Cbl Negatively Regulates NLRP3 Inflammasome Activation through GLUT1-Dependent Glycolysis Inhibition



 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
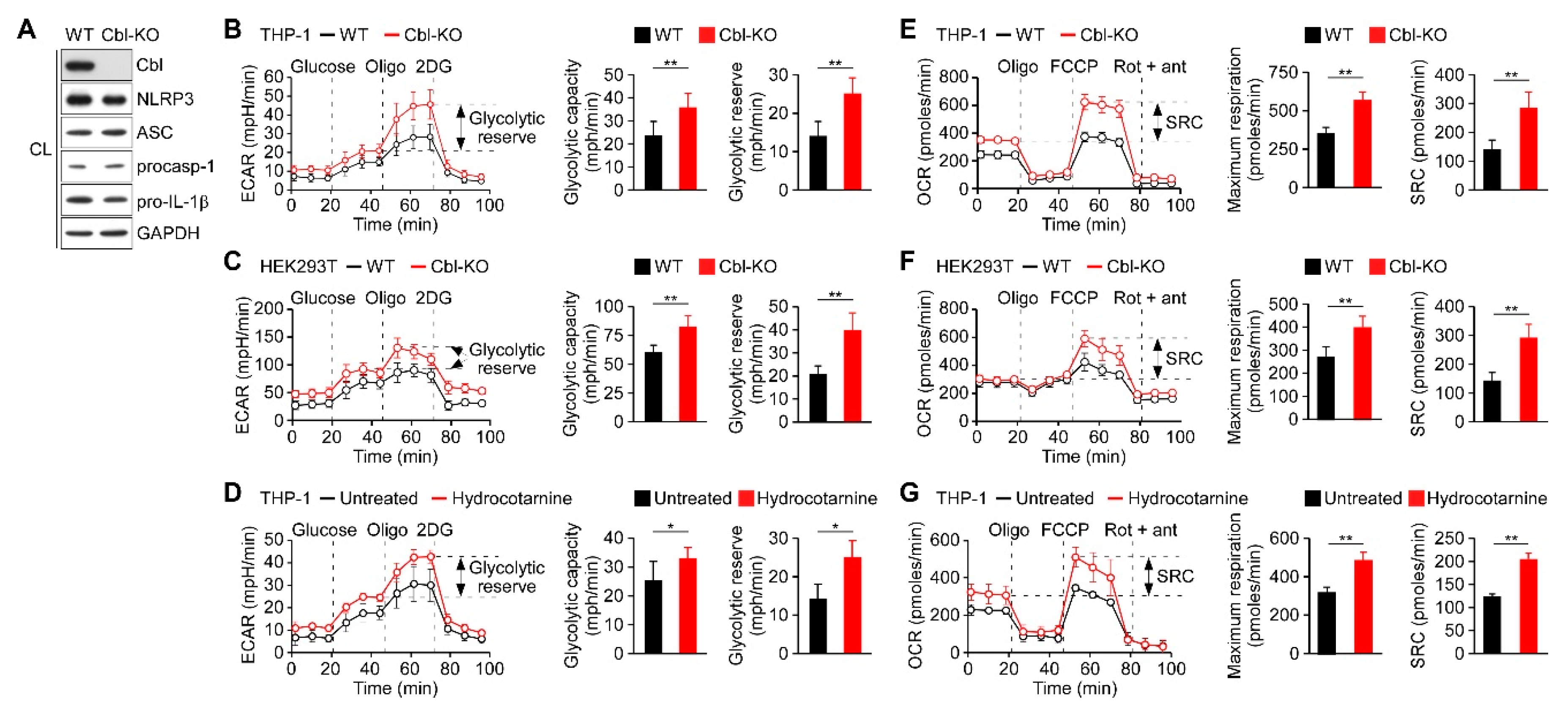
2.1. Cbl Inhibition Increased the Rate of Glycolysis and Oxidative Phosphorylation
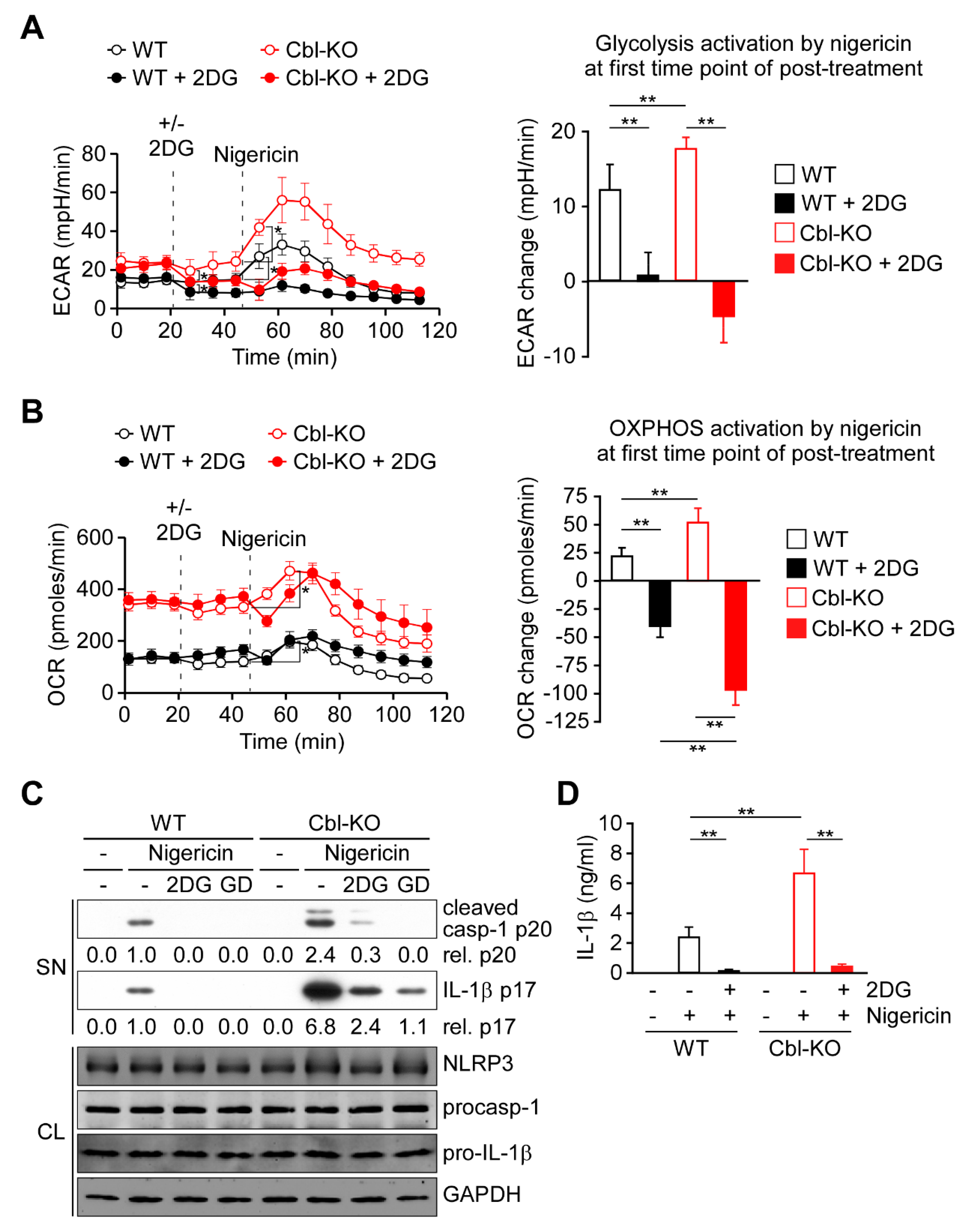
2.2. Glycolysis Upregulation by Cbl Inhibition Enhanced NLRP3 Inflammasome Activation
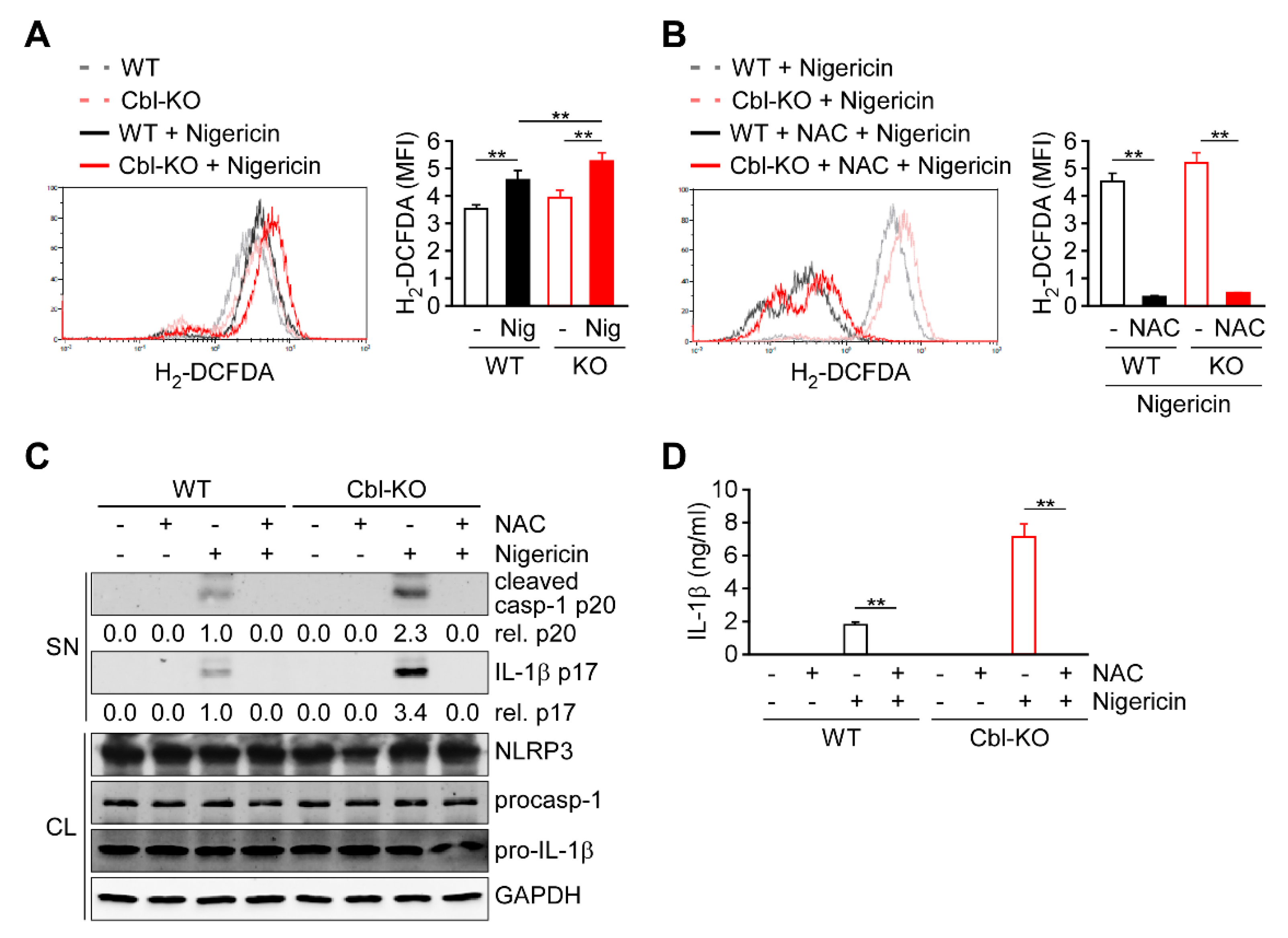
2.3. Regulation of Cellular ROS Production by Cbl Enhanced NLRP3 Inflammasome Activation
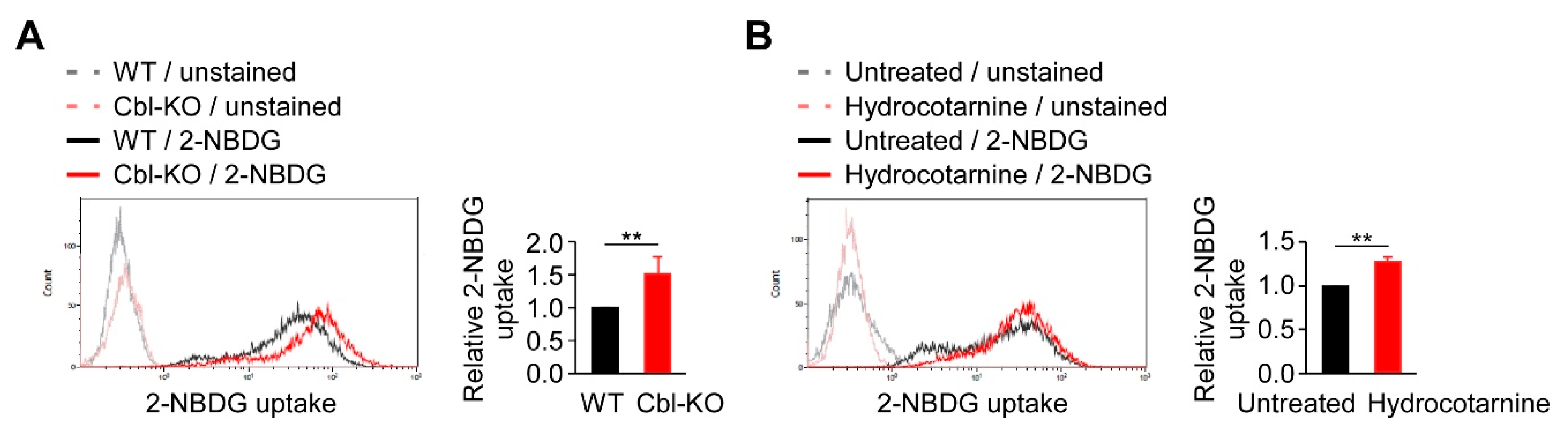
2.4. Inhibition of Cbl Increased Cellular Glucose Uptake
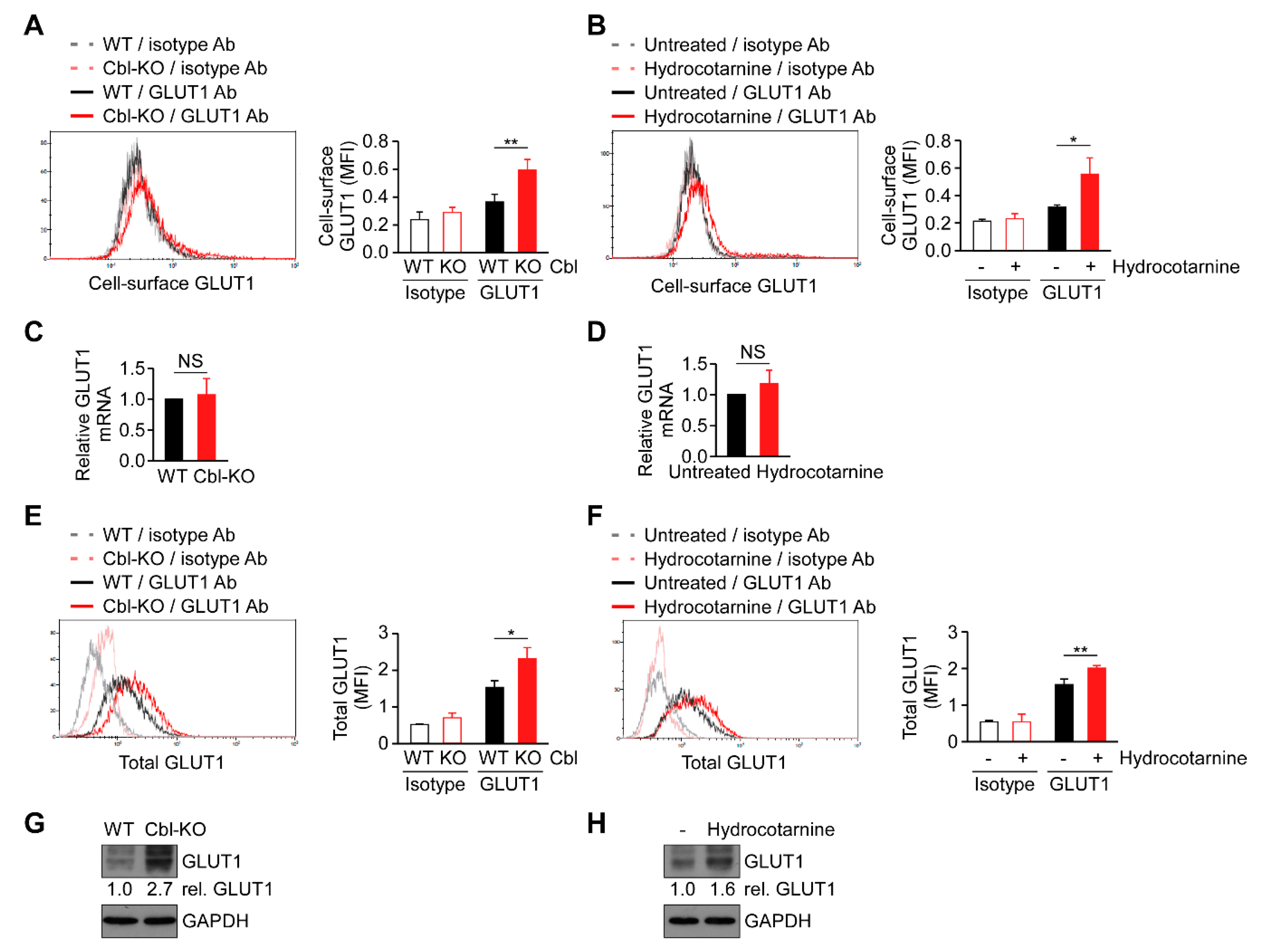
2.5. Surface Expression of GLUT1 Protein Was Post-Transcriptionally Regulated by Cbl
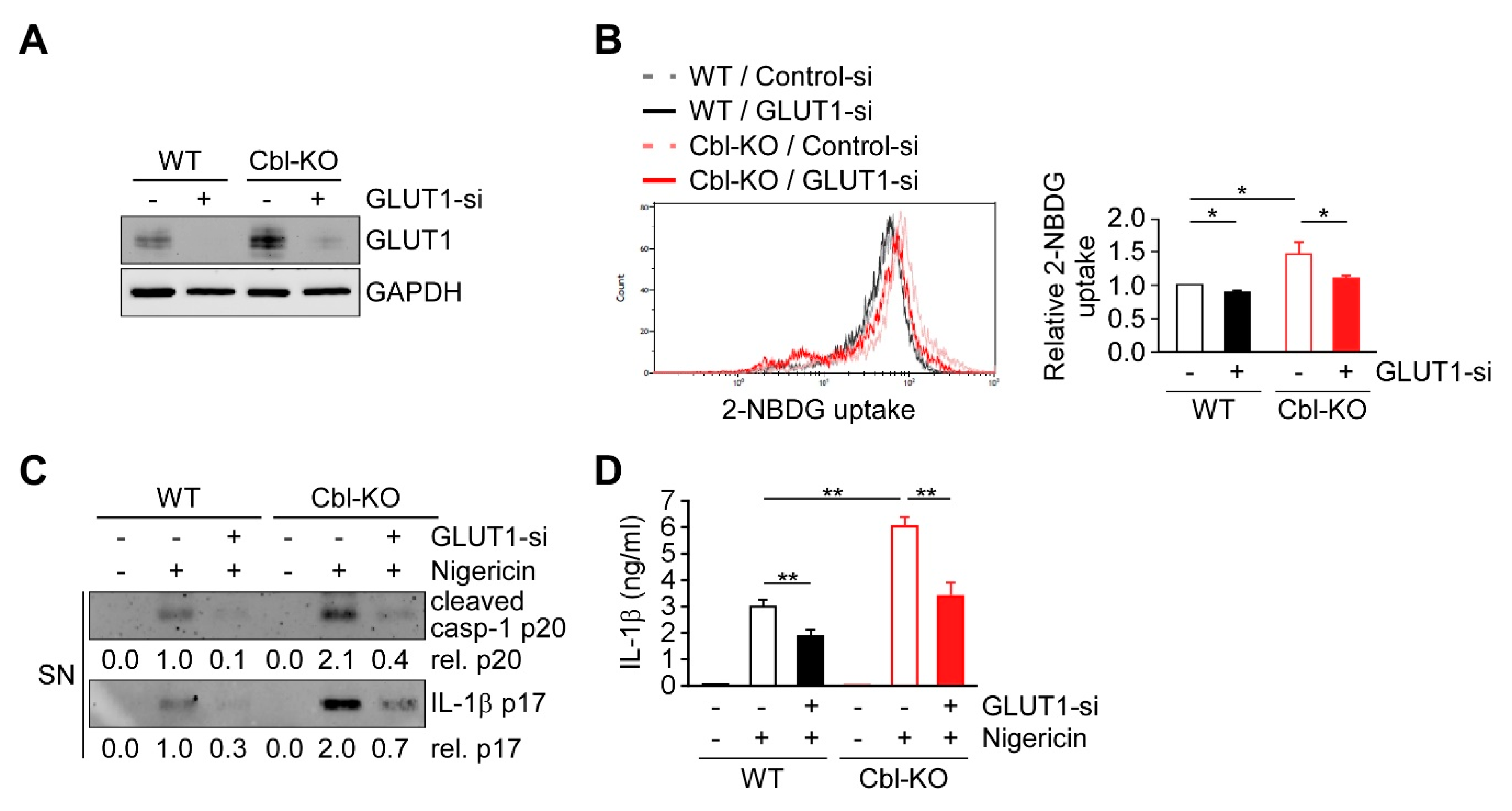
2.6. Cbl Regulation of GLUT1 Modulated Cellular Glucose Uptake and NLRP3 Inflammasome Activation
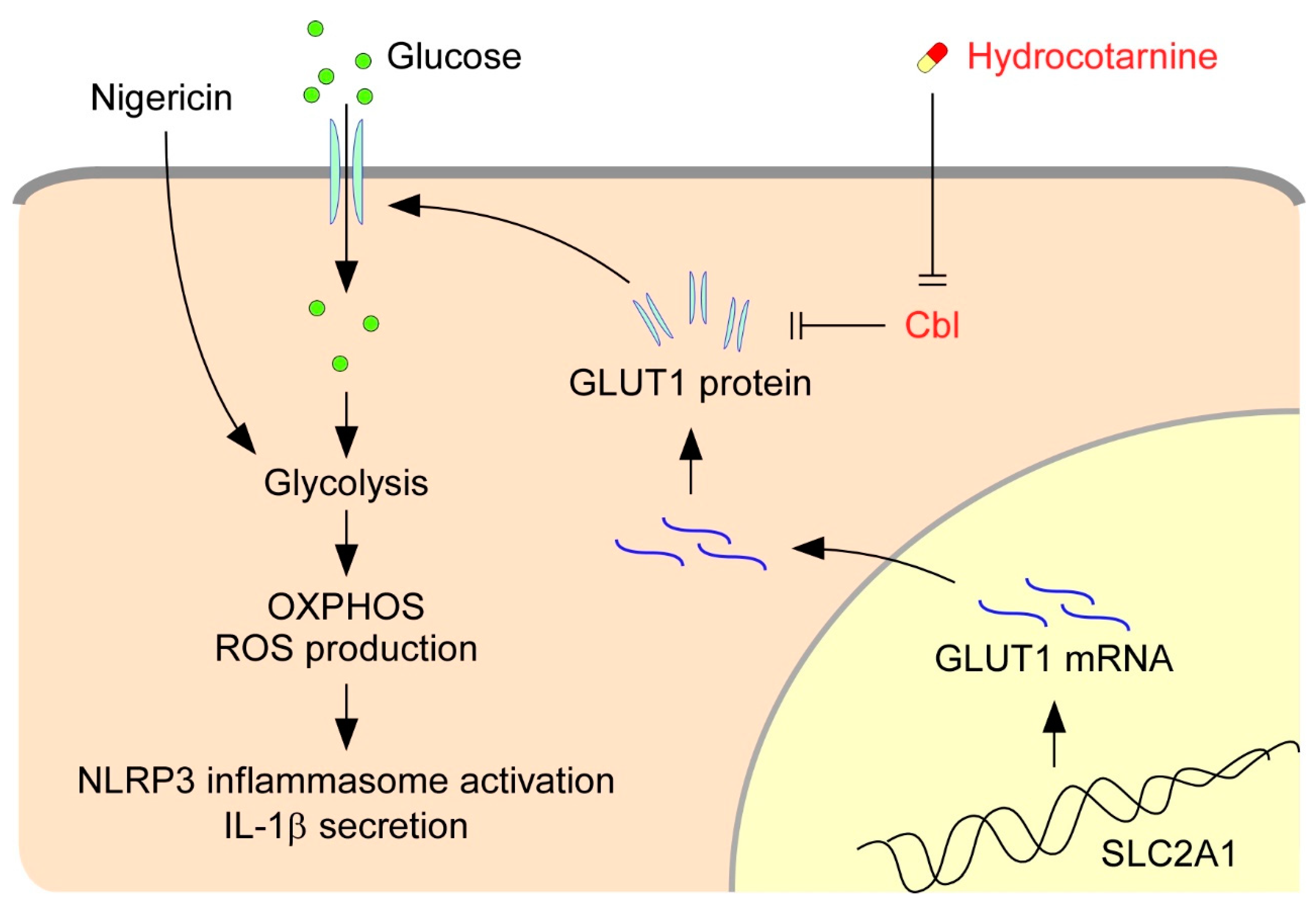
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Seahorse Analysis
4.3. Immunoblot Analysis
4.4. Glucose Uptake Assay
4.5. Assessment of GLUT1 Expression by Flow Cytometry
4.6. Quantitative RT-PCR
4.7. RNA Interference
4.8. IL-1β Enzyme-Linked Immunosorbent Assay
4.9. Cellular ROS Production Assay
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| NLRP3 | nod-like receptor 3 |
| ASC | apoptosis associated speck-like protein containing a CARD |
| GLUT1 | glucose transporter 1 |
| PAMPs | pathogen-associated molecule patterns |
| DAMPs | damage-associated molecular patterns |
| ROS | reactive oxygen species |
| OXPHOS | oxidative phosphorylation |
| ECAR | extracellular acidification rate |
| OCR | oxygen consumption rate |
| SRC | spare respiratory capacity |
References
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.; Magupalli, V.G.; Ruan, J.; Yin, Q.; Atianand, M.K.; Vos, M.R.; Schroder, G.F.; Fitzgerald, K.A.; Wu, H.; Egelman, E.H. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 2014, 156, 1193–1206. [Google Scholar] [CrossRef] [Green Version]
- Munoz-Planillo, R.; Kuffa, P.; Martinez-Colon, G.; Smith, B.L.; Rajendiran, T.M.; Nunez, G. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anand, P.K.; Malireddi, R.K.; Kanneganti, T.D. Role of the nlrp3 inflammasome in microbial infection. Front. Microbiol. 2011, 2, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.M.; Kim, J.J.; Kim, H.J.; Shong, M.; Ku, B.J.; Jo, E.K. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes 2013, 62, 194–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Nakahira, K.; Dalli, J.; Siempos, I.I.; Norris, P.C.; Colas, R.A.; Moon, J.S.; Shinohara, M.; Hisata, S.; Howrylak, J.A.; et al. NLRP3 Inflammasome Deficiency Protects against Microbial Sepsis via Increased Lipoxin B4 Synthesis. Am. J. Respir. Crit. Care Med. 2017, 196, 713–726. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Petrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef] [Green Version]
- Grebe, A.; Hoss, F.; Latz, E. NLRP3 Inflammasome and the IL-1 Pathway in Atherosclerosis. Circ. Res. 2018, 122, 1722–1740. [Google Scholar] [CrossRef]
- Mathews, R.J.; Robinson, J.I.; Battellino, M.; Wong, C.; Taylor, J.C.; Biologics in Rheumatoid Arthritis, G.; Genomics Study, S.; Eyre, S.; Churchman, S.M.; Wilson, A.G.; et al. Evidence of NLRP3-inflammasome activation in rheumatoid arthritis (RA); genetic variants within the NLRP3-inflammasome complex in relation to susceptibility to RA and response to anti-TNF treatment. Ann. Rheum. Dis. 2014, 73, 1202–1210. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Cordero, M.D.; Alcocer-Gomez, E.; Ryffel, B. Gain of function mutation and inflammasome driven diseases in human and mouse models. J. Autoimmun. 2018, 91, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Moossavi, M.; Parsamanesh, N.; Bahrami, A.; Atkin, S.L.; Sahebkar, A. Role of the NLRP3 inflammasome in cancer. Mol. Cancer 2018, 17, 158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz, C.M.; Rinna, A.; Forman, H.J.; Ventura, A.L.; Persechini, P.M.; Ojcius, D.M. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J. Biol. Chem. 2007, 282, 2871–2879. [Google Scholar] [CrossRef] [Green Version]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Ragheb, K.; Lawler, G.; Sturgis, J.; Rajwa, B.; Melendez, J.A.; Robinson, J.P. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J. Biol. Chem. 2003, 278, 8516–8525. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.S.; Cobessi, D.; Tung, E.Y.; Berry, E.A. Binding of the respiratory chain inhibitor antimycin to the mitochondrial bc1 complex: A new crystal structure reveals an altered intramolecular hydrogen-bonding pattern. J. Mol. Biol. 2005, 351, 573–597. [Google Scholar] [CrossRef] [Green Version]
- Heid, M.E.; Keyel, P.A.; Kamga, C.; Shiva, S.; Watkins, S.C.; Salter, R.D. Mitochondrial reactive oxygen species induces NLRP3-dependent lysosomal damage and inflammasome activation. J. Immunol. 2013, 191, 5230–5238. [Google Scholar] [CrossRef] [Green Version]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Z.; Liang, S.; Sanchez-Lopez, E.; He, F.; Shalapour, S.; Lin, X.J.; Wong, J.; Ding, S.; Seki, E.; Schnabl, B.; et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 2018, 560, 198–203. [Google Scholar] [CrossRef]
- Moon, J.S.; Hisata, S.; Park, M.A.; DeNicola, G.M.; Ryter, S.W.; Nakahira, K.; Choi, A.M.K. mTORC1-Induced HK1-Dependent Glycolysis Regulates NLRP3 Inflammasome Activation. Cell Rep. 2015, 12, 102–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, M.; Yu, Y.; Kang, R.; Zhu, S.; Yang, L.; Zeng, L.; Sun, X.; Yang, M.; Billiar, T.R.; Wang, H.; et al. PKM2-dependent glycolysis promotes NLRP3 and AIM2 inflammasome activation. Nat. Commun. 2016, 7, 13280. [Google Scholar] [CrossRef] [PubMed]
- Mohapatra, B.; Ahmad, G.; Nadeau, S.; Zutshi, N.; An, W.; Scheffe, S.; Dong, L.; Feng, D.; Goetz, B.; Arya, P.; et al. Protein tyrosine kinase regulation by ubiquitination: Critical roles of Cbl-family ubiquitin ligases. Biochim. Biophys. Acta 2013, 1833, 122–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, I.C.; Yuan, S.N.; OuYang, C.N.; Lin, H.C.; Huang, K.Y.; Chen, Y.J.; Chung, A.K.; Chu, C.L.; Ojcius, D.M.; Chang, Y.S.; et al. Src-family kinase-Cbl axis negatively regulates NLRP3 inflammasome activation. Cell Death Dis. 2018, 9, 1109. [Google Scholar] [CrossRef]
- Chung, I.C.; OuYang, C.N.; Yuan, S.N.; Li, H.P.; Chen, J.T.; Shieh, H.R.; Chen, Y.J.; Ojcius, D.M.; Chu, C.L.; Yu, J.S.; et al. Pyk2 activates the NLRP3 inflammasome by directly phosphorylating ASC and contributes to inflammasome-dependent peritonitis. Sci. Rep. 2016, 6, 36214. [Google Scholar] [CrossRef]
- Hara, H.; Tsuchiya, K.; Kawamura, I.; Fang, R.; Hernandez-Cuellar, E.; Shen, Y.; Mizuguchi, J.; Schweighoffer, E.; Tybulewicz, V.; Mitsuyama, M. Phosphorylation of the adaptor ASC acts as a molecular switch that controls the formation of speck-like aggregates and inflammasome activity. Nat. Immunol. 2013, 14, 1247–1255. [Google Scholar] [CrossRef] [Green Version]
- Molero, J.C.; Jensen, T.E.; Withers, P.C.; Couzens, M.; Herzog, H.; Thien, C.B.; Langdon, W.Y.; Walder, K.; Murphy, M.A.; Bowtell, D.D.; et al. c-Cbl-deficient mice have reduced adiposity, higher energy expenditure, and improved peripheral insulin action. J. Clin. Investig. 2004, 114, 1326–1333. [Google Scholar] [CrossRef] [Green Version]
- Dror, E.; Dalmas, E.; Meier, D.T.; Wueest, S.; Thevenet, J.; Thienel, C.; Timper, K.; Nordmann, T.M.; Traub, S.; Schulze, F.; et al. Postprandial macrophage-derived IL-1beta stimulates insulin, and both synergistically promote glucose disposal and inflammation. Nat. Immunol. 2017, 18, 283–292. [Google Scholar] [CrossRef] [Green Version]
- Freemerman, A.J.; Johnson, A.R.; Sacks, G.N.; Milner, J.J.; Kirk, E.L.; Troester, M.A.; Macintyre, A.N.; Goraksha-Hicks, P.; Rathmell, J.C.; Makowski, L. Metabolic reprogramming of macrophages: Glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J. Biol. Chem. 2014, 289, 7884–7896. [Google Scholar] [CrossRef] [Green Version]
- Shirai, T.; Nazarewicz, R.R.; Wallis, B.B.; Yanes, R.E.; Watanabe, R.; Hilhorst, M.; Tian, L.; Harrison, D.G.; Giacomini, J.C.; Assimes, T.L.; et al. The glycolytic enzyme PKM2 bridges metabolic and inflammatory dysfunction in coronary artery disease. J. Exp. Med. 2016, 213, 337–354. [Google Scholar] [CrossRef] [Green Version]
- Moon, J.S.; Nakahira, K.; Chung, K.P.; DeNicola, G.M.; Koo, M.J.; Pabon, M.A.; Rooney, K.T.; Yoon, J.H.; Ryter, S.W.; Stout-Delgado, H.; et al. NOX4-dependent fatty acid oxidation promotes NLRP3 inflammasome activation in macrophages. Nat. Med. 2016, 22, 1002–1012. [Google Scholar] [CrossRef] [PubMed]
- Cader, M.Z.; Boroviak, K.; Zhang, Q.; Assadi, G.; Kempster, S.L.; Sewell, G.W.; Saveljeva, S.; Ashcroft, J.W.; Clare, S.; Mukhopadhyay, S.; et al. C13orf31 (FAMIN) is a central regulator of immunometabolic function. Nat. Immunol. 2016, 17, 1046–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carey, I.M.; Critchley, J.A.; DeWilde, S.; Harris, T.; Hosking, F.J.; Cook, D.G. Risk of Infection in Type 1 and Type 2 Diabetes Compared With the General Population: A Matched Cohort Study. Diabetes Care 2018, 41, 513–521. [Google Scholar] [CrossRef] [Green Version]
- Pearson-Stuttard, J.; Blundell, S.; Harris, T.; Cook, D.G.; Critchley, J. Diabetes and infection: Assessing the association with glycaemic control in population-based studies. Lancet Diabetes Endocrinol. 2016, 4, 148–158. [Google Scholar] [CrossRef]
- Muller, L.M.; Gorter, K.J.; Hak, E.; Goudzwaard, W.L.; Schellevis, F.G.; Hoepelman, A.I.; Rutten, G.E. Increased risk of common infections in patients with type 1 and type 2 diabetes mellitus. Clin. Infect. Dis. 2005, 41, 281–288. [Google Scholar] [CrossRef] [Green Version]
- Raff, A.B.; Kroshinsky, D. Cellulitis: A Review. JAMA 2016, 316, 325–337. [Google Scholar] [CrossRef]
- Gunderson, C.G.; Martinello, R.A. A systematic review of bacteremias in cellulitis and erysipelas. J. Infect. 2012, 64, 148–155. [Google Scholar] [CrossRef]
- Harder, J.; Franchi, L.; Munoz-Planillo, R.; Park, J.H.; Reimer, T.; Nunez, G. Activation of the Nlrp3 inflammasome by Streptococcus pyogenes requires streptolysin O and NF-kappa B activation but proceeds independently of TLR signaling and P2 × 7 receptor. J. Immunol. 2009, 183, 5823–5829. [Google Scholar] [CrossRef] [Green Version]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef]
- Wang, L.J.; Hsu, C.W.; Chen, C.C.; Liang, Y.; Chen, L.C.; Ojcius, D.M.; Tsang, N.M.; Hsueh, C.; Wu, C.C.; Chang, Y.S. Interactome-wide analysis identifies end-binding protein 1 as a crucial component for the speck-like particle formation of activated absence in melanoma 2 (AIM2) inflammasomes. Mol. Cell Proteomics 2012, 11, 1230–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, I.C.; OuYang, C.N.; Yuan, S.N.; Lin, H.C.; Huang, K.Y.; Wu, P.S.; Liu, C.Y.; Tsai, K.J.; Loi, L.K.; Chen, Y.J.; et al. Pretreatment with a Heat-Killed Probiotic Modulates the NLRP3 Inflammasome and Attenuates Colitis-Associated Colorectal Cancer in Mice. Nutrients 2019, 11, 516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, I.C.; Yuan, S.N.; OuYang, C.N.; Hu, S.I.; Lin, H.C.; Huang, K.Y.; Lin, W.N.; Chuang, Y.T.; Chen, Y.J.; Ojcius, D.M.; et al. EFLA 945 restricts AIM2 inflammasome activation by preventing DNA entry for psoriasis treatment. Cytokine 2019, 127, 154951. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, H.-C.; Chen, Y.-J.; Wei, Y.-H.; Chuang, Y.-T.; Hsieh, S.-H.; Hsieh, J.-Y.; Hsieh, Y.-L.; Ojcius, D.M.; Huang, K.-Y.; Chung, I.-C.; et al. Cbl Negatively Regulates NLRP3 Inflammasome Activation through GLUT1-Dependent Glycolysis Inhibition. Int. J. Mol. Sci. 2020, 21, 5104. https://doi.org/10.3390/ijms21145104
Lin H-C, Chen Y-J, Wei Y-H, Chuang Y-T, Hsieh S-H, Hsieh J-Y, Hsieh Y-L, Ojcius DM, Huang K-Y, Chung I-C, et al. Cbl Negatively Regulates NLRP3 Inflammasome Activation through GLUT1-Dependent Glycolysis Inhibition. International Journal of Molecular Sciences. 2020; 21(14):5104. https://doi.org/10.3390/ijms21145104
Chicago/Turabian StyleLin, Hsin-Chung, Yu-Jen Chen, Yau-Huei Wei, Yu-Ting Chuang, Su-Heng Hsieh, Jing-Yu Hsieh, Yi-Lin Hsieh, David M. Ojcius, Kuo-Yang Huang, I.-Che Chung, and et al. 2020. "Cbl Negatively Regulates NLRP3 Inflammasome Activation through GLUT1-Dependent Glycolysis Inhibition" International Journal of Molecular Sciences 21, no. 14: 5104. https://doi.org/10.3390/ijms21145104
APA StyleLin, H. -C., Chen, Y. -J., Wei, Y. -H., Chuang, Y. -T., Hsieh, S. -H., Hsieh, J. -Y., Hsieh, Y. -L., Ojcius, D. M., Huang, K. -Y., Chung, I. -C., Yuan, S. -N., Chang, Y. -S., & Chen, L. -C. (2020). Cbl Negatively Regulates NLRP3 Inflammasome Activation through GLUT1-Dependent Glycolysis Inhibition. International Journal of Molecular Sciences, 21(14), 5104. https://doi.org/10.3390/ijms21145104