Identification of RNA-Binding Proteins as Targetable Putative Oncogenes in Neuroblastoma


 ,
,  and
and 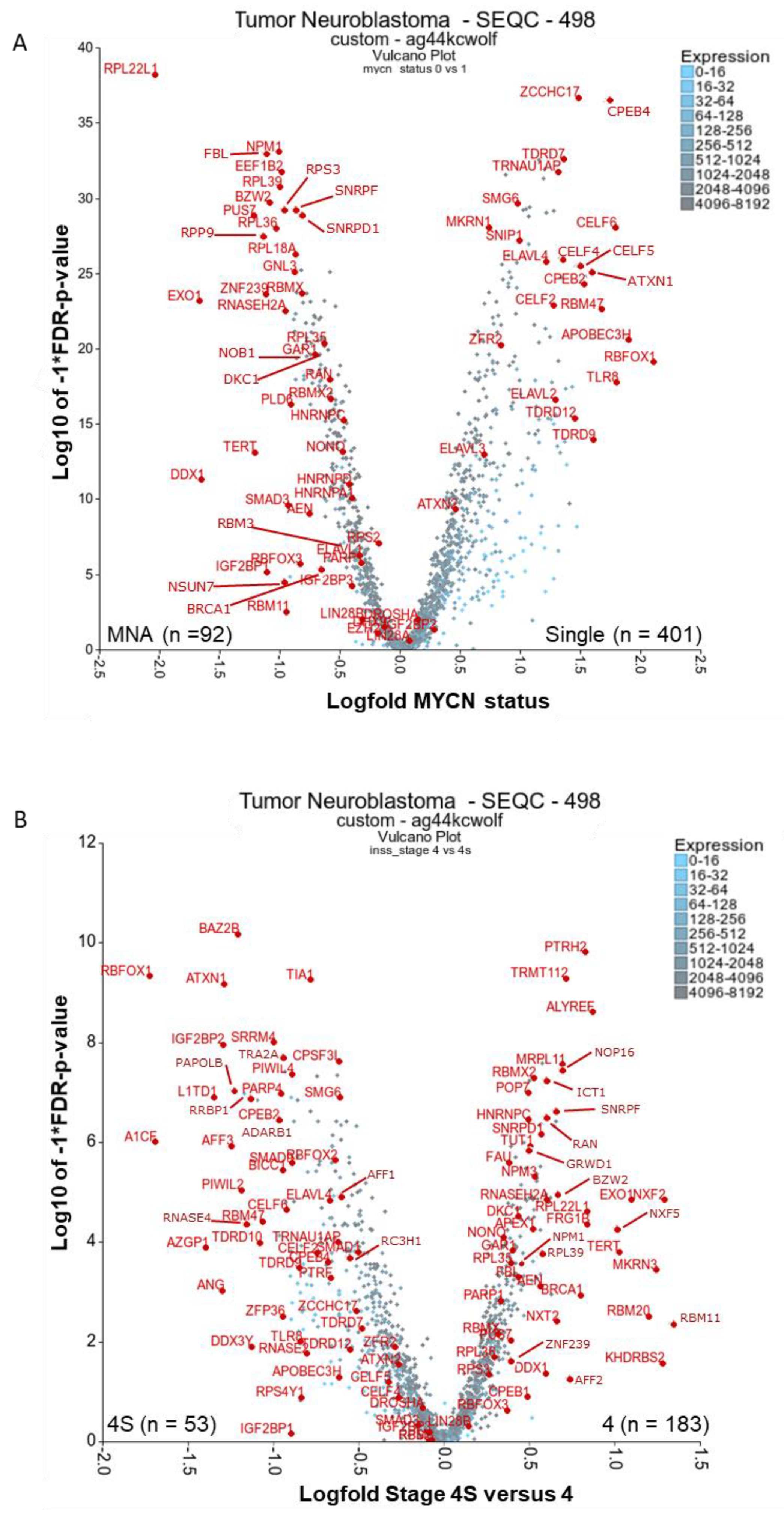{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. There Are Many Novel and Reported RBPs Associated with MYCN-Amplified Neuroblastoma
2.1.1. Ribosomal Proteins: RPL22L1, RPL39, RPL18A, RPL36, RPL35, RPS3
2.1.2. Ribosome Biogenesis: NPM1 and FBL
2.1.3. RBPs Involved in Replication Stress and Telomere Maintenance: RNASEH2A, EXO1, TERT, DDX1, DKC1, GAR1, BRCA1 and PARP1
2.1.4. Other Reported MYCN-Associated RBPs: NONO, IGF2BP1, IGF2BP3, and EZH2
2.2. RBPs Associated with Lower-Risk Groupings and Involved in Neuronal Homeostasis and Splicing: ELAVLs, CPEBs, CELFs, TDRDs, ZCCHC17, ATXN1/2 and RBFOX1
2.3. There Are Several Novel and Reported Genes that are Expressed with Significant Differences for Stages 4 and 4S Comparisons: PTRH2, TRMT112, KHDRBS2, MKRN3, RBM11, RBMX2, NXF2 (NXF1)
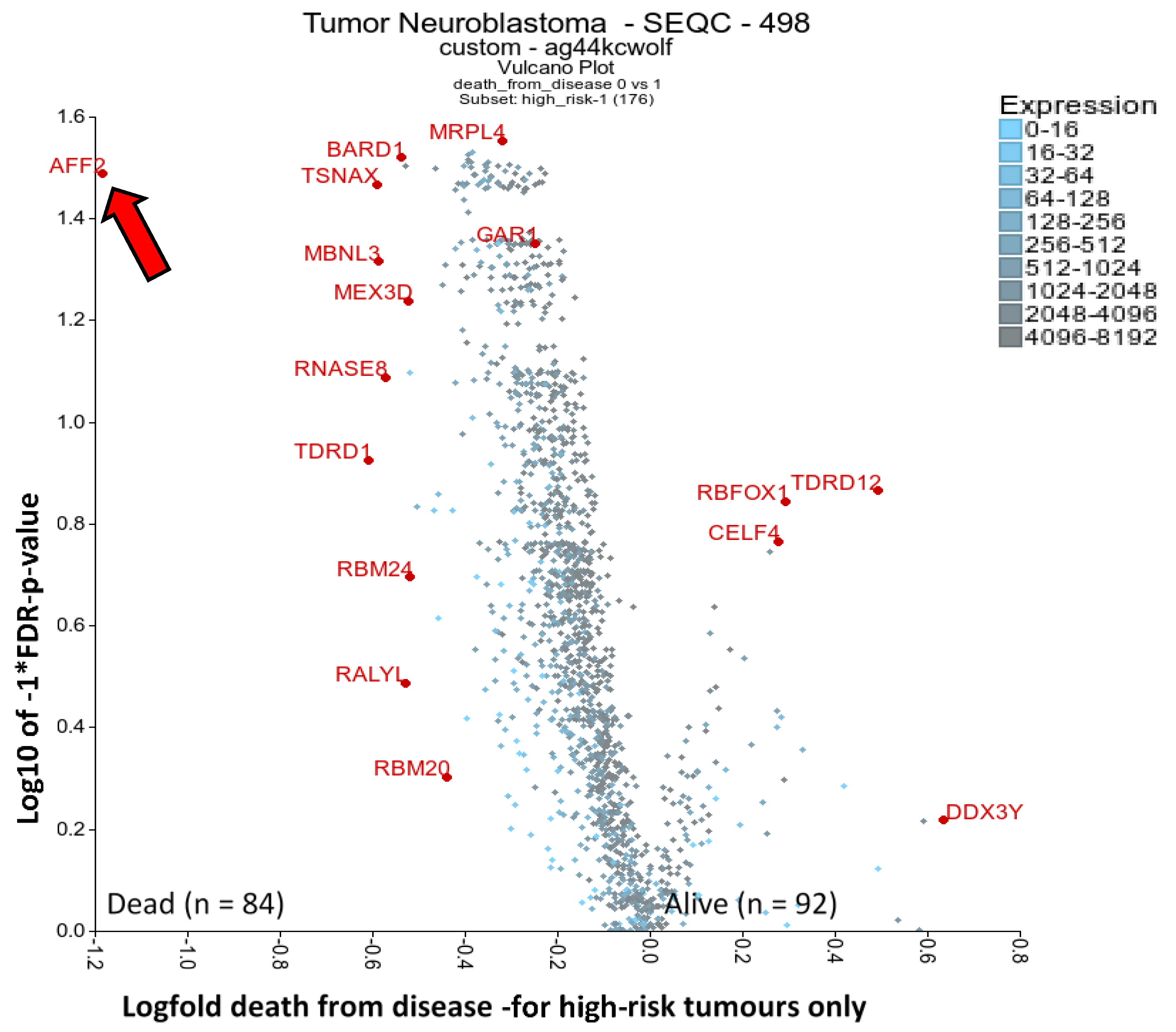
2.4. RBPs Associated with High-Risk Patient Death: AFF2 and BARD1
2.5. One-Third of RBPs Have Prognostic Value in Neuroblastoma, Including MRPL11, RAN and HNRNPC
2.6. Other RBPs Reported in Neuroblastoma Involved in miRNA Biogenesis: LIN28B, DROSHA, DICER and DHX9
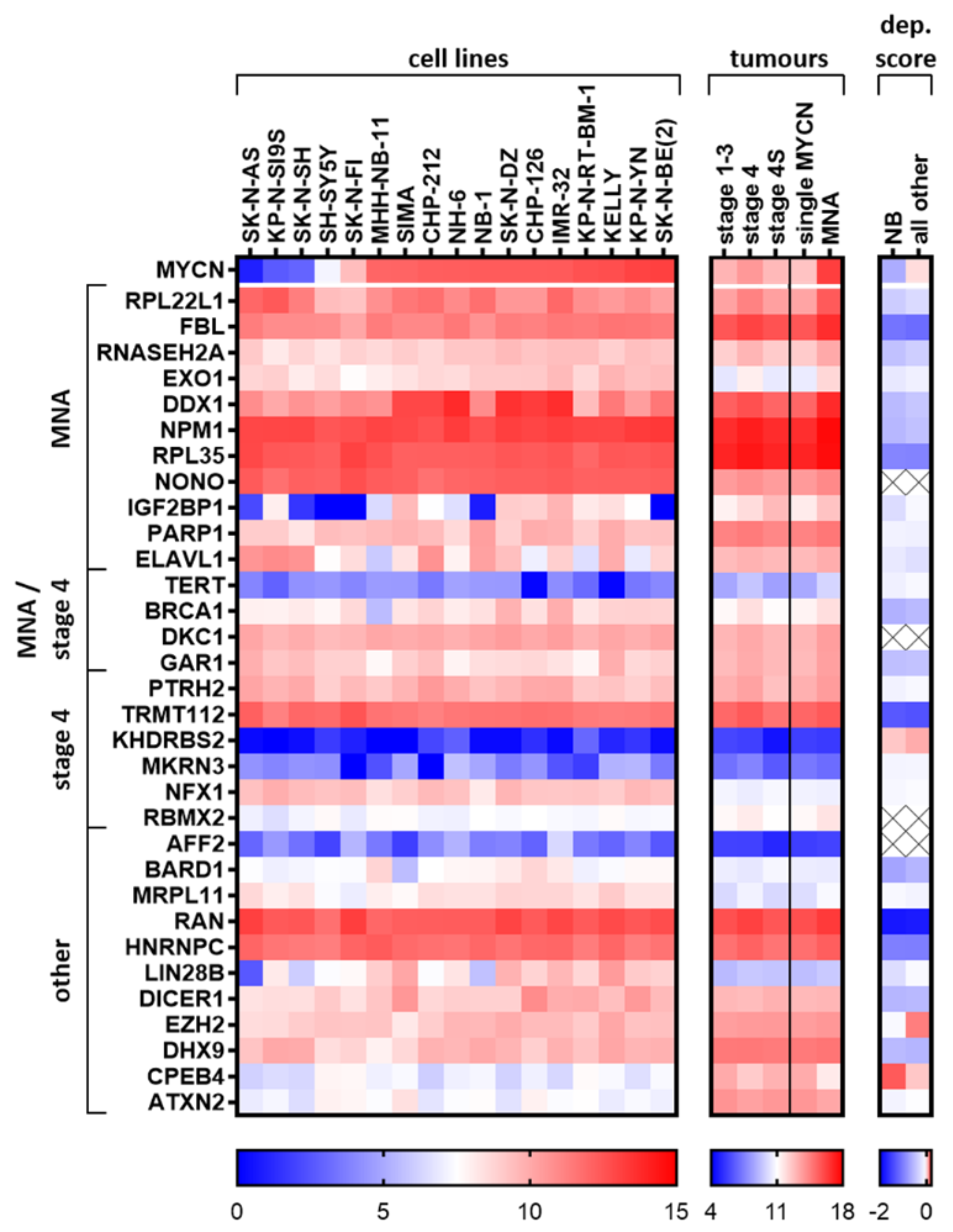
2.7. Elucidated and Reported Oncogenic-Like RBPs Are Highly Expressed in Neuroblastoma Cell Lines
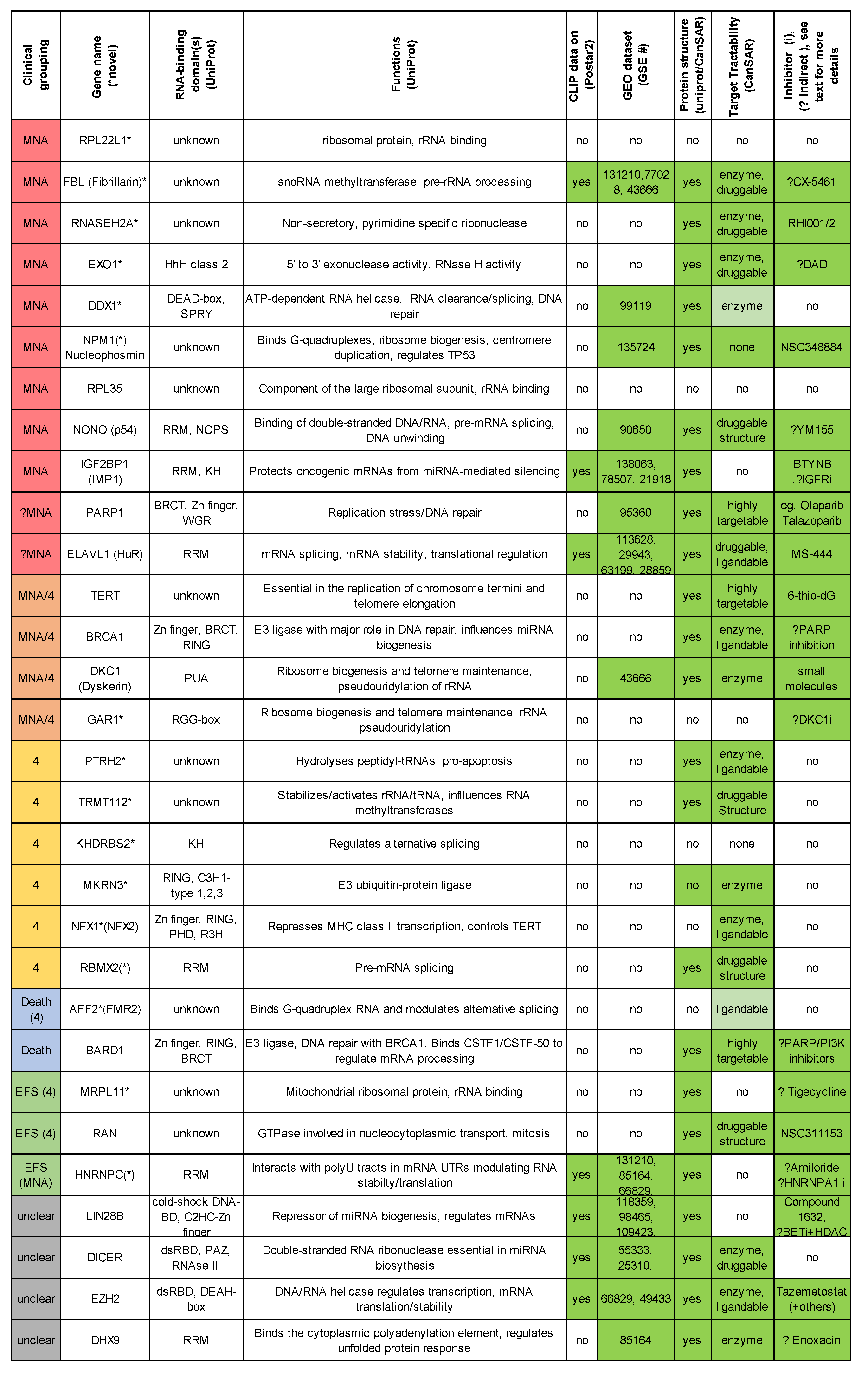
2.8. Summary of Top Oncogenic-Like RBP Regarding Available Structures and Inhibitors
3. Conclusions
4. Materials and Methods
4.1. Differential Expression Analysis
4.2. Kaplan–Meier Significance Scanning with the RBP Gene List
4.3. Heat Map of Expression and Dependency in Neuroblastoma Cell Lines
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Matthay, K.K.; Maris, J.M.; Schleiermacher, G.; Nakagawara, A.; Mackall, C.L.; Diller, L.; Weiss, W.A. Neuroblastoma. Nat. Rev. Dis. Primers 2016, 2, 16078. [Google Scholar] [CrossRef] [PubMed]
- Laverdiere, C.; Liu, Q.; Yasui, Y.; Nathan, P.C.; Gurney, J.G.; Stovall, M.; Diller, L.R.; Cheung, N.K.; Wolden, S.; Robison, L.L.; et al. Long-term outcomes in survivors of neuroblastoma: A report from the Childhood Cancer Survivor Study. J. Natl. Cancer Inst. 2009, 101, 1131–1140. [Google Scholar] [CrossRef]
- Koach, J.; Holien, J.K.; Massudi, H.; Carter, D.R.; Ciampa, O.C.; Herath, M.; Lim, T.; Seneviratne, J.A.; Milazzo, G.; Murray, J.E.; et al. Drugging MYCN oncogenic signalling through the MYCN-PA2G4 binding interface. Cancer Res. 2019, 79, 5652–5667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boeva, V.; Louis-Brennetot, C.; Peltier, A.; Durand, S.; Pierre-Eugene, C.; Raynal, V.; Etchevers, H.C.; Thomas, S.; Lermine, A.; Daudigeos-Dubus, E.; et al. Heterogeneity of neuroblastoma cell identity defined by transcriptional circuitries. Nat. Genet. 2017, 49, 1408–1413. [Google Scholar] [CrossRef] [PubMed]
- Warner, K.D.; Hajdin, C.E.; Weeks, K.M. Principles for targeting RNA with drug-like small molecules. Nat. Rev. Drug Discov. 2018, 17, 547–558. [Google Scholar] [CrossRef]
- Molenaar, J.J.; Koster, J.; Zwijnenburg, D.A.; van Sluis, P.; Valentijn, L.J.; van der Ploeg, I.; Hamdi, M.; van Nes, J.; Westerman, B.A.; van Arkel, J.; et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature 2012, 483, 589–593. [Google Scholar] [CrossRef]
- Hermann, T. Rational ligand design for RNA: The role of static structure and conformational flexibility in target recognition. Biochimie 2002, 84, 869–875. [Google Scholar] [CrossRef]
- Disney, M.D.; Dwyer, B.G.; Childs-Disney, J.L. Drugging the RNA World. Cold Spring Harb Perspect. Biol. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Donlic, A.; Hargrove, A.E. Targeting RNA in mammalian systems with small molecules. Wiley Interdiscip. Rev. RNA 2018, 9, e1477. [Google Scholar] [CrossRef]
- Schaadt, R.; Sweeney, D.; Shinabarger, D.; Zurenko, G. In vitro activity of TR-700, the active ingredient of the antibacterial prodrug TR-701, a novel oxazolidinone antibacterial agent. Antimicrob. Agents Chemother. 2009, 53, 3236–3239. [Google Scholar] [CrossRef] [Green Version]
- Ford, C.W.; Zurenko, G.E.; Barbachyn, M.R. The discovery of linezolid, the first oxazolidinone antibacterial agent. Curr. Drug Targets Infect. Disord. 2001, 1, 181–199. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.C. The Process of Structure-Based Drug Design. Chem. Biol. 2003, 10, 787–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mavromoustakos, T.; Durdagi, S.; Koukoulitsa, C.; Simcic, M.; Papadopoulos, M.G.; Hodoscek, M.; Grdadolnik, S.G. Strategies in the rational drug design. Curr. Med. Chem. 2011, 18, 2517–2530. [Google Scholar] [CrossRef] [PubMed]
- Zhi, F.; Wang, R.; Wang, Q.; Xue, L.; Deng, D.; Wang, S.; Yang, Y. MicroRNAs in Neuroblastoma: Small-Sized Players with a Large Impact. Neurochem. Res. 2014, 39, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Lee, F.C.Y.; Ule, J. Advances in CLIP Technologies for Studies of Protein-RNA Interactions. Mol. Cell 2018, 69, 354–369. [Google Scholar] [CrossRef] [Green Version]
- Lan, Q.; Liu, P.Y.; Haase, J.; Bell, J.L.; Huttelmaier, S.; Liu, T. The Critical Role of RNA m(6)A Methylation in Cancer. Cancer Res. 2019, 79, 1285–1292. [Google Scholar] [CrossRef] [Green Version]
- Gerstberger, S.; Hafner, M.; Tuschl, T. A census of human RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 829–845. [Google Scholar] [CrossRef]
- Brodeur, G.M.; Pritchard, J.; Berthold, F.; Carlsen, N.L.; Castel, V.; Castelberry, R.P.; De Bernardi, B.; Evans, A.E.; Favrot, M.; Hedborg, F.; et al. Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1993, 11, 1466–1477. [Google Scholar] [CrossRef]
- Boon, K.; Caron, H.N.; van Asperen, R.; Valentijn, L.; Hermus, M.C.; van Sluis, P.; Roobeek, I.; Weis, I.; Voûte, P.A.; Schwab, M.; et al. N-myc enhances the expression of a large set of genes functioning in ribosome biogenesis and protein synthesis. EMBO J. 2001, 20, 1383–1393. [Google Scholar] [CrossRef]
- Hald, Ø.H.; Olsen, L.; Gallo-Oller, G.; Elfman, L.H.M.; Løkke, C.; Kogner, P.; Sveinbjörnsson, B.; Flægstad, T.; Johnsen, J.I.; Einvik, C. Inhibitors of ribosome biogenesis repress the growth of MYCN-amplified neuroblastoma. Oncogene 2019, 38, 2800–2813. [Google Scholar] [CrossRef]
- Pelletier, J.; Thomas, G.; Volarević, S. Ribosome biogenesis in cancer: New players and therapeutic avenues. Nat. Rev. Cancer 2018, 18, 51–63. [Google Scholar] [CrossRef]
- Wu, N.; Wei, J.; Wang, Y.; Yan, J.; Qin, Y.; Tong, D.; Pang, B.; Sun, D.; Sun, H.; Yu, Y.; et al. Ribosomal L22-like1 (RPL22L1) Promotes Ovarian Cancer Metastasis by Inducing Epithelial-to-Mesenchymal Transition. PLoS ONE 2015, 10, e0143659. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.Y.; Tee, A.E.; Milazzo, G.; Hannan, K.M.; Maag, J.; Mondal, S.; Atmadibrata, B.; Bartonicek, N.; Peng, H.; Ho, N.; et al. The long noncoding RNA lncNB1 promotes tumorigenesis by interacting with ribosomal protein RPL35. Nat. Commun. 2019, 10, 5026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeid, R.; Lawlor, M.A.; Poon, E.; Reyes, J.M.; Fulciniti, M.; Lopez, M.A.; Scott, T.G.; Nabet, B.; Erb, M.A.; Winter, G.E.; et al. Enhancer invasion shapes MYCN-dependent transcriptional amplification in neuroblastoma. Nat. Genet. 2018, 50, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Grisendi, S.; Mecucci, C.; Falini, B.; Pandolfi, P.P. Nucleophosmin and cancer. Nat. Rev. Cancer 2006, 6, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Shakalya, K.; Stejskal, A.; Goldman, A.; Beeck, S.; Cooke, L.; Mahadevan, D. NSC348884, a nucleophosmin inhibitor disrupts oligomer formation and induces apoptosis in human cancer cells. Oncogene 2008, 27, 4210–4220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Hassouni, B.; Sarkisjan, D.; Vos, J.C.; Giovannetti, E.; Peters, G.J. Targeting the Ribosome Biogenesis Key Molecule Fibrillarin to Avoid Chemoresistance. Curr. Med. Chem. 2019, 26, 6020–6032. [Google Scholar] [CrossRef]
- Abel, F.; Dalevi, D.; Nethander, M.; Jornsten, R.; De Preter, K.; Vermeulen, J.; Stallings, R.; Kogner, P.; Maris, J.; Nilsson, S. A 6-gene signature identifies four molecular subgroups of neuroblastoma. Cancer Cell Int. 2011, 11, 9. [Google Scholar] [CrossRef] [Green Version]
- Herold, S.; Kalb, J.; Buchel, G.; Ade, C.P.; Baluapuri, A.; Xu, J.; Koster, J.; Solvie, D.; Carstensen, A.; Klotz, C.; et al. Recruitment of BRCA1 limits MYCN-driven accumulation of stalled RNA polymerase. Nature 2019, 567, 545–549. [Google Scholar] [CrossRef]
- Colicchia, V.; Petroni, M.; Guarguaglini, G.; Sardina, F.; Sahun-Roncero, M.; Carbonari, M.; Ricci, B.; Heil, C.; Capalbo, C.; Belardinilli, F.; et al. PARP inhibitors enhance replication stress and cause mitotic catastrophe in MYCN-dependent neuroblastoma. Oncogene 2017, 36, 4682–4691. [Google Scholar] [CrossRef]
- Petroni, M.; Sardina, F.; Heil, C.; Sahun-Roncero, M.; Colicchia, V.; Veschi, V.; Albini, S.; Fruci, D.; Ricci, B.; Soriani, A.; et al. The MRN complex is transcriptionally regulated by MYCN during neural cell proliferation to control replication stress. Cell Death Differ. 2016, 23, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogarty, M.D. The requirement for evasion of programmed cell death in neuroblastomas with MYCN amplification. Cancer Lett. 2003, 197, 173–179. [Google Scholar] [CrossRef]
- Amon, J.D.; Koshland, D. RNase H enables efficient repair of R-loop induced DNA damage. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Keijzers, G.; Bakula, D.; Petr, M.A.; Madsen, N.G.K.; Teklu, A.; Mkrtchyan, G.; Osborne, B.; Scheibye-Knudsen, M. Human Exonuclease 1 (EXO1) Regulatory Functions in DNA Replication with Putative Roles in Cancer. Int. J. Mol. Sci. 2018, 20, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, C.H.; Leu, Y.L.; Wang, T.H.; Tseng, W.C.; Feng, C.H.; Wang, S.H.; Chen, C.C. A novel DNA repair inhibitor, diallyl disulfide (DADS), impairs DNA resection during DNA double-strand break repair by reducing Sae2 and Exo1 levels. DNA Repair (Amst.) 2019, 82, 102690. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Yoon, J.; Ju, M.; Lee, Y.; Kim, T.-H.; Kim, J.; Sommer, P.; No, Z.; Cechetto, J.; Han, S.-J. Identification of two HIV inhibitors that also inhibit human RNaseH2. Mol. Cells 2013, 36, 212–218. [Google Scholar] [CrossRef] [Green Version]
- Weber, A.; Imisch, P.; Bergmann, E.; Christiansen, H. Coamplification of DDX1 Correlates With an Improved Survival Probability in Children With MYCN-Amplified Human Neuroblastoma. J. Clin. Oncol. 2004, 22, 2681–2690. [Google Scholar] [CrossRef]
- Han, C.; Liu, Y.; Wan, G.; Choi, H.J.; Zhao, L.; Ivan, C.; He, X.; Sood, A.K.; Zhang, X.; Lu, X. The RNA-binding protein DDX1 promotes primary microRNA maturation and inhibits ovarian tumor progression. Cell Rep. 2014, 8, 1447–1460. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Khadijah, S.; Fang, S.; Wang, L.; Tay, F.P.; Liu, D.X. The cellular RNA helicase DDX1 interacts with coronavirus nonstructural protein 14 and enhances viral replication. J. Virol. 2010, 84, 8571–8583. [Google Scholar] [CrossRef] [Green Version]
- Yu, E.Y.; Cheung, I.Y.; Feng, Y.; Rabie, M.O.; Roboz, G.J.; Guzman, M.L.; Cheung, N.-K.V.; Lue, N.F. Telomere Trimming and DNA Damage as Signatures of High Risk Neuroblastoma. Neoplasia 2019, 21, 689–701. [Google Scholar] [CrossRef]
- Hiyama, E.; Hiyama, K.; Yokoyama, T.; Matsuura, Y.; Piatyszek, M.A.; Shay, J.W. Correlating telomerase activity levels with human neuroblastoma outcomes. Nat. Med. 1995, 1, 249–255. [Google Scholar] [CrossRef]
- Huang, M.; Zeki, J.; Sumarsono, N.; Coles, G.L.; Taylor, J.S.; Danzer, E.; Bruzoni, M.; Hazard, F.K.; Lacayo, N.J.; Sakamoto, K.M.; et al. Epigenetic Targeting of Tert-Associated Gene Expression Signature in Human Neuroblastoma with Tert Overexpression. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Valentijn, L.J.; Koster, J.; Zwijnenburg, D.A.; Hasselt, N.E.; van Sluis, P.; Volckmann, R.; van Noesel, M.M.; George, R.E.; Tytgat, G.A.; Molenaar, J.J.; et al. TERT rearrangements are frequent in neuroblastoma and identify aggressive tumors. Nat. Genet. 2015, 47, 1411–1414. [Google Scholar] [CrossRef]
- Peifer, M.; Hertwig, F.; Roels, F.; Dreidax, D.; Gartlgruber, M.; Menon, R.; Kramer, A.; Roncaioli, J.L.; Sand, F.; Heuckmann, J.M.; et al. Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature 2015, 526, 700–704. [Google Scholar] [CrossRef]
- Reyes-Uribe, P.; Adrianzen-Ruesta, M.P.; Deng, Z.; Echevarria-Vargas, I.; Mender, I.; Saheb, S.; Liu, Q.; Altieri, D.C.; Murphy, M.E.; Shay, J.W.; et al. Exploiting TERT dependency as a therapeutic strategy for NRAS-mutant melanoma. Oncogene 2018, 37, 4058–4072. [Google Scholar] [CrossRef]
- Brien, R.; Tran, S.L.; Maritz, M.F.; Liu, B.; Kong, C.F.; Purgato, S.; Yang, C.; Murray, J.; Russell, A.J.; Flemming, C.L.; et al. Driven Neuroblastomas Are Addicted to a Telomerase-Independent Function of Dyskerin. Cancer Res. 2016, 76, 3604. [Google Scholar] [CrossRef] [Green Version]
- Quan Wang, J.; Di Yang, M.; Chen, X.; Wang, Y.; Zeng Chen, L.; Cheng, X.; Hua Liu, X. Discovery of new chromen-4-one derivatives as telomerase inhibitors through regulating expression of dyskerin. J. Enzym. Inhib. Med. Chem. 2018, 33, 1199–1211. [Google Scholar] [CrossRef] [Green Version]
- Rocchi, L.; Barbosa, A.J.M.; Onofrillo, C.; Del Rio, A.; Montanaro, L. Inhibition of Human Dyskerin as a New Approach to Target Ribosome Biogenesis. PLoS ONE 2014, 9, e101971. [Google Scholar] [CrossRef]
- Sanmartin, E.; Munoz, L.; Piqueras, M.; Sirerol, J.A.; Berlanga, P.; Canete, A.; Castel, V.; Font de Mora, J. Deletion of 11q in Neuroblastomas Drives Sensitivity to PARP Inhibition. Clin. Cancer Res. 2017, 23, 6875–6887. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.Y.; Atmadibrata, B.; Mondal, S.; Tee, A.E.; Liu, T. NCYM is upregulated by lncUSMycN and modulates N-Myc expression. Int. J. Oncol. 2016, 49, 2464–2470. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.Y.; Erriquez, D.; Marshall, G.M.; Tee, A.E.; Polly, P.; Wong, M.; Liu, B.; Bell, J.L.; Zhang, X.D.; Milazzo, G.; et al. Effects of a novel long noncoding RNA, lncUSMycN, on N-Myc expression and neuroblastoma progression. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef]
- Cobbold, L.C.; Spriggs, K.A.; Haines, S.J.; Dobbyn, H.C.; Hayes, C.; de Moor, C.H.; Lilley, K.S.; Bushell, M.; Willis, A.E. Identification of internal ribosome entry segment (IRES)-trans-acting factors for the Myc family of IRESs. Mol. Cell Biol. 2008, 28, 40–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, W.; Liang, X.H.; Sun, H.; De Hoyos, C.L.; Crooke, S.T. Depletion of NEAT1 lncRNA attenuates nucleolar stress by releasing sequestered P54nrb and PSF to facilitate c-Myc translation. PLoS ONE 2017, 12, e0173494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petti, E.; Buemi, V.; Zappone, A.; Schillaci, O.; Broccia, P.V.; Dinami, R.; Matteoni, S.; Benetti, R.; Schoeftner, S. SFPQ and NONO suppress RNA:DNA-hybrid-related telomere instability. Nat. Commun. 2019, 10, 1001. [Google Scholar] [CrossRef] [Green Version]
- Yamauchi, T.; Nakamura, N.; Hiramoto, M.; Yuri, M.; Yokota, H.; Naitou, M.; Takeuchi, M.; Yamanaka, K.; Kita, A.; Nakahara, T.; et al. Sepantronium Bromide (YM155) induces disruption of the ILF3/p54nrb complex, which is required for survivin expression. Biochem. Biophys. Res. Commun. 2012, 425, 711–716. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.L.; Wachter, K.; Muhleck, B.; Pazaitis, N.; Kohn, M.; Lederer, M.; Huttelmaier, S. Insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs): Post-transcriptional drivers of cancer progression? Cell Mol. Life Sci. 2013, 70, 2657–2675. [Google Scholar] [CrossRef] [Green Version]
- Degrauwe, N.; Suva, M.L.; Janiszewska, M.; Riggi, N.; Stamenkovic, I. IMPs: An RNA-binding protein family that provides a link between stem cell maintenance in normal development and cancer. Genes Dev. 2016, 30, 2459–2474. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Weng, H.; Sun, W.; Qin, X.; Shi, H.; Wu, H.; Zhao, B.S.; Mesquita, A.; Liu, C.; Yuan, C.L.; et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat. Cell Biol. 2018, 20, 285–295. [Google Scholar] [CrossRef]
- Bell, J.L.; Turlapati, R.; Liu, T.; Schulte, J.H.; Hüttelmaier, S. IGF2BP1 Harbors Prognostic Significance by Gene Gain and Diverse Expression in Neuroblastoma. J. Clin. Oncol. 2015, 33, 1285–1293. [Google Scholar] [CrossRef] [Green Version]
- Burdelski, C.; Jakani-Karimi, N.; Jacobsen, F.; Moller-Koop, C.; Minner, S.; Simon, R.; Sauter, G.; Steurer, S.; Clauditz, T.S.; Wilczak, W. IMP3 overexpression occurs in various important cancer types and is linked to aggressive tumor features: A tissue microarray study on 8,877 human cancers and normal tissues. Oncol. Rep. 2018, 39, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.T.; Jeng, Y.M.; Chang, C.C.; Chang, H.H.; Huang, M.C.; Juan, H.F.; Hsu, C.H.; Lee, H.; Liao, Y.F.; Lee, Y.L.; et al. Insulin-like growth factor II mRNA-binding protein 3 expression predicts unfavorable prognosis in patients with neuroblastoma. Cancer Sci. 2011, 102, 2191–2198. [Google Scholar] [CrossRef]
- Narbonne-Reveau, K.; Lanet, E.; Dillard, C.; Foppolo, S.; Chen, C.-H.; Parrinello, H.; Rialle, S.; Sokol, N.S.; Maurange, C. Neural stem cell-encoded temporal patterning delineates an early window of malignant susceptibility in Drosophila. eLife 2016, 5, e13463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panebianco, F.; Kelly, L.M.; Liu, P.; Zhong, S.; Dacic, S.; Wang, X.; Singhi, A.D.; Dhir, R.; Chiosea, S.I.; Kuan, S.-F.; et al. THADA fusion is a mechanism of IGF2BP3 activation and IGF1R signaling in thyroid cancer. Proc. Natl. Acad. Sci. USA 2017, 114, 2307–2312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahapatra, L.; Andruska, N.; Mao, C.; Le, J.; Shapiro, D.J. A Novel IMP1 Inhibitor, BTYNB, Targets c-Myc and Inhibits Melanoma and Ovarian Cancer Cell Proliferation. Transl. Oncol. 2017, 10, 818–827. [Google Scholar] [CrossRef] [PubMed]
- Himoto, T.; Kuriyama, S.; Zhang, J.Y.; Chan, E.K.; Nishioka, M.; Tan, E.M. Significance of autoantibodies against insulin-like growth factor II mRNA-binding proteins in patients with hepatocellular carcinoma. Int. J. Oncol. 2005, 26, 311–317. [Google Scholar] [CrossRef]
- Zhang, J.-Y.; Chan, E.K.L.; Peng, X.-X.; Tan, E.M. A Novel Cytoplasmic Protein with RNA-binding Motifs Is an Autoantigen in Human Hepatocellular Carcinoma. J. Exp. Med. 1999, 189, 1101–1110. [Google Scholar] [CrossRef]
- Pandey, G.K.; Mitra, S.; Subhash, S.; Hertwig, F.; Kanduri, M.; Mishra, K.; Fransson, S.; Ganeshram, A.; Mondal, T.; Bandaru, S.; et al. The Risk-Associated Long Noncoding RNA NBAT-1 Controls Neuroblastoma Progression by Regulating Cell Proliferation and Neuronal Differentiation. Cancer Cell 2014, 26, 722–737. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Xu, F.; Chang, C.-K.; He, Q.; Wu, L.-Y.; Zhang, Z.; Li, X. MYCN contributes to the malignant characteristics of erythroleukemia through EZH2-mediated epigenetic repression of p21. Cell Death Dis. 2017, 8, e3126. [Google Scholar] [CrossRef] [Green Version]
- Pascale, A.; Amadio, M.; Quattrone, A. Defining a neuron: Neuronal ELAV proteins. Cell. Mol. Life Sci. 2007, 65, 128. [Google Scholar] [CrossRef]
- Takemoto, J.; Kuda, M.; Kohashi, K.; Yamada, Y.; Koga, Y.; Kinoshita, I.; Souzaki, R.; Taguchi, T.; Oda, Y. HuC/D expression in small round cell tumors and neuroendocrine tumors: A useful tool for distinguishing neuroblastoma from childhood small round cell tumors. Hum. Pathol. 2019, 85, 162–167. [Google Scholar] [CrossRef]
- Lazarova, D.L.; Spengler, B.A.; Biedler, J.L.; Ross, R.A. HuD, a neuronal-specific RNA-binding protein, is a putative regulator of N-myc pre-mRNA processing/stability in malignant human neuroblasts. Oncogene 1999, 18, 2703–2710. [Google Scholar] [CrossRef] [PubMed]
- Samaraweera, L.; Spengler, B.A.; Ross, R.A. Reciprocal antagonistic regulation of N-myc mRNA by miR-17 and the neuronal-specific RNA-binding protein HuD. Oncol. Rep. 2017, 38, 545–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grandinetti, K.B.; Spengler, B.A.; Biedler, J.L.; Ross, R.A. Loss of one HuD allele on chromosome #1p selects for amplification of the N-myc proto-oncogene in human neuroblastoma cells. Oncogene 2006, 25, 706–712. [Google Scholar] [CrossRef] [PubMed]
- Amadio, M.; Scapagnini, G.; Laforenza, U.; Intrieri, M.; Romeo, L.; Govoni, S.; Pascale, A. Post-transcriptional regulation of HSP70 expression following oxidative stress in SH-SY5Y cells: The potential involvement of the RNA-binding protein HuR. Curr. Pharm. Des. 2008, 14, 2651–2658. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, F.; Fang, E.; Xiao, W.; Mei, H.; Li, H.; Li, D.; Song, H.; Wang, J.; Hong, M.; et al. Circular RNA circAGO2 drives cancer progression through facilitating HuR-repressed functions of AGO2-miRNA complexes. Cell Death Differ. 2019, 26, 1346–1364. [Google Scholar] [CrossRef]
- Wang, J.; Hjelmeland, A.B.; Nabors, L.B.; King, P.H. Anti-cancer effects of the HuR inhibitor, MS-444, in malignant glioma cells. Cancer Biol. Ther. 2019, 20, 979–988. [Google Scholar] [CrossRef]
- Underwood, J.G.; Boutz, P.L.; Dougherty, J.D.; Stoilov, P.; Black, D.L. Homologues of the Caenorhabditis elegans Fox-1 protein are neuronal splicing regulators in mammals. Mol. Cell. Biol. 2005, 25, 10005–10016. [Google Scholar] [CrossRef] [Green Version]
- Tomljanovic, Z.; Patel, M.; Shin, W.; Califano, A.; Teich, A.F. ZCCHC17 is a master regulator of synaptic gene expression in Alzheimer’s disease. Bioinformatics (Oxf. Engl.) 2018, 34, 367–371. [Google Scholar] [CrossRef]
- Parras, A.; Anta, H.; Santos-Galindo, M.; Swarup, V.; Elorza, A.; Nieto-González, J.L.; Picó, S.; Hernández, I.H.; Díaz-Hernández, J.I.; Belloc, E.; et al. Autism-like phenotype and risk gene mRNA deadenylation by CPEB4 mis-splicing. Nature 2018, 560, 441–446. [Google Scholar] [CrossRef]
- Hägele, S.; Kühn, U.; Böning, M.; Katschinski, D.M. Cytoplasmic polyadenylation-element-binding protein (CPEB)1 and 2 bind to the HIF-1alpha mRNA 3’-UTR and modulate HIF-1alpha protein expression. Biochem. J. 2009, 417, 235–246. [Google Scholar] [CrossRef]
- Liu, G.; Zhang, Q.; Xia, L.; Shi, M.; Cai, J.; Zhang, H.; Li, J.; Lin, G.; Xie, W.; Zhang, Y.; et al. RNA-binding protein CELF6 is cell cycle regulated and controls cancer cell proliferation by stabilizing p21. Cell Death Dis. 2019, 10, 688. [Google Scholar] [CrossRef] [PubMed]
- Gan, B.; Chen, S.; Liu, H.; Min, J.; Liu, K. Structure and function of eTudor domain containing TDRD proteins. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Wiedemeyer, R.; Westermann, F.; Wittke, I.; Nowock, J.; Schwab, M. Ataxin-2 promotes apoptosis of human neuroblastoma cells. Oncogene 2003, 22, 401–411. [Google Scholar] [CrossRef] [Green Version]
- Zaman, S.; Chobrutskiy, B.I.; Blanck, G. MAPT (Tau) expression is a biomarker for an increased rate of survival in pediatric neuroblastoma. Cell Cycle (Georget. Tex.) 2018, 17, 2474–2483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, H.; Matter, M.L.; Issa-Jahns, L.; Jijiwa, M.; Kraemer, N.; Musante, L.; de la Vega, M.; Ninnemann, O.; Schindler, D.; Damatova, N.; et al. Mutations in PTRH2 cause novel infantile-onset multisystem disease with intellectual disability, microcephaly, progressive ataxia, and muscle weakness. Ann. Clin. Transl. Neurol. 2014, 1, 1024–1035. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois, G.; Létoquart, J.; van Tran, N.; Graille, M. Trm112, a Protein Activator of Methyltransferases Modifying Actors of the Eukaryotic Translational Apparatus. Biomolecules 2017, 7, 7. [Google Scholar] [CrossRef]
- Zhu, X.; Bührer, C.; Wellmann, S. Cold-inducible proteins CIRP and RBM3, a unique couple with activities far beyond the cold. Cell. Mol. Life Sci. 2016, 73, 3839–3859. [Google Scholar] [CrossRef] [Green Version]
- Pedrotti, S.; Busà, R.; Compagnucci, C.; Sette, C. The RNA recognition motif protein RBM11 is a novel tissue-specific splicing regulator. Nucleic Acids Res. 2011, 40, 1021–1032. [Google Scholar] [CrossRef] [Green Version]
- Adamson, B.; Smogorzewska, A.; Sigoillot, F.D.; King, R.W.; Elledge, S.J. A genome-wide homologous recombination screen identifies the RNA-binding protein RBMX as a component of the DNA-damage response. Nat. Cell Biol. 2012, 14, 318–328. [Google Scholar] [CrossRef] [Green Version]
- Munschauer, M.; Nguyen, C.T.; Sirokman, K.; Hartigan, C.R.; Hogstrom, L.; Engreitz, J.M.; Ulirsch, J.C.; Fulco, C.P.; Subramanian, V.; Chen, J.; et al. The NORAD lncRNA assembles a topoisomerase complex critical for genome stability. Nature 2018, 561, 132–136. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Hackett, C.S.; Zhang, S.; Song, Y.K.; Bell, R.J.A.; Molinaro, A.M.; Quigley, D.A.; Balmain, A.; Song, J.S.; Costello, J.F.; et al. The genetics of splicing in neuroblastoma. Cancer Discov. 2015, 5, 380–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Schafer, S.; Greaser, M.L.; Radke, M.H.; Liss, M.; Govindarajan, T.; Maatz, H.; Schulz, H.; Li, S.; Parrish, A.M.; et al. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat. Med. 2012, 18, 766–773. [Google Scholar] [CrossRef]
- Radine, C.; Peters, D.; Reese, A.; Neuwahl, J.; Budach, W.; Jänicke, R.U.; Sohn, D. The RNA-binding protein RBM47 is a novel regulator of cell fate decisions by transcriptionally controlling the p53-p21-axis. Cell Death Differ. 2019. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.-J.; Zhuang, R.-J.; Li, Y.-B.; Li, T.; Yuan, X.; Lei, B.-B.; Xie, Y.-F.; Wang, M. Cold-inducible protein RBM3 mediates hypothermic neuroprotection against neurotoxin rotenone via inhibition on MAPK signalling. J. Cell Mol. Med. 2019, 23, 7010–7020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.-J.; Shi, X.; Ju, F.; Hao, B.-N.; Ma, S.-P.; Wang, L.; Cheng, B.-F.; Wang, M. Cold Shock Induced Protein RBM3 but Not Mild Hypothermia Protects Human SH-SY5Y Neuroblastoma Cells From MPP+-Induced Neurotoxicity. Front. Neurosci. 2018, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.-J.; Ju, F.; Guo, X.-X.; Ma, S.-P.; Wang, L.; Cheng, B.-F.; Zhuang, R.-J.; Zhang, B.-B.; Shi, X.; Feng, Z.-W.; et al. RNA-binding protein RBM3 prevents NO-induced apoptosis in human neuroblastoma cells by modulating p38 signaling and miR-143. Sci. Rep. 2017, 7, 41738. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, R.-J.; Ma, J.; Shi, X.; Ju, F.; Ma, S.-P.; Wang, L.; Cheng, B.-F.; Ma, Y.-W.; Wang, M.; Li, T.; et al. Cold-Inducible Protein RBM3 Protects UV Irradiation-Induced Apoptosis in Neuroblastoma Cells by Affecting p38 and JNK Pathways and Bcl2 Family Proteins. J. Mol. Neurosci. 2017, 63, 142–151. [Google Scholar] [CrossRef]
- Zhu, X.; Yan, J.; Bregere, C.; Zelmer, A.; Goerne, T.; Kapfhammer, J.P.; Guzman, R.; Wellmann, S. RBM3 promotes neurogenesis in a niche-dependent manner via IMP2-IGF2 signaling pathway after hypoxic-ischemic brain injury. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Grupp, K.; Hofmann, B.; Kutup, A.; Bachmann, K.; Bogoevski, D.; Melling, N.; Uzunoglu, F.G.; El Gammal, A.T.; Koop, C.; Simon, R.; et al. Reduced RBM3 expression is associated with aggressive tumor features in esophageal cancer but not significantly linked to patient outcome. BMC Cancer 2018, 18, 1106. [Google Scholar] [CrossRef]
- Hagen, C.P.; Sørensen, K.; Mieritz, M.G.; Johannsen, T.H.; Almstrup, K.; Juul, A. Circulating MKRN3 Levels Decline Prior to Pubertal Onset and Through Puberty: A Longitudinal Study of Healthy Girls. J. Clin. Endocrinol. Metab. 2015, 100, 1920–1926. [Google Scholar] [CrossRef] [Green Version]
- Heras, V.; Sangiao-Alvarellos, S.; Manfredi-Lozano, M.; Sanchez-Tapia, M.J.; Ruiz-Pino, F.; Roa, J.; Lara-Chica, M.; Morrugares-Carmona, R.; Jouy, N.; Abreu, A.P.; et al. Hypothalamic miR-30 regulates puberty onset via repression of the puberty-suppressing factor, Mkrn3. PLoS Biol. 2019, 17, e3000532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yellapragada, V.; Liu, X.; Lund, C.; Känsäkoski, J.; Pulli, K.; Vuoristo, S.; Lundin, K.; Tuuri, T.; Varjosalo, M.; Raivio, T. MKRN3 Interacts With Several Proteins Implicated in Puberty Timing but Does Not Influence GNRH1 Expression. Front. Endocrinol. (Lausanne) 2019, 10, 48. [Google Scholar] [CrossRef] [PubMed]
- Iijima, T.; Wu, K.; Witte, H.; Hanno-Iijima, Y.; Glatter, T.; Richard, S.; Scheiffele, P. SAM68 regulates neuronal activity-dependent alternative splicing of neurexin-1. Cell 2011, 147, 1601–1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Li, Z.; He, B.; Liu, J.; Li, S.; Zhou, L.; Pan, C.; Yu, Z.; Xu, Z. Sam68 is a novel marker for aggressive neuroblastoma. OncoTargets Ther. 2013, 6, 1751–1760. [Google Scholar] [CrossRef] [Green Version]
- Melko, M.; Douguet, D.; Bensaid, M.; Zongaro, S.; Verheggen, C.; Gecz, J.; Bardoni, B. Functional characterization of the AFF (AF4/FMR2) family of RNA-binding proteins: Insights into the molecular pathology of FRAXE intellectual disability. Hum. Mol. Genet. 2011, 20, 1873–1885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuva-Aydemir, Y.; Almeida, S.; Krishnan, G.; Gendron, T.F.; Gao, F.-B. Transcription elongation factor AFF2/FMR2 regulates expression of expanded GGGGCC repeat-containing C9ORF72 allele in ALS/FTD. Nat. Commun. 2019, 10, 5466. [Google Scholar] [CrossRef] [PubMed]
- Bosse, K.R.; Diskin, S.J.; Cole, K.A.; Wood, A.C.; Schnepp, R.W.; Norris, G.; Nguyen, L.B.; Jagannathan, J.; Laquaglia, M.; Winter, C.; et al. Common variation at BARD1 results in the expression of an oncogenic isoform that influences neuroblastoma susceptibility and oncogenicity. Cancer Res. 2012, 72, 2068–2078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Liu, Y.; Zhang, C.; Chu, J.; Wu, Y.; Li, Y.; Liu, J.; Li, Q.; Li, S.; Shi, Q.; et al. Tamoxifen-resistant breast cancer cells are resistant to DNA-damaging chemotherapy because of upregulated BARD1 and BRCA1. Nat. Commun. 2018, 9, 1595. [Google Scholar] [CrossRef]
- Ozden, O.; Bishehsari, F.; Bauer, J.; Park, S.-H.; Jana, A.; Baik, S.H.; Sporn, J.C.; Staudacher, J.J.; Yazici, C.; Krett, N.; et al. Expression of an Oncogenic BARD1 Splice Variant Impairs Homologous Recombination and Predicts Response to PARP-1 Inhibitor Therapy in Colon Cancer. Sci. Rep. 2016, 6, 26273. [Google Scholar] [CrossRef]
- Koc, E.C.; Haciosmanoglu, E.; Claudio, P.P.; Wolf, A.; Califano, L.; Friscia, M.; Cortese, A.; Koc, H. Impaired mitochondrial protein synthesis in head and neck squamous cell carcinoma. Mitochondrion 2015, 24, 113–121. [Google Scholar] [CrossRef]
- Hu, B.; Guo, Y. Inhibition of mitochondrial translation as a therapeutic strategy for human ovarian cancer to overcome chemoresistance. Biochem. Biophys. Res. Commun. 2019, 509, 373–378. [Google Scholar] [CrossRef]
- Fischl, H.; Neve, J.; Wang, Z.; Patel, R.; Louey, A.; Tian, B.; Furger, A. hnRNPC regulates cancer-specific alternative cleavage and polyadenylation profiles. Nucleic Acids Res. 2019, 47, 7580–7591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anantha, R.W.; Alcivar, A.L.; Ma, J.; Cai, H.; Simhadri, S.; Ule, J.; König, J.; Xia, B. Requirement of heterogeneous nuclear ribonucleoprotein C for BRCA gene expression and homologous recombination. PLoS ONE 2013, 8, e61368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.K.; Kim, H.H.; Kuwano, Y.; Abdelmohsen, K.; Srikantan, S.; Subaran, S.S.; Gleichmann, M.; Mughal, M.R.; Martindale, J.L.; Yang, X.; et al. hnRNP C promotes APP translation by competing with FMRP for APP mRNA recruitment to P bodies. Nat. Struct. Mol. Biol. 2010, 17, 732–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Wei, J.S.; Li, S.Q.; Badgett, T.C.; Song, Y.K.; Agarwal, S.; Coarfa, C.; Tolman, C.; Hurd, L.; Liao, H.; et al. MYCN controls an alternative RNA splicing program in high-risk metastatic neuroblastoma. Cancer Lett. 2016, 371, 214–224. [Google Scholar] [CrossRef] [Green Version]
- Carabet, L.A.; Leblanc, E.; Lallous, N.; Morin, H.; Ghaidi, F.; Lee, J.; Rennie, P.S.; Cherkasov, A. Computer-Aided Discovery of Small Molecules Targeting the RNA Splicing Activity of hnRNP A1 in Castration-Resistant Prostate Cancer. Molecules 2019, 24, 763. [Google Scholar] [CrossRef] [Green Version]
- Chang, W.H.; Liu, T.C.; Yang, W.K.; Lee, C.C.; Lin, Y.H.; Chen, T.Y.; Chang, J.G. Amiloride modulates alternative splicing in leukemic cells and resensitizes Bcr-AblT315I mutant cells to imatinib. Cancer Res. 2011, 71, 383–392. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Hennchen, M.; Stubbusch, J.; Abarchan-El Makhfi, I.; Kramer, M.; Deller, T.; Pierre-Eugene, C.; Janoueix-Lerosey, I.; Delattre, O.; Ernsberger, U.; Schulte, J.B.; et al. Lin28B and Let-7 in the Control of Sympathetic Neurogenesis and Neuroblastoma Development. J. Neurosci. 2015, 35, 16531–16544. [Google Scholar] [CrossRef] [Green Version]
- Viswanathan, S.R.; Daley, G.Q.; Gregory, R.I. Selective Blockade of MicroRNA Processing by Lin28. Science 2008, 320, 97. [Google Scholar] [CrossRef] [Green Version]
- Diskin, S.J.; Capasso, M.; Schnepp, R.W.; Cole, K.A.; Attiyeh, E.F.; Hou, C.; Diamond, M.; Carpenter, E.L.; Winter, C.; Lee, H.; et al. Common variation at 6q16 within HACE1 and LIN28B influences susceptibility to neuroblastoma. Nat. Genet. 2012, 44, 1126–1130. [Google Scholar] [CrossRef] [PubMed]
- Depuydt, P.; Koster, J.; Boeva, V.; Hocking, T.D.; Speleman, F.; Schleiermacher, G.; De Preter, K. Meta-mining of copy number profiles of high-risk neuroblastoma tumors. Sci. Data 2018, 5, 180240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Roy, N.; Van Der Linden, M.; Menten, B.; Dheedene, A.; Vandeputte, C.; Van Dorpe, J.; Laureys, G.; Renard, M.; Sante, T.; Lammens, T.; et al. Shallow Whole Genome Sequencing on Circulating Cell-Free DNA Allows Reliable Noninvasive Copy-Number Profiling in Neuroblastoma Patients. Clin. Cancer Res. 2017, 23, 6305–6314. [Google Scholar] [CrossRef] [Green Version]
- Molenaar, J.J.; Domingo-Fernández, R.; Ebus, M.E.; Lindner, S.; Koster, J.; Drabek, K.; Mestdagh, P.; van Sluis, P.; Valentijn, L.J.; van Nes, J.; et al. LIN28B induces neuroblastoma and enhances MYCN levels via let-7 suppression. Nat. Genet. 2012, 4, 1199–1206. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.; Potenza, N. Antiproliferative Activity of microRNA-125a and its Molecular Targets. Microrna 2019, 8, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Beckers, A.; Van Peer, G.; Carter, D.R.; Gartlgruber, M.; Herrmann, C.; Agarwal, S.; Helsmoortel, H.H.; Althoff, K.; Molenaar, J.J.; Cheung, B.B.; et al. MYCN-driven regulatory mechanisms controlling LIN28B in neuroblastoma. Cancer Lett. 2015, 366, 123–132. [Google Scholar] [CrossRef] [Green Version]
- Cotterman, R.; Knoepfler, P.S. N-Myc regulates expression of pluripotency genes in neuroblastoma including lif, klf2, klf4, and lin28b. PLoS ONE 2009, 4, e5799. [Google Scholar] [CrossRef] [Green Version]
- Corallo, D.; Donadon, M.; Pantile, M.; Sidarovich, V.; Cocchi, S.; Ori, M.; De Sarlo, M.; Candiani, S.; Frasson, C.; Distel, M.; et al. LIN28B increases neural crest cell migration and leads to transformation of trunk sympathoadrenal precursors. Cell Death Differ. 2020, 27, 1225–1242. [Google Scholar] [CrossRef]
- Ustianenko, D.; Chiu, H.-S.; Treiber, T.; Weyn-Vanhentenryck, S.M.; Treiber, N.; Meister, G.; Sumazin, P.; Zhang, C. LIN28 Selectively Modulates a Subclass of Let-7 MicroRNAs. Mol. Cell 2018, 71, 271–283.e275. [Google Scholar] [CrossRef] [Green Version]
- Schnepp, R.W.; Khurana, P.; Attiyeh, E.F.; Raman, P.; Chodosh, S.E.; Oldridge, D.A.; Gagliardi, M.E.; Conkrite, K.L.; Asgharzadeh, S.; Seeger, R.C.; et al. A LIN28B-RAN-AURKA Signaling Network Promotes Neuroblastoma Tumorigenesis. Cancer Cell 2015, 28, 599–609. [Google Scholar] [CrossRef] [Green Version]
- Roos, M.; Pradère, U.; Ngondo, R.P.; Behera, A.; Allegrini, S.; Civenni, G.; Zagalak, J.A.; Marchand, J.R.; Menzi, M.; Towbin, H.; et al. A Small-Molecule Inhibitor of Lin28. ACS Chem. Biol. 2016, 11, 2773–2781. [Google Scholar] [CrossRef] [PubMed]
- Shahbazi, J.; Liu, P.Y.; Atmadibrata, B.; Bradner, J.E.; Marshall, G.M.; Lock, R.B.; Liu, T. The Bromodomain Inhibitor JQ1 and the Histone Deacetylase Inhibitor Panobinostat Synergistically Reduce N-Myc Expression and Induce Anticancer Effects. Clin. Cancer Res. 2016, 22, 2534–2544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treiber, T.; Treiber, N.; Meister, G. Regulation of microRNA biogenesis and its crosstalk with other cellular pathways. Nat. Rev. Mol. Cell Biol. 2019, 20, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.-J.; Lin, Y.-C.; Chen, J.; Kuo, H.-H.; Chen, Y.-Y.; Diccianni, M.B.; London, W.B.; Chang, C.-H.; Yu, A.L. microRNA signature and expression of Dicer and Drosha can predict prognosis and delineate risk groups in neuroblastoma. Cancer Res. 2010, 70, 7841–7850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, P.M.; Seifried, B.A.; Kyemba, S.K.; Jensen, S.J.; Guo, C.; Maris, J.M.; Brodeur, G.M.; Stram, D.O.; Seeger, R.C.; Gerbing, R.; et al. Loss of heterozygosity for chromosome 14q in neuroblastoma. Med. Pediatr. Oncol. 2001, 36, 28–31. [Google Scholar] [CrossRef]
- Hill, D.A.; Ivanovich, J.; Priest, J.R.; Gurnett, C.A.; Dehner, L.P.; Desruisseau, D.; Jarzembowski, J.A.; Wikenheiser-Brokamp, K.A.; Suarez, B.K.; Whelan, A.J.; et al. DICER1 Mutations in Familial Pleuropulmonary Blastoma. Science 2009, 325, 965. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.; Liu, Y.; Huang, M.; Zhao, X.; Cheng, L. Wnt1-cre-mediated conditional loss of Dicer results in malformation of the midbrain and cerebellum and failure of neural crest and dopaminergic differentiation in mice. J. Mol. Cell Biol. 2010, 2, 152–163. [Google Scholar] [CrossRef] [Green Version]
- Calissano, M.; Diss, J.K.J.; Latchman, D.S. Post-transcriptional regulation of the Brn-3b transcription factor in differentiating neuroblastoma cells. FEBS Lett. 2007, 581, 2490–2496. [Google Scholar] [CrossRef] [Green Version]
- Potenza, N.; Papa, U.; Russo, A. Differential expression of Dicer and Argonaute genes duringthe differentiation of human neuroblastoma cells. Cell Biol. Int. 2009, 33, 734–738. [Google Scholar] [CrossRef]
- Jauhari, A.; Singh, T.; Pandey, A.; Singh, P.; Singh, N.; Srivastava, A.K.; Pant, A.B.; Parmar, D.; Yadav, S. Differentiation Induces Dramatic Changes in miRNA Profile, Where Loss of Dicer Diverts Differentiating SH-SY5Y Cells Toward Senescence. Mol. Neurobiol. 2017, 54, 4986–4995. [Google Scholar] [CrossRef]
- Potenza, N.; Papa, U.; Scaruffi, P.; Mosca, N.; Tonini, G.P.; Russo, A. A novel splice variant of the human dicer gene is expressed in neuroblastoma cells. FEBS Lett. 2010, 584, 3452–3457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosca, N.; Starega-Roslan, J.; Castiello, F.; Russo, A.; Krzyzosiak, W.J.; Potenza, N. Characterization of a naturally occurring truncated Dicer. Mol. Biol. Rep. 2015, 42, 1333–1340. [Google Scholar] [CrossRef] [PubMed]
- Velagapudi, S.P.; Costales, M.G.; Vummidi, B.R.; Nakai, Y.; Angelbello, A.J.; Tran, T.; Haniff, H.S.; Matsumoto, Y.; Wang, Z.F.; Chatterjee, A.K.; et al. Approved Anti-cancer Drugs Target Oncogenic Non-coding RNAs. Cell Chem. Biol. 2018, 25, 1086–1094.e1087. [Google Scholar] [CrossRef] [PubMed]
- Geretto, M.; Pulliero, A.; Rosano, C.; Zhabayeva, D.; Bersimbaev, R.; Izzotti, A. Resistance to cancer chemotherapeutic drugs is determined by pivotal microRNA regulators. Am. J. Cancer Res. 2017, 7, 1350–1371. [Google Scholar] [PubMed]
- Kawai, S.; Amano, A. BRCA1 regulates microRNA biogenesis via the DROSHA microprocessor complex. J. Cell Biol. 2012, 197, 201–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weidensdorfer, D.; Stohr, N.; Baude, A.; Lederer, M.; Kohn, M.; Schierhorn, A.; Buchmeier, S.; Wahle, E.; Huttelmaier, S. Control of c-myc mRNA stability by IGF2BP1-associated cytoplasmic RNPs. RNA 2009, 15, 104–115. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.X.; Wallis, K.; Fell, S.M.; Sobrado, V.R.; Hemmer, M.C.; Ramsköld, D.; Hellman, U.; Sandberg, R.; Kenchappa, R.S.; Martinson, T.; et al. RNA Helicase A Is a Downstream Mediator of KIF1Bβ Tumor-Suppressor Function in Neuroblastoma. Cancer Discov. 2014, 4, 434. [Google Scholar] [CrossRef] [Green Version]
- Cao, S.; Sun, R.; Wang, W.; Meng, X.; Zhang, Y.; Zhang, N.; Yang, S. RNA helicase DHX9 may be a therapeutic target in lung cancer and inhibited by enoxacin. Am. J. Transl. Res. 2017, 9, 674–682. [Google Scholar]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Meyers, R.M.; Bryan, J.G.; McFarland, J.M.; Weir, B.A.; Sizemore, A.E.; Xu, H.; Dharia, N.V.; Montgomery, P.G.; Cowley, G.S.; Pantel, S.; et al. Computational correction of copy number effect improves specificity of CRISPR–Cas9 essentiality screens in cancer cells. Nat. Genet. 2017, 49, 1779–1784. [Google Scholar] [CrossRef] [Green Version]
- Powers, J.T.; Tsanov, K.M.; Pearson, D.S.; Roels, F.; Spina, C.S.; Ebright, R.; Seligson, M.; de Soysa, Y.; Cahan, P.; Theißen, J.; et al. Multiple mechanisms disrupt the let-7 microRNA family in neuroblastoma. Nature 2016, 535, 246–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seiler, M.; Yoshimi, A.; Darman, R.; Chan, B.; Keaney, G.; Thomas, M.; Agrawal, A.A.; Caleb, B.; Csibi, A.; Sean, E.; et al. H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat. Med. 2018, 24, 497–504. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, V.G.; Sighel, D.; Zucal, C.; Bonomo, I.; Micaelli, M.; Lolli, G.; Provenzani, A.; Quattrone, A.; Adami, V. Screening Approaches for Targeting Ribonucleoprotein Complexes: A New Dimension for Drug Discovery. SLAS Discov. 2019, 24, 314–331. [Google Scholar] [CrossRef] [PubMed]
- Consortium, T.U. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2018, 47, D506–D515. [Google Scholar] [CrossRef] [Green Version]
- Tym, J.E.; Mitsopoulos, C.; Coker, E.A.; Razaz, P.; Schierz, A.C.; Antolin, A.A.; Al-Lazikani, B. canSAR: An updated cancer research and drug discovery knowledgebase. Nucleic Acids Res. 2015, 44, D938–D943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, S.; Glaß, M.; Singh, A.K.; Haase, J.; Bley, N.; Fuchs, T.; Lederer, M.; Dahl, A.; Huang, H.; Chen, J.; et al. IGF2BP1 promotes SRF-dependent transcription in cancer in a m6A- and miRNA-dependent manner. Nucleic Acids Res. 2019, 47, 375–390. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bell, J.L.; Hagemann, S.; Holien, J.K.; Liu, T.; Nagy, Z.; Schulte, J.H.; Misiak, D.; Hüttelmaier, S. Identification of RNA-Binding Proteins as Targetable Putative Oncogenes in Neuroblastoma. Int. J. Mol. Sci. 2020, 21, 5098. https://doi.org/10.3390/ijms21145098
Bell JL, Hagemann S, Holien JK, Liu T, Nagy Z, Schulte JH, Misiak D, Hüttelmaier S. Identification of RNA-Binding Proteins as Targetable Putative Oncogenes in Neuroblastoma. International Journal of Molecular Sciences. 2020; 21(14):5098. https://doi.org/10.3390/ijms21145098
Chicago/Turabian StyleBell, Jessica L., Sven Hagemann, Jessica K. Holien, Tao Liu, Zsuzsanna Nagy, Johannes H. Schulte, Danny Misiak, and Stefan Hüttelmaier. 2020. "Identification of RNA-Binding Proteins as Targetable Putative Oncogenes in Neuroblastoma" International Journal of Molecular Sciences 21, no. 14: 5098. https://doi.org/10.3390/ijms21145098
APA StyleBell, J. L., Hagemann, S., Holien, J. K., Liu, T., Nagy, Z., Schulte, J. H., Misiak, D., & Hüttelmaier, S. (2020). Identification of RNA-Binding Proteins as Targetable Putative Oncogenes in Neuroblastoma. International Journal of Molecular Sciences, 21(14), 5098. https://doi.org/10.3390/ijms21145098