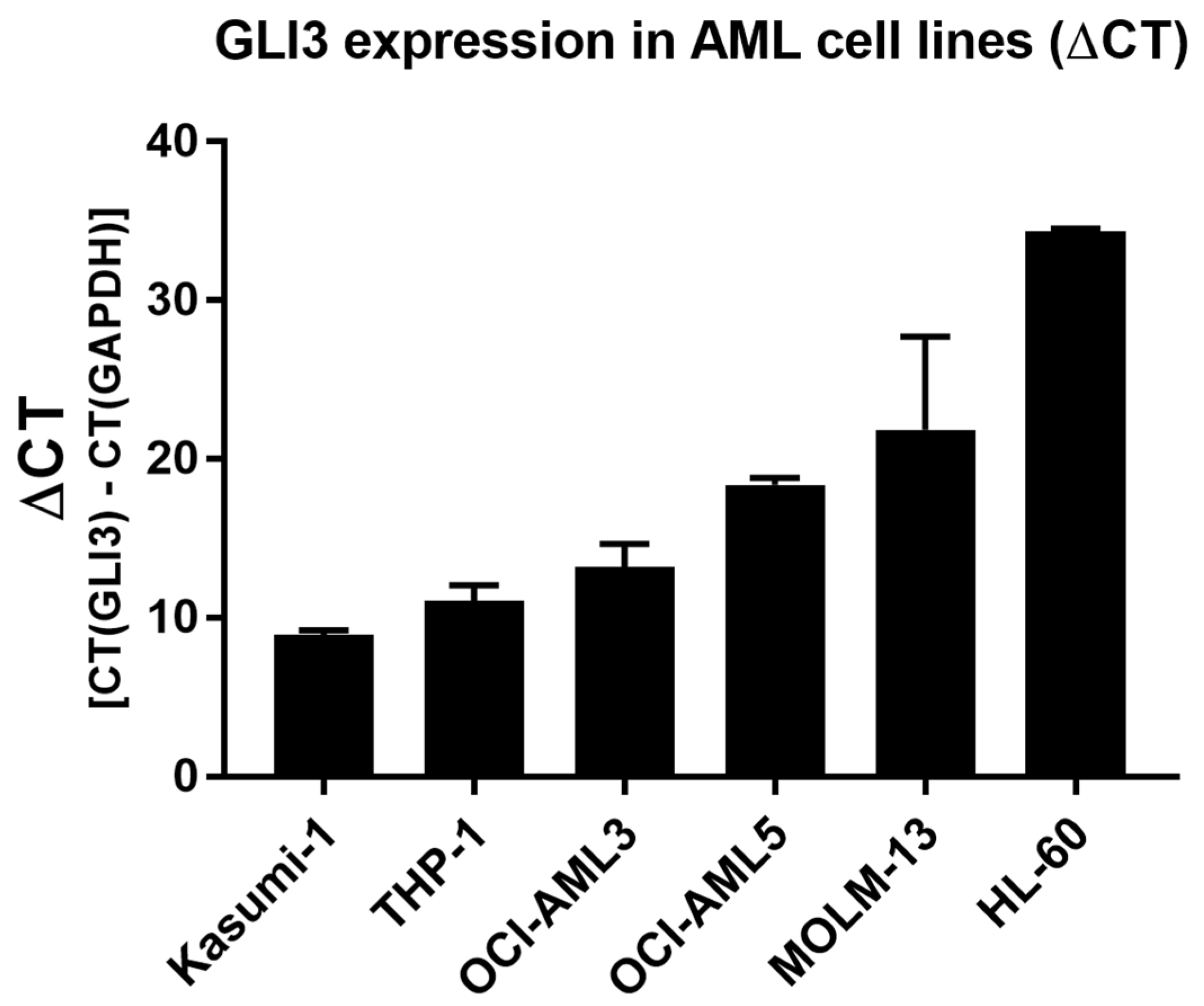
2.1. GLI3 Expression Silenced in Cytarabine (Ara-C)-Resistant Cell Lines
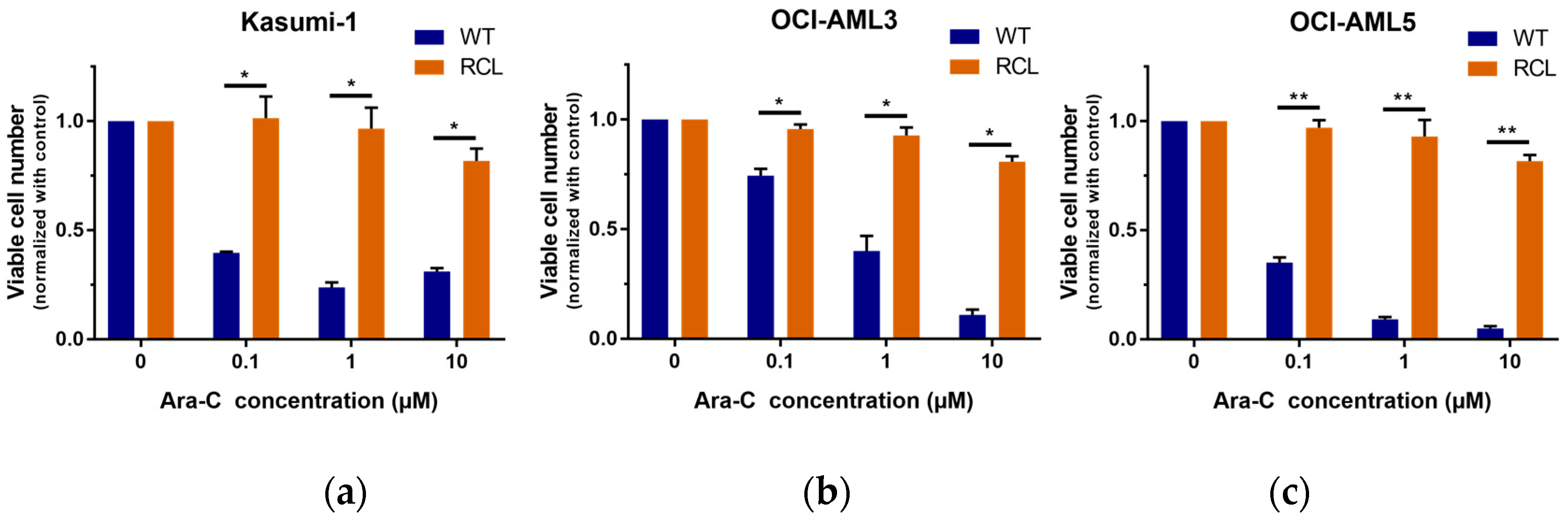
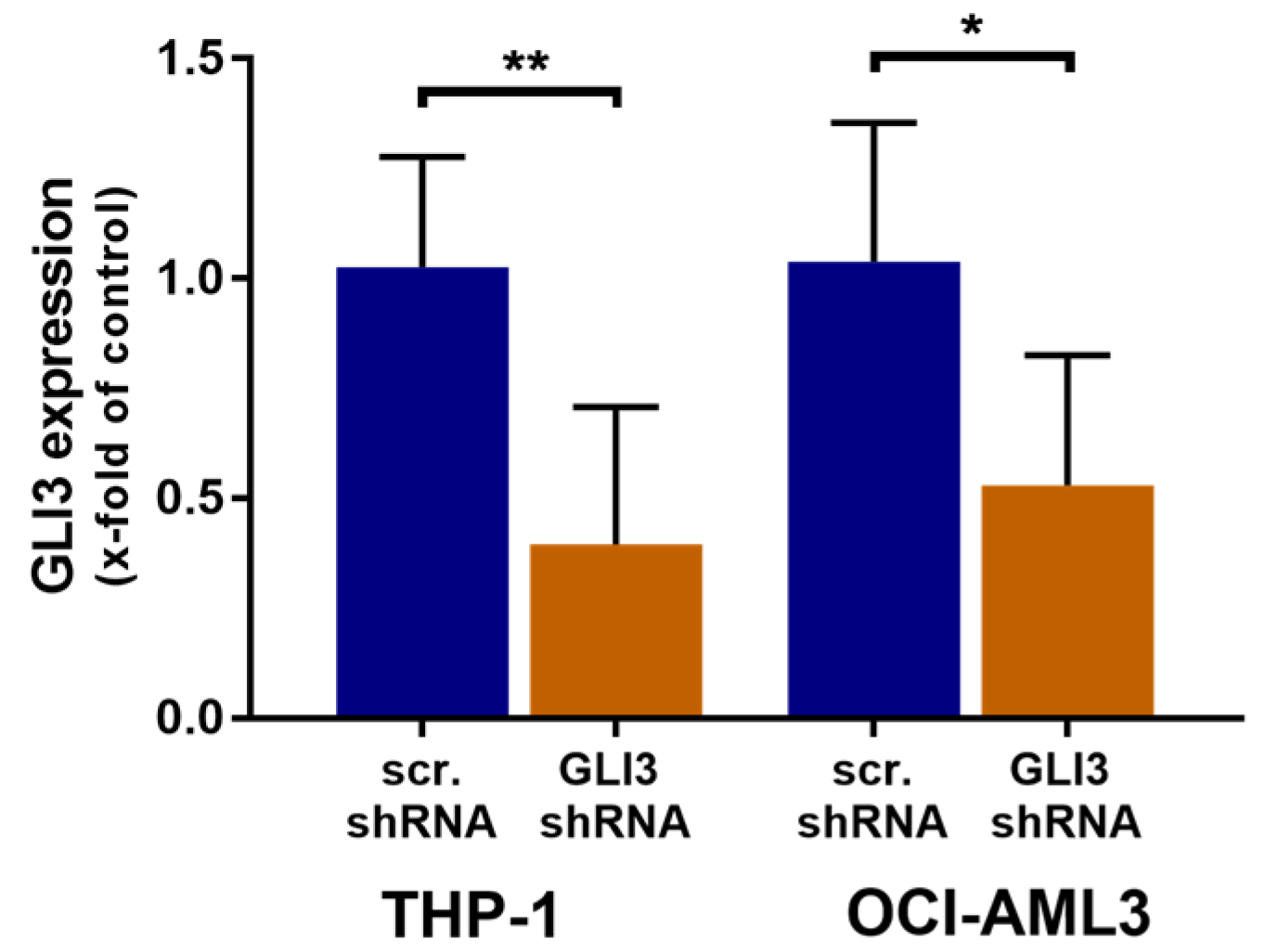
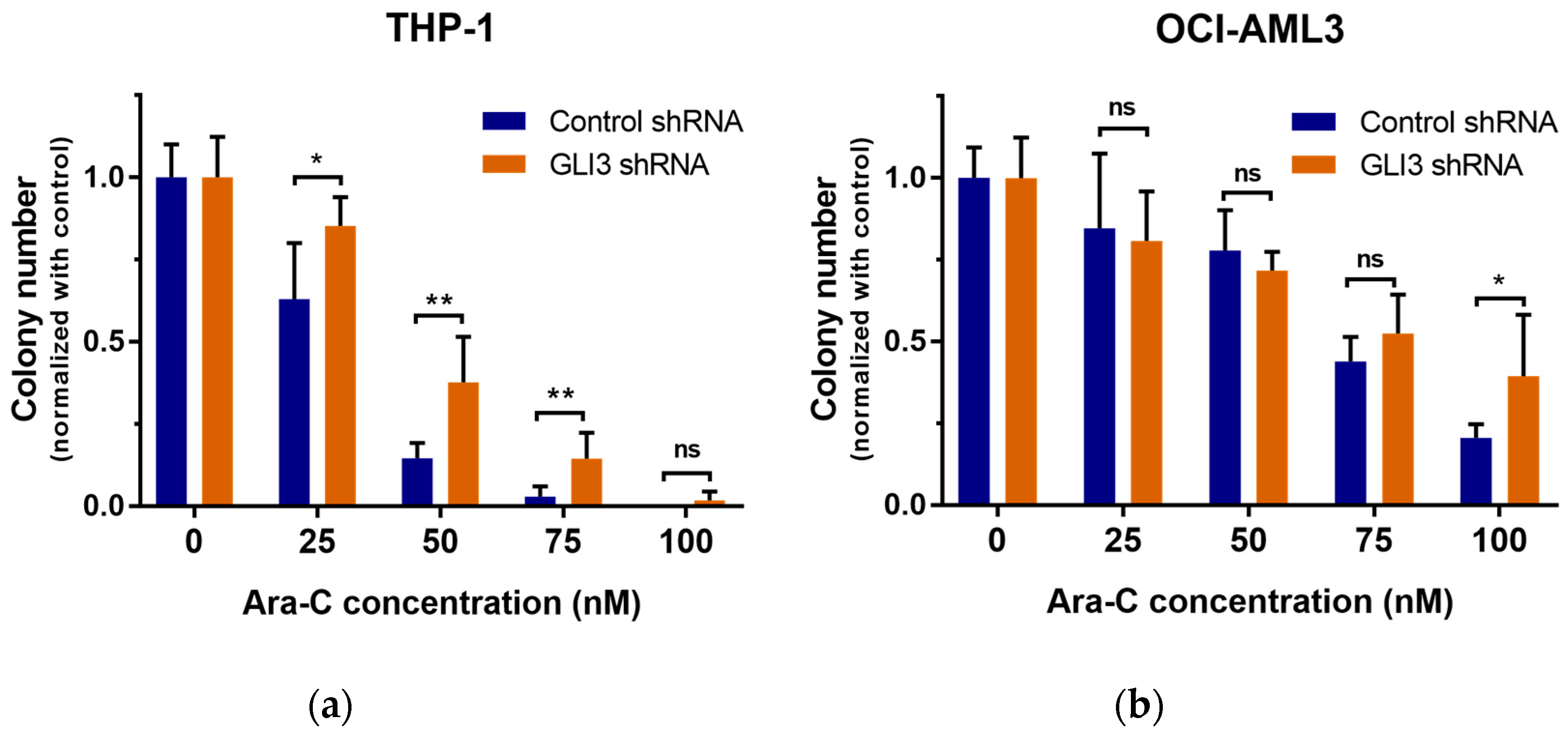
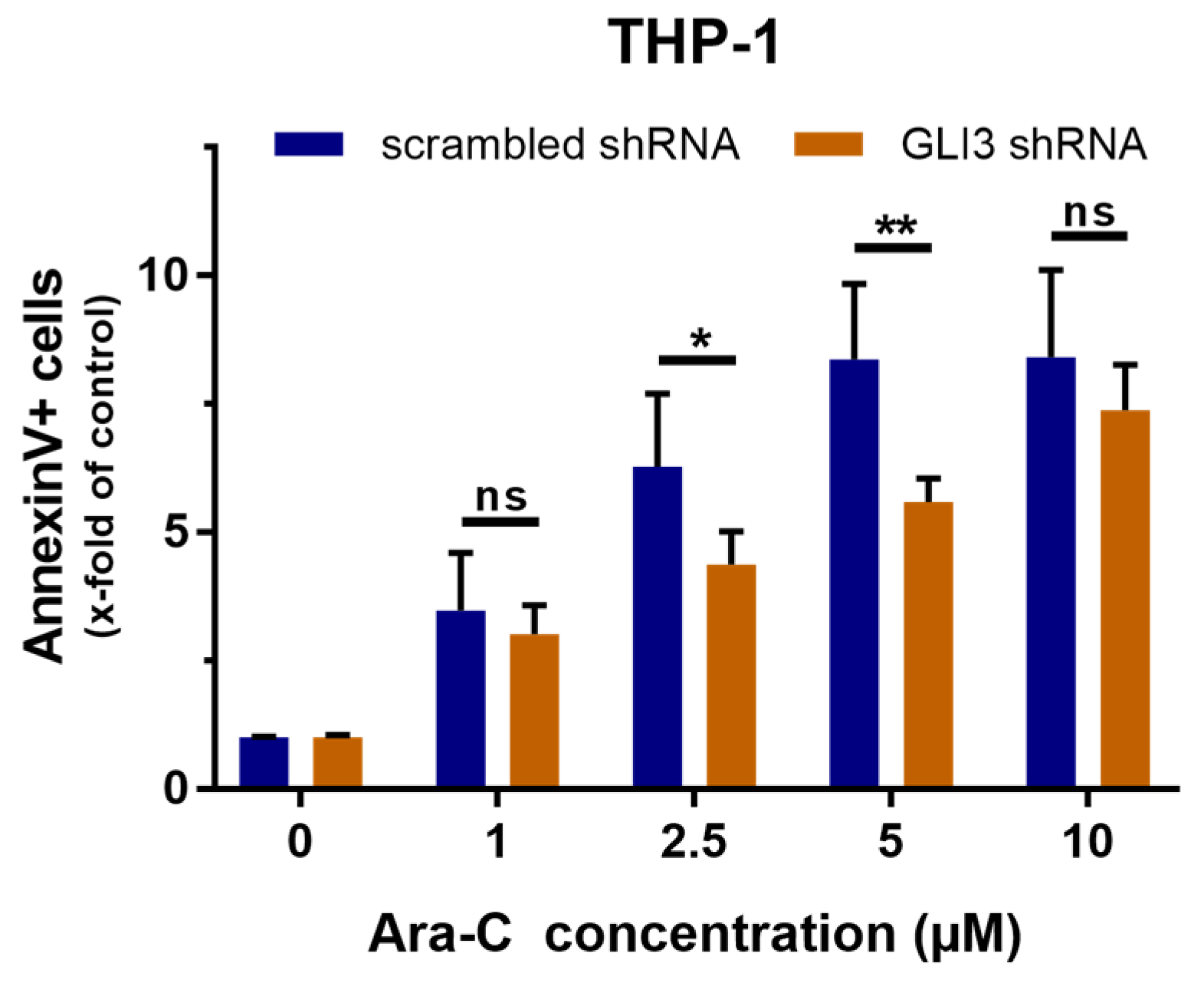
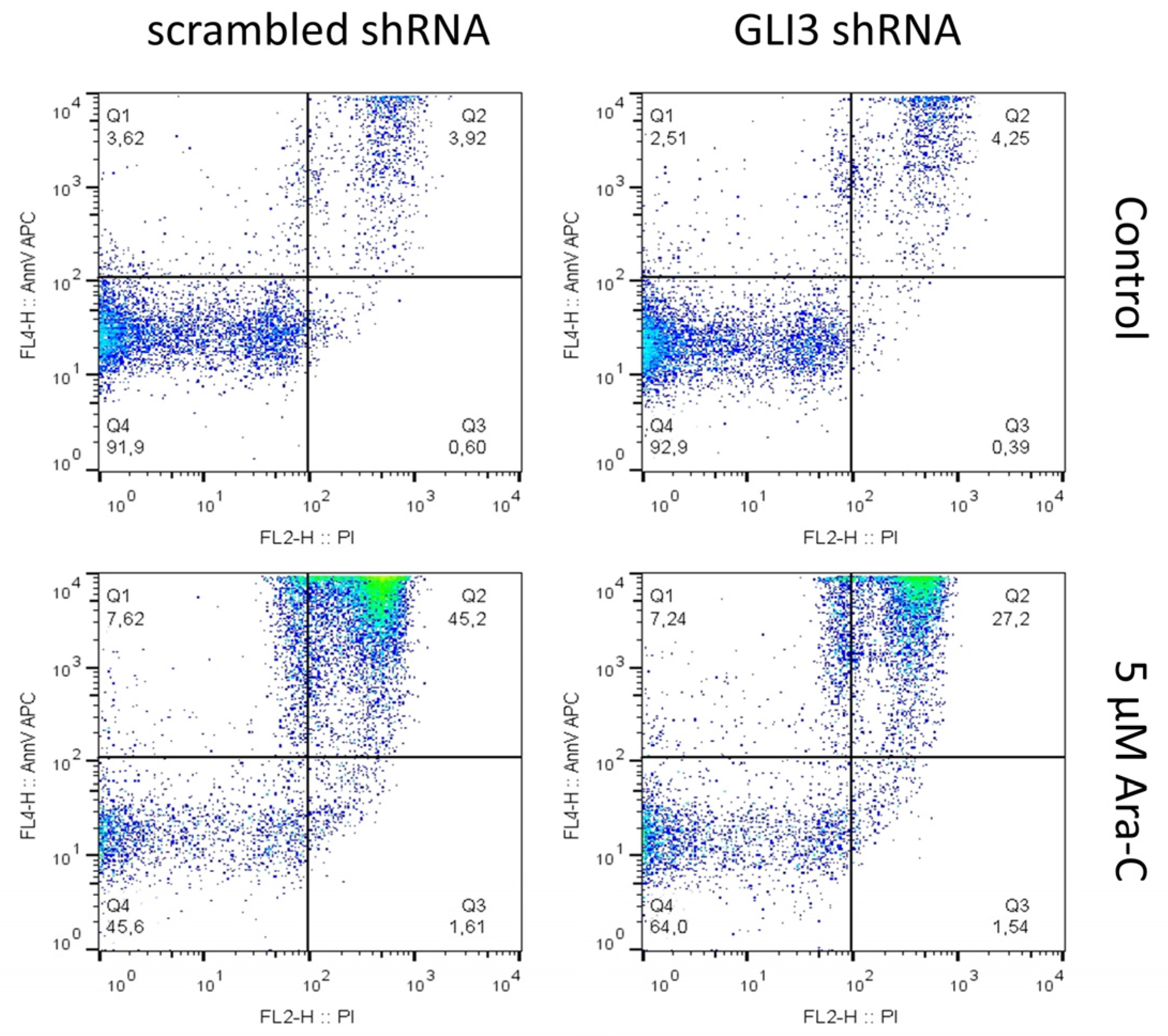
2.2. GLI3 Knockdown Promotes Resistance to Chemotherapy
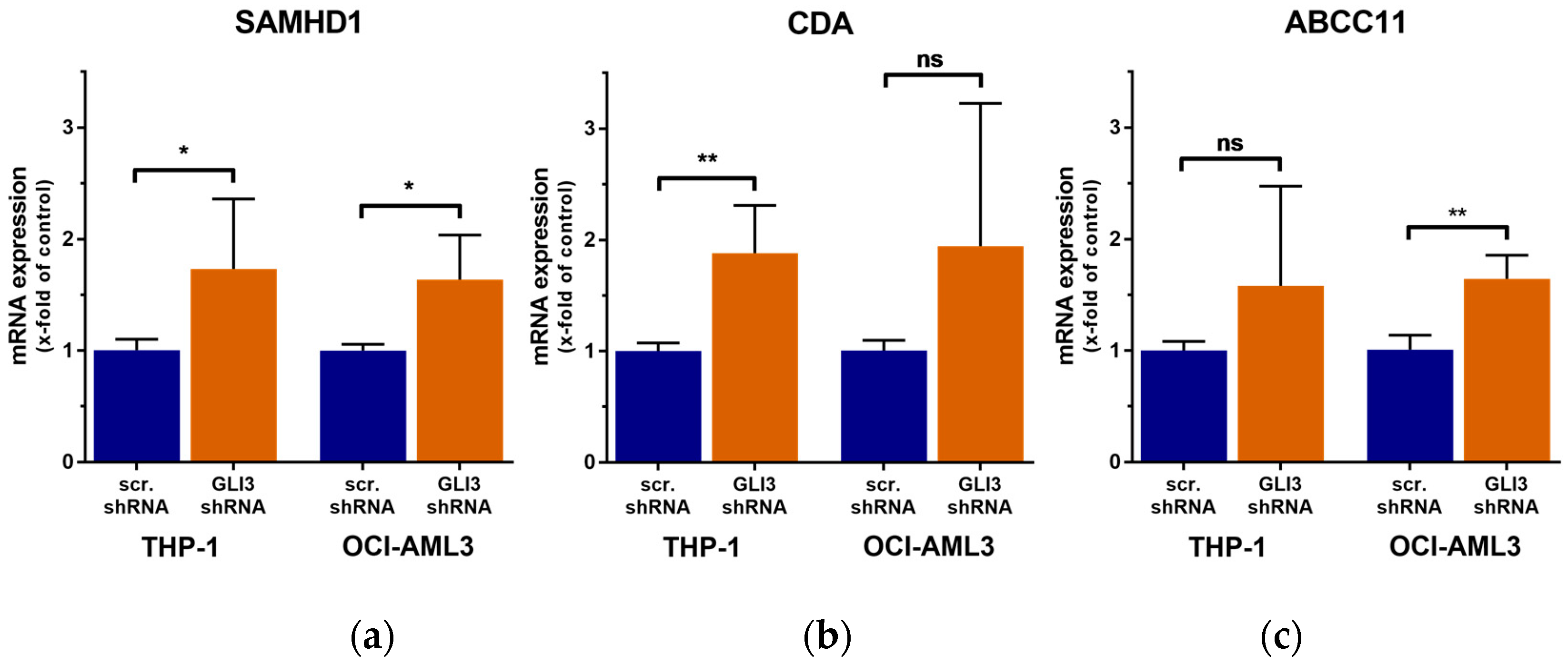
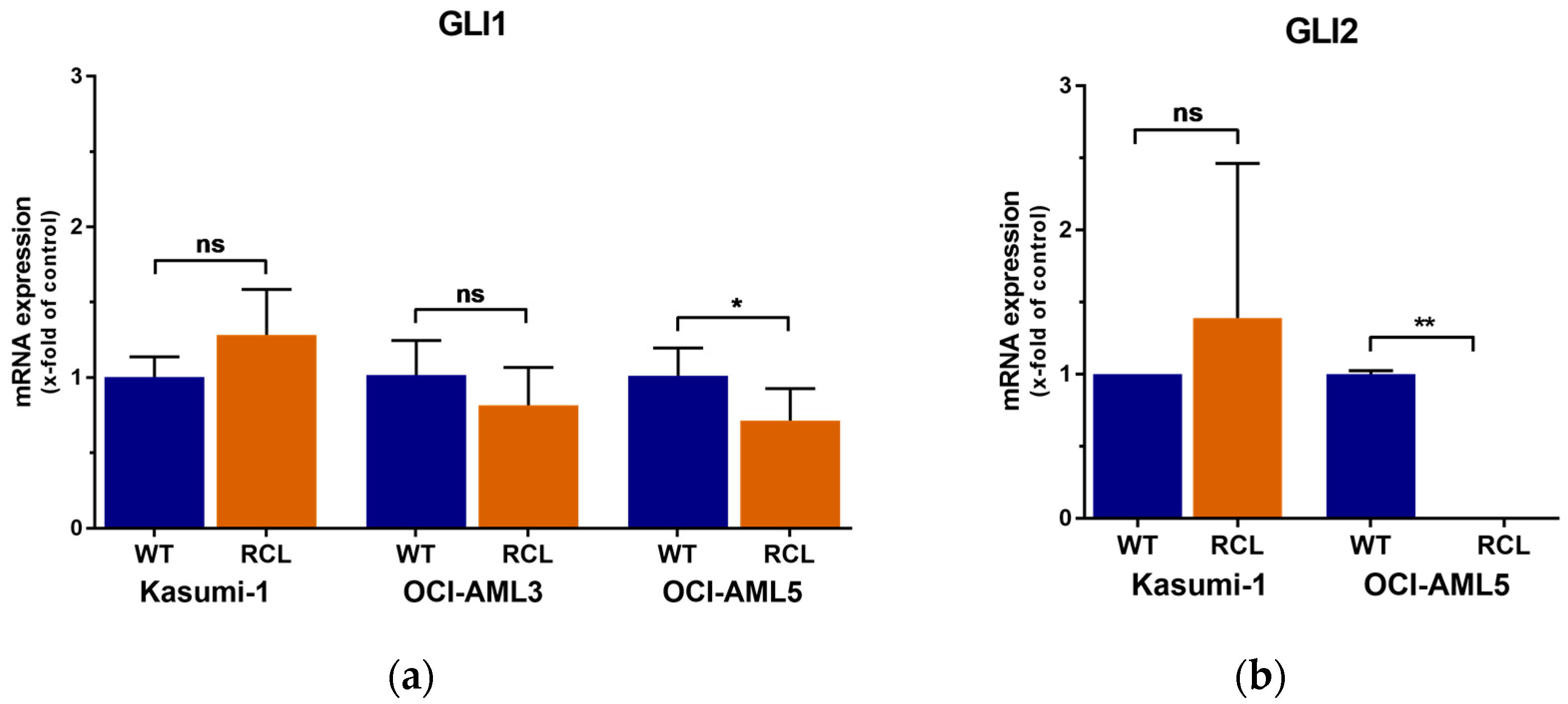
2.3. GLI3 Knockdown Impacts the Expression of Ara-C Resistance Genes
3. Discussion
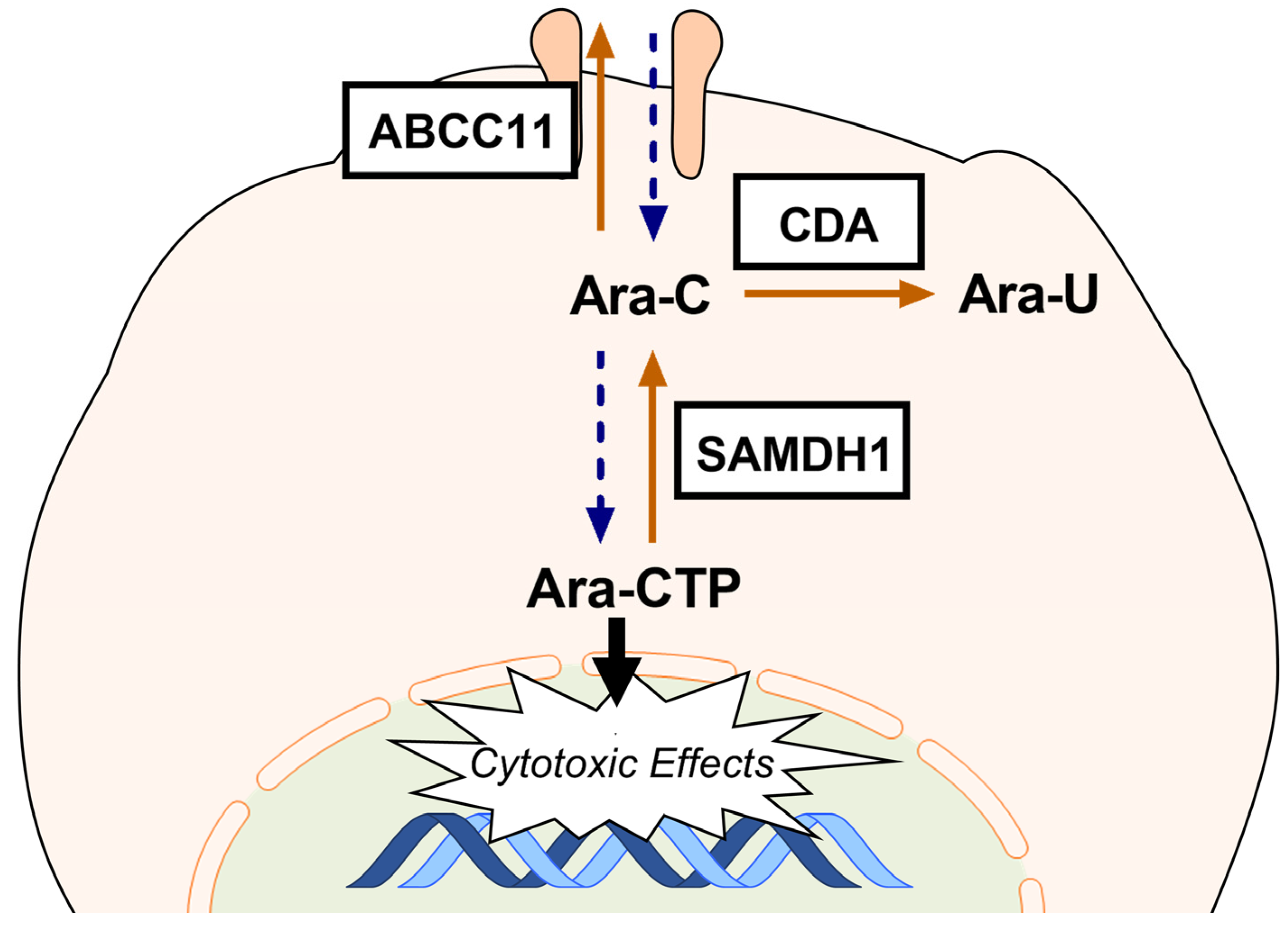
4. Materials and Methods
4.1. Cell Lines and Cell Culture
4.2. Generation of Ara-C-Resistant Cell Lines
4.3. Lentiviral Transduction of AML Cell Lines with GLI3-Specific shRNA
4.4. Proliferation Assay
4.5. Apoptosis Assay
4.6. Colony Formation Assay
4.7. Protein Isolation and Western Blot Analysis
4.8. Reverse Transcription and Quantitative PCR
4.9. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AML | acute myeloid leukemia |
| LSC | leukemic stem cells |
| HSC | hematopoietic stem cells |
| HH | hedgehog (signaling pathway) |
| GLI | glioma-associated oncogene homolog family zinc finger protein |
| GLI3FL | GLI3, full length |
| GLI3R | GLI3, repressor form |
| Ara-C | cytarabine, cytosine arabinoside |
| Ara-U | uracil arabinoside |
| Ara-CTP | aracytidine-5′-triphosphate |
| SAMHD1 | SAM and HD domain-containing protein 1 |
| CDA | cytidine deaminase |
| ABCC11 | ATP-binding cassette C11 |
| MRP8 | multidrug resistance-associated protein 8 |
| WT | wildtype |
| RCL | resistant cell line |
| shRNA | small hairpin RNA |
| APC | allophycocyanin |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sense | Anti-Sense |
|---|---|---|
| GAPDH | GTCAGTGGTGGACCTGACCT | TGCTGTAGCCAAATTCGTTG |
| GLI1 | CTACATCAACTCCGGCCAAT | CGGCTGACAGTATAGGCAGA |
| GLI2 | GGCCATCCACATGGAATATC | TGAAGAGCTGCTACGGGAAT |
| GLI3 | GGCCATCCACATGGAATATC | TGAAGAGCTGCTACGGGAAT |
| SAMHD1 | ATTGAAAGACGCACGAGAG | AAGAGATTCATAGTCCTCCCT |
| CDA | GAGAATCTTCAAAGGGTGCA | TTGTACCCTTCTGAGACGG |
| ABCC11 | CCTACTTCATTATTGGATACACTGC | CTTGTCATGAATACCGCCAG |




References
- Löwenberg, B.; Downing, J.R.; Burnett, A. Acute Myeloid Leukemia. N. Engl. J. Med. 1999, 341, 1051–1062. [Google Scholar] [CrossRef] [PubMed]
- Shipley, J.L.; Butera, J.N. Acute myelogenous leukemia. Exp. Hematol. 2009, 37, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Gentles, A.J.; Plevritis, S.K.; Majeti, R.; Alizadeh, A.A. Association of a leukemic stem cell gene expression signature with clinical outcomes in acute myeloid leukemia. JAMA J. Am. Med. Assoc. 2010, 304, 2706–2715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Dick, J.E. Stem cell concepts renew cancer research. Blood 2008, 112, 4793–4807. [Google Scholar] [CrossRef] [Green Version]
- Lane, S.W.; Scadden, D.T.; Gilliland, D.G. The leukemic stem cell niche: Current concepts and therapeutic opportunities. Blood 2009, 114, 1150–1157. [Google Scholar] [CrossRef] [Green Version]
- Irvine, D.A.; Copland, M. Targeting hedgehog in hematologic malignancy. Blood 2012, 119, 2196–2204. [Google Scholar] [CrossRef] [Green Version]
- Hui, C.; Angers, S. Gli Proteins in Development and Disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 513–537. [Google Scholar] [CrossRef] [Green Version]
- Teglund, S.; Toftgård, R. Hedgehog beyond medulloblastoma and basal cell carcinoma. Biochim. Biophys. Acta Rev. Cancer 2010, 1805, 181–208. [Google Scholar] [CrossRef]
- Wellbrock, J.; Latuske, E.; Kohler, J.; Wagner, K.; Stamm, H.; Vettorazzi, E.; Vohwinkel, G.; Klokow, M.; Uibeleisen, R.; Ehm, P.; et al. Expression of hedgehog pathway mediator GLI represents a negative prognostic marker in human acute myeloid leukemia and its inhibition exerts Antileukemic effects. Clin. Cancer Res. 2015, 21, 2388–2398. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Wang, B. A novel protein-processing domain in Gli2 and Gli3 differentially blocks complete protein degradation by the proteasome. J. Biol. Chem. 2007, 282, 10846–10852. [Google Scholar] [CrossRef] [Green Version]
- Kinzler, K.W.; Vogelstein, B. The GLI gene encodes a nuclear protein which binds specific sequences in the human genome. Mol. Cell. Biol. 1990, 10, 634–642. [Google Scholar] [CrossRef] [Green Version]
- Niewiadomski, P.; Niedziółka, S.M.; Markiewicz, Ł.; Uśpieński, T.; Baran, B.; Chojnowska, K. Gli Proteins: Regulation in Development and Cancer. Cells 2019, 8, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipinski, R.J.; Gipp, J.J.; Zhang, J.; Doles, J.D.; Bushman, W. Unique and complimentary activities of the Gli transcription factors in Hedgehog signaling. Exp. Cell Res. 2006, 312, 1925–1938. [Google Scholar] [CrossRef]
- Petrova, R.; Garcia, D.R.; Joyner, A.L. Titration of GLI3 repressor activity by sonic hedgehog signaling is critical for maintaining multiple adult neural stem cell and astrocyte functions. J. Neurosci. 2013, 33, 17490–17505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, D.; Xie, J. Non-canonical Hh signaling in cancer—Current understanding and future directions. Cancers 2015, 7, 1684–1698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latuske, E.M.; Stamm, H.; Klokow, M.; Vohwinkel, G.; Muschhammer, J.; Bokemeyer, C.; Jücker, M.; Kebenko, M.; Fiedler, W.; Wellbrock, J. Combined inhibition of GLI and FLT3 signaling leads to effective anti-leukemic effects in human acute myeloid leukemia. Oncotarget 2017, 8, 29187–29201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhry, P.; Singh, M.; Triche, T.J.; Guzman, M.; Merchant, A.A. GLI3 repressor determines Hedgehog pathway activation and is required for response to SMO antagonist glasdegib in AML. Blood 2017, 129, 3465–3475. [Google Scholar] [CrossRef]
- Zhang, W.; Yu, F.; Wang, Y.; Zhang, Y.; Meng, L.; Chi, Y. Rab23 promotes the cisplatin resistance of ovarian cancer via the Shh-Gli-ABCG2 signaling pathway. Oncol. Lett. 2018, 15, 5155–5160. [Google Scholar] [CrossRef] [Green Version]
- Yoon, C.; Park, D.J.; Schmidt, B.; Thomas, N.J.; Lee, H.J.; Kim, T.S.; Janjigian, Y.Y.; Cohen, D.J.; Yoon, S.S. CD44 expression denotes a subpopulation of gastric cancer cells in which Hedgehog signaling promotes chemotherapy resistance. Clin. Cancer Res. 2014, 20, 3974–3988. [Google Scholar] [CrossRef] [Green Version]
- Kobune, M.; Takimoto, R.; Murase, K.; Iyama, S.; Sato, T.; Kikuchi, S.; Kawano, Y.; Miyanishi, K.; Sato, Y.; Niitsu, Y.; et al. Drug resistance is dramatically restored by hedgehog inhibitors in CD34+ leukemic cells. Cancer Sci. 2009, 100, 948–955. [Google Scholar] [CrossRef] [PubMed]
- Zahreddine, H.A.; Culjkovic-Kraljacic, B.; Assouline, S.; Gendron, P.; Romeo, A.A.; Morris, S.J.; Cormack, G.; Jaquith, J.B.; Cerchietti, L.; Cocolakis, E.; et al. The sonic hedgehog factor GLI1 imparts drug resistance through inducible glucuronidation. Nature 2014, 511, 90–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, B.; Gu, D.; Zhang, X.; Liu, B.; Xie, J. The role of GLI2-ABCG2 signaling axis for 5Fu resistance in gastric cancer. J. Genet. Genom. 2017, 44, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Terwijn, M.; Zeijlemaker, W.; Kelder, A.; Rutten, A.P.; Snel, A.N.; Scholten, W.J.; Pabst, T.; Verhoef, G.; Löwenberg, B.; Zweegman, S.; et al. Leukemic stem cell frequency: A strong biomarker for clinical outcome in acute myeloid leukemia. PLoS ONE 2014, 9, e107587. [Google Scholar] [CrossRef] [Green Version]
- Jordan, C.T. The leukemic stem cell. Best Pr. Res. Clin. Haematol. 2007, 20, 13–18. [Google Scholar] [CrossRef]
- Carpenter, R.L.; Lo, H.W. Identification, Functional Characterization, and Pathobiological Significance of GLI1 Isoforms in Human Cancers; Elsevier: Amsterdam, The Netherlands, 2012; Volume 88, ISBN 9780123946225. [Google Scholar]
- Sasaki, H.; Nishizaki, Y.; Hui, C.C.; Nakafuku, M.; Kondoh, H. Regulation of Gli2 and Gli3 activities by an amino-terminal repression domain: Implication of Gli2 and Gli3 as primary mediators of Shh signaling. Development 1999, 126, 3915–3924. [Google Scholar]
- Niewiadomski, P.; Kong, J.H.; Ahrends, R.; Ma, Y.; Humke, E.W.; Khan, S.; Teruel, M.N.; Novitch, B.G.; Rohatgi, R. Gli protein activity is controlled by multisite phosphorylation in vertebrate hedgehog signaling. Cell Rep. 2014, 6, 168–181. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.G.; Kim, H.J.; Dlugosz, A.A.; Ellison, D.W.; Gilbertson, R.J.; Alvarez-Buylla, A. Dual and opposing roles of primary cilia in medulloblastoma development. Nat. Med. 2009, 15, 1062–1065. [Google Scholar] [CrossRef] [Green Version]
- Laclef, C.; Anselme, I.; Besse, L.; Catala, M.; Palmyre, A.; Baas, D.; Paschaki, M.; Pedraza, M.; Métin, C.; Durand, B.; et al. The role of primary cilia in corpus callosum formation is mediated by production of the Gli3 repressor. Hum. Mol. Genet. 2015, 24, 4997–5014. [Google Scholar] [CrossRef] [Green Version]
- Singh, M.; Chaudhry, P.; Merchant, A.A. Primary cilia are present on human blood and bone marrow cells and mediate Hedgehog signaling. Exp. Hematol. 2016, 44, 1181–1187. [Google Scholar] [CrossRef] [Green Version]
- Queiroz, K.C.S.; Ruela-De-Sousa, R.R.; Fuhler, G.M.; Aberson, H.L.; Ferreira, C.V.; Peppelenbosch, M.P.; Spek, C.A. Hedgehog signaling maintains chemoresistance in myeloid leukemic cells. Oncogene 2010, 29, 6314–6322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, H.; Zheng, Q.L.; Fang, P.; Zhang, J.; Zhang, T.; Liu, W.; Guo, M.; Robinson, C.L.; Chen, S.B.; Chen, X.P.; et al. Targeting the PI3K/AKT pathway via GLI1 inhibition enhanced the drug sensitivity of acute myeloid leukemia cells. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Long, B.; Wang, L.X.; Zheng, F.M.; Lai, S.P.; Xu, D.R.; Hu, Y.; Lin, D.J.; Zhang, X.Z.; Dong, L.; Long, Z.J.; et al. Targeting GLI1 suppresses cell growth and enhances chemosensitivity in CD34 + enriched acute myeloid leukemia progenitor cells. Cell. Physiol. Biochem. 2016, 38, 1288–1302. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Sun, Z.; Ding, B.; Jiang, X.; Wang, Z.; Zhu, Y.; Meng, F. Suppressing hedgehog signaling reverses drug resistance of refractory acute myeloid leukemia. Onco Targets. Ther. 2019, 12, 7477–7488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.T.; Ding, J.; Li, H.Y.; Zuo, J.L.; Ge, S.Y.; Jia, H.L.; Wu, J. Hedgehog signalling mediates drug resistance through targeting TAP1 in hepatocellular carcinoma. J. Cell. Mol. Med. 2020, 24, 4298–4311. [Google Scholar] [CrossRef] [Green Version]
- Meng, E.; Hanna, A.; Samant, R.S.; Shevde, L.A. The impact of hedgehog signaling pathway on DNA repair mechanisms in human cancer. Cancers 2015, 7, 1333–1348. [Google Scholar] [CrossRef]
- Chen, Y.; Bieber, M.M.; Teng, N.N.H. Hedgehog signaling regulates drug sensitivity by targeting ABC transporters ABCB1 and ABCG2 in epithelial ovarian cancer. Mol. Carcinog. 2014, 53, 625–634. [Google Scholar] [CrossRef]
- Guo, Y.; Kotova, E.; Chen, Z.S.; Lee, K.; Hopper-Borge, E.; Belinsky, M.G.; Kruh, G.D. MRP8, ATP-binding cassette C11 (ABCC11), is a cyclic nucleotide efflux pump and a resistance factor for fluoropyrimidines 2′,3′-dideoxycytidine and 9′-(2′-phosphonylmethoxyethyl)adenine. J. Biol. Chem. 2003, 278, 29509–29514. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Kock, K.; Ritter, C.A.; Chen, Z.S.; Grube, M.; Jedlitschky, G.; Illmer, T.; Ayres, M.; Beck, J.F.; Siegmund, W.; et al. Expression of ABCC-type nucleotide exporters in blasts of adult acute myeloid leukemia: Relation to long-term survival. Clin. Cancer Res. 2009, 15, 1762–1769. [Google Scholar] [CrossRef] [Green Version]
- Lamba, J.K. Genetic factors influencing cytarabine therapy. Pharmacogenomics 2009, 10, 1657–1674. [Google Scholar] [CrossRef] [Green Version]
- Franzolin, E.; Pontarin, G.; Rampazzo, C.; Miazzi, C.; Ferraro, P.; Palumbo, E.; Reichard, P.; Bianchi, V. The deoxynucleotide triphosphohydrolase SAMHD1 is a major regulator of DNA precursor pools in mammalian cells. Proc. Natl. Acad. Sci. USA 2013, 110, 14272–14277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollenbaugh, J.A.; Shelton, J.; Tao, S.; Amiralaei, S.; Liu, P.; Lu, X.; Goetze, R.W.; Zhou, L.; Nettles, J.H.; Schinazi, R.F.; et al. Substrates and inhibitors of SAMHD1. PLoS ONE 2017, 12, e0169052. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.; Oellerich, T.; Baldauf, H.M.; Schwarz, S.M.; Thomas, D.; Flick, R.; Bohnenberger, H.; Kaderali, L.; Stegmann, L.; Cremer, A.; et al. SAMHD1 is a biomarker for cytarabine response and a therapeutic target in acute myeloid leukemia. Nat. Med. 2017, 23, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S. The mechanisms of lethal action of arabinosyl cytosine (araC) and arabinosyl adenine (araA). Cancer 1977, 40, 509–518. [Google Scholar] [CrossRef]
- Ohta, T.; Hori, H.; Ogawa, M.; Miyahara, M.; Kawasaki, H.; Taniguchi, N.; Komada, Y. Impact of cytidine deaminase activity on intrinsic resistance to cytarabine in carcinoma cells. Oncol. Rep. 2004, 12, 1115–1120. [Google Scholar] [CrossRef]
- Abraham, A.; Varatharajan, S.; Karathedath, S.; Philip, C.; Lakshmi, K.M.; Jayavelu, A.K.; Mohanan, E.; Janet, N.B.; Srivastava, V.M.; Shaji, R.V.; et al. RNA expression of genes involved in cytarabine metabolism and transport. Pharmacogenomics 2015, 16, 877–890. [Google Scholar] [CrossRef] [Green Version]
- Jahns-streubel, G.; Reuter, C.; Auf der Landwehr, U.; Unterhalt, M.; Schleyer, E.; Wörmann, B.; Büchner, T.; Hiddemann, W. Activity of Thymidine Kinase and of Polymerase a as Well as Activity and Gene Expression of Deoxycytidine Deaminase in Leukemic Blasts Are Correlated With Clinical Response in the Setting of Granulocyte- Macrophage Colony-Stimulating Factor—Based Priming. Blood 1997, 90, 1968–1976. [Google Scholar] [CrossRef] [Green Version]
- Schröder, J.K.; Kirch, C.; Seeber, S.; Schütte, J. Structural and functional analysis of the cytidine deaminase gene in patients with acute myeloid leukaemia. Br. J. Haematol. 1998, 103, 1096–1103. [Google Scholar] [CrossRef]
- Weber, K.; Mock, U.; Petrowitz, B.; Bartsch, U.; Fehse, B. Lentiviral gene ontology (LeGO) vectors equipped with novel drug-selectable fluorescent proteins: New building blocks for cell marking and multi-gene analysis. Gene Ther. 2010, 17, 511–520. [Google Scholar] [CrossRef] [Green Version]
- David, F.P.A.; Rougemont, J.; Deplancke, B. GETPrime 2.0: Gene- and transcript-specific qPCR primers for 13 species including polymorphisms. Nucleic Acids Res. 2017, 45, D56–D60. [Google Scholar] [CrossRef] [Green Version]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT–PCR. Nucleic Acids Res. 2001, 29, 2002–2007. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Freisleben, F.; Behrmann, L.; Thaden, V.; Muschhammer, J.; Bokemeyer, C.; Fiedler, W.; Wellbrock, J. Downregulation of GLI3 Expression Mediates Chemotherapy Resistance in Acute Myeloid Leukemia. Int. J. Mol. Sci. 2020, 21, 5084. https://doi.org/10.3390/ijms21145084
Freisleben F, Behrmann L, Thaden V, Muschhammer J, Bokemeyer C, Fiedler W, Wellbrock J. Downregulation of GLI3 Expression Mediates Chemotherapy Resistance in Acute Myeloid Leukemia. International Journal of Molecular Sciences. 2020; 21(14):5084. https://doi.org/10.3390/ijms21145084
Chicago/Turabian StyleFreisleben, Fabian, Lena Behrmann, Vanessa Thaden, Jana Muschhammer, Carsten Bokemeyer, Walter Fiedler, and Jasmin Wellbrock. 2020. "Downregulation of GLI3 Expression Mediates Chemotherapy Resistance in Acute Myeloid Leukemia" International Journal of Molecular Sciences 21, no. 14: 5084. https://doi.org/10.3390/ijms21145084
APA StyleFreisleben, F., Behrmann, L., Thaden, V., Muschhammer, J., Bokemeyer, C., Fiedler, W., & Wellbrock, J. (2020). Downregulation of GLI3 Expression Mediates Chemotherapy Resistance in Acute Myeloid Leukemia. International Journal of Molecular Sciences, 21(14), 5084. https://doi.org/10.3390/ijms21145084