Generation of Glioblastoma Patient-Derived Intracranial Xenografts for Preclinical Studies

Abstract
:1. Introduction
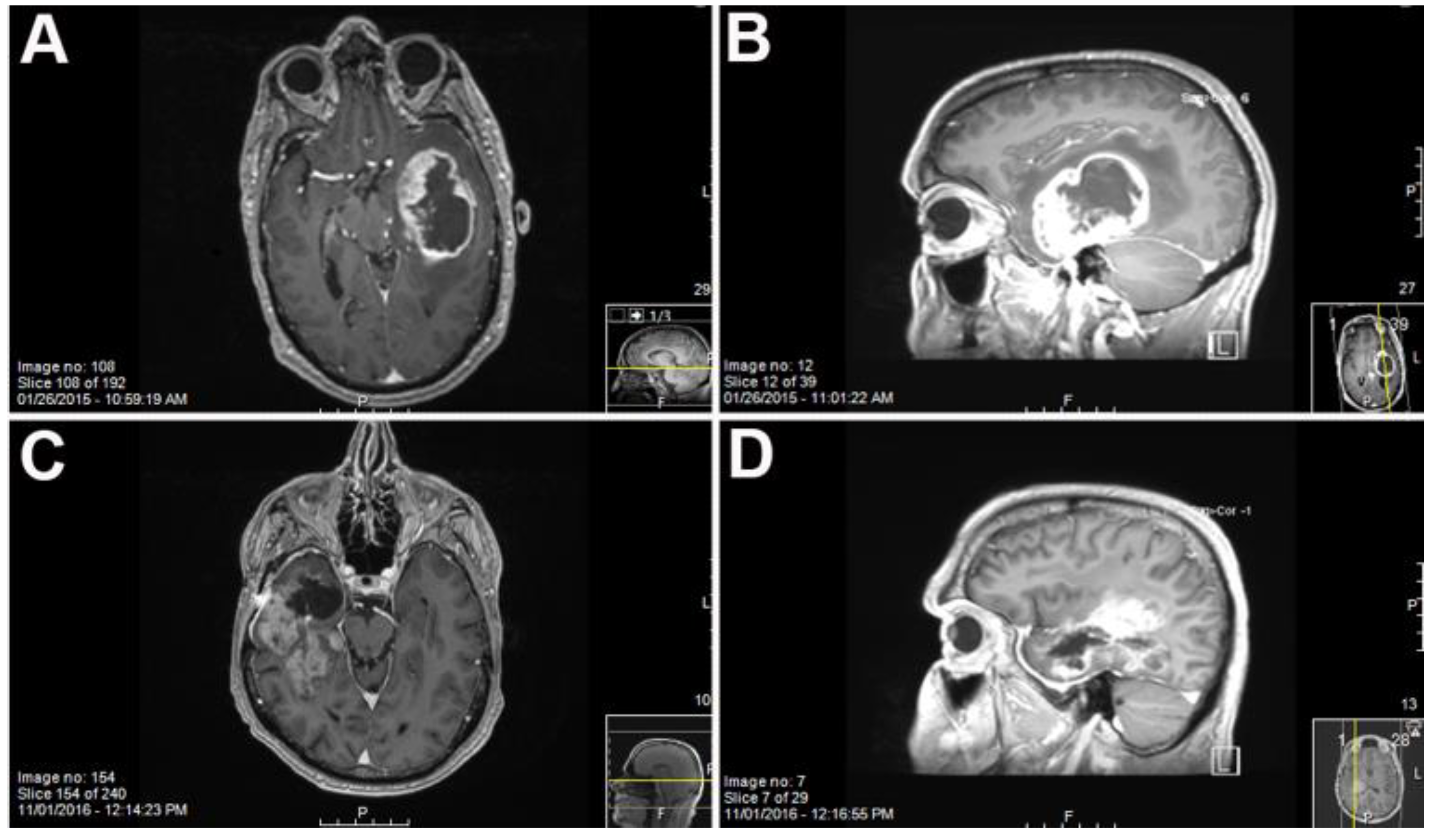
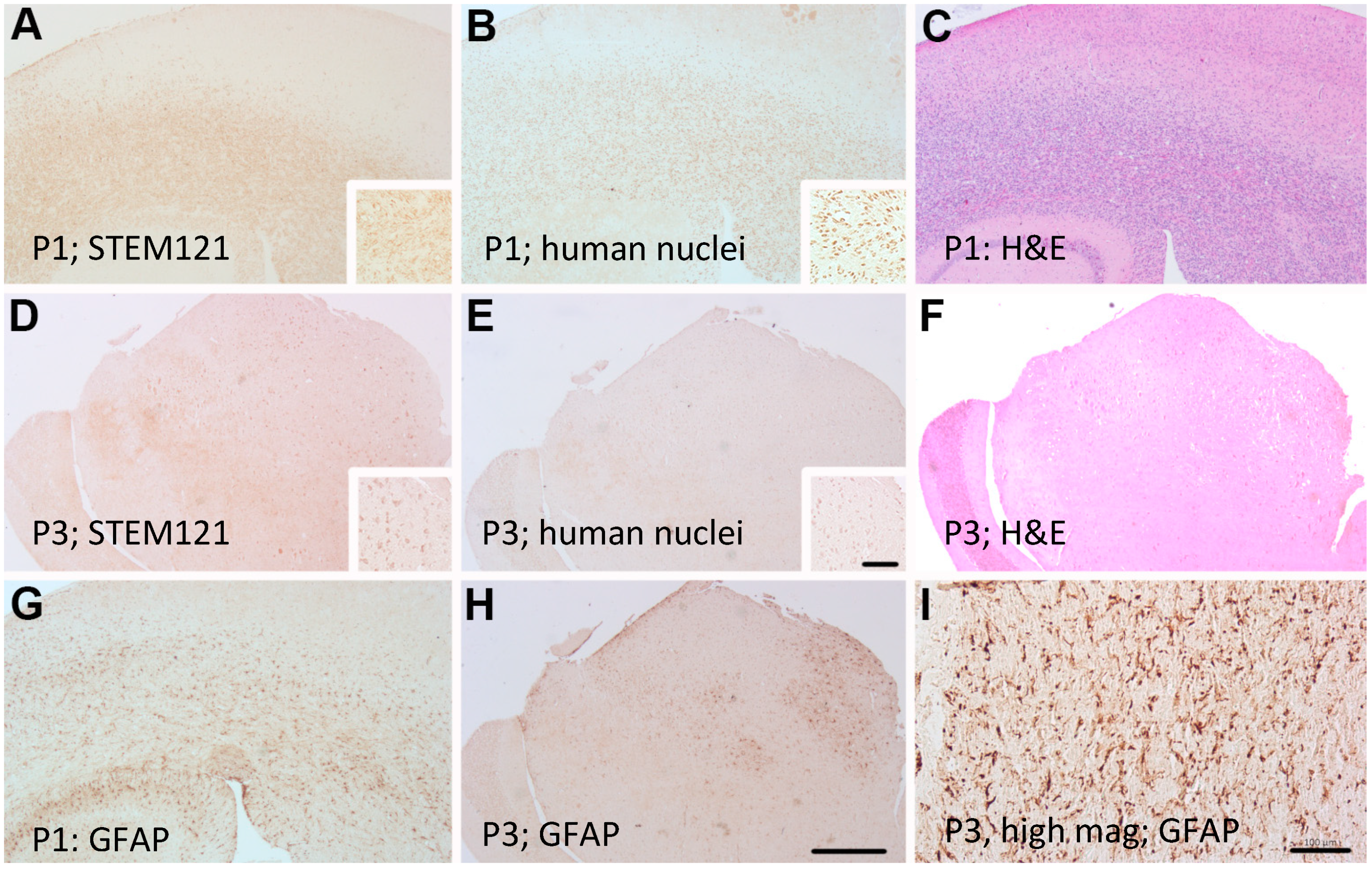
2. Results
Primary GBM and PDX Derivatives Morphology After In Vivo Passaging
3. Materials and Methods
3.1. Tumor Collection
3.2. Xenografting
3.3. Immunohistochemistry and H&E Staining
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.; Gittleman, H.; Fulop, J.; Liu, M.; Blanda, R.; Kromer, C.; Wolinksy, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2011–2015. Neuro Oncol. 2015, 17, iv1–iv62. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; Mclendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma Stem Cells Promote radioresistance by Preferential Activation of the DNA Damage Response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, Y.; Yu, T.; Mckay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A Restricted Cell Population Propagates Glioblastoma Growth after Chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundar, S.J.; Hseih, J.K.; Manjila, S.; Lathia, J.; Sloan, A.E. The role of cancer stem cells in Glioblastoma. Neurosurg. Focus 2014, 37, E6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erasimus, H.; Gobin, M.; Niclou, S.; Van Dyck, E. DNA Repair Mechanisms and their Clinical Impact in Glioblastoma. Mutat. Res. 2016, 769, 19–35. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumor initiating cells. Nature 2004, 18, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Hemmati, H.D.; Nakano, I.; Lazareff, J.A.; Mastermansmith, M.; Geschwind, D.H.; Bronnerfraser, M.; Kornblum, H.I. Cancerous stem cells arise from pediatric brain tumors. Proc. Natl. Acad. Sci. USA 2003, 100, 15178–15183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ignatova, T.N.; Kukekov, V.G.; Laywell, E.D.; Suslov, O.N.; Vrionis, F.D.; Steindler, D.A. Human Cortical Glial Tumors Contain Neural Stem-Like Cells Expressing Astroglial and Neuronal Markers in vitro. Glia 2002, 39, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Kim, J.L.Y.; Wu, A.; Wallace, L.C.; Prager, B.C.; Sanvoranart, T.; Gimple, R.C.; Gimple, R.C.; Wang, X.; Mack, S.C.; et al. Targeting glioma stem cells through combined BMI1 and EXH2 inhibition. Nat. Med. 2017, 23, 1352–1361. [Google Scholar] [CrossRef] [PubMed]
- Vargo-Gogola, T.; Rosen, J.M. Modelling Breast Cancer: One Size does not Fit All. Nat. Rev. Cancer 2007, 7, 659–672. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kotliarova, S.; Kotliarov, Y.; Li, A.; Su, Q.; Donin, N.M.; Pastorino, S.; Purow, B.W.; Christopher, N.; Zhang, W.; et al. Tumor Stem Cells Derived from Glioblastomas cultured in bFGF and EGF more Closely Mirror the Phenotype and Genotype of Primary Tumors than do Serum-Cultured Cell Lines. Cancer Cell 2006, 9, 391–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Y.; Elahi, A.; Denley, R.C.; Rao, P.H.; Brennan, M.F.; Jhanwar, S.C. Molecular Characterization of Permanent Cell Lines from Primary, Metastatic and Recurrent Malignant Peripheral Nerve Sheath Tumors (MPNST) with Underlying Neurofibromatosis-1. Anticancer Res. 2009, 29, 1255–1262. [Google Scholar]
- Gazdar, A.F.; Girard, L.; Lockwood, W.W.; Lam, W.L.; Minna, J.D. Lung Cancer Cell Lines as Tools for Biomedical Discovery and Research. J. Natl. Cancer Inst. 2010, 102, 1310–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, P.; Fillmore, C.M.; Jiang, G.; Shapira, S.; Tao, K.; Kuperwasser, C.; Lander, E.S. Stochastic State Transitions Give Rise to Phenotypic equilibrium in Populations of Cancer Cells. Cell 2011, 146, 633–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassidy, J.W.; Caldas, C.; Bruna, A. Maintaining Tumor Heterogeneity in Patient-Derived Tumor Xenografts. Cancer Res. 2015, 75, 2963–2968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, X.; Chen, S.; Guo, Y.; Li, W.; Qi, X.; Yang, H.; Xiao, S.; Fang, G.; Hu, J.; Wen, C.; et al. Establishment and Evaluation of Four Different Types of Patient-Derived Xenograft Models. Cancer Cell Int. 2017, 17, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- William, D.; Mullins, C.S.; Schneider, B.; Orthmann, A.; Lamp, N.; Krohn, M.; Hoffmann, A.; Classen, C.; Linnebacher, M. Optimized creation of glioblastoma patient derived xenografts for the use in preclinical studies. J. Transl. Med. 2017, 15, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piaskowski, S.; Bienkowski, M.; Stoczynska-Fidelus, E.; Strawski, R.; Sieruta, M.; Szybka, M.; Papierz, W.; Wolanczyk, M.; Jaskolski, D.J.; Liberski, P.P.; et al. Glioma cells showing IDH1 mutation cannot be propagated in standard culture conditions. Br. J. Cancer 2011, 104, 968. [Google Scholar] [CrossRef] [PubMed]
- Sternberger, L.A.; Sternberger, N.H. The Unlabeled Antibody Method: Comparison of Peroxidase-Anti-Peroxidase with Avidin-Biotin complex by a New Method of Quantification. J. Histochem. Cytochem. 1986, 34, 599–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case | Patient Age at Diagnosis, Gender | Molecular Status of Patient Tumor (If Known) | Survival: Original PDX (in Days Post Inoculation) | Survival 2nd Generation PDX (in Days Post Inoculation) | Survival 3rd Generation PDX (in Days Post Inoculation) | Percent Decrease in Time to Moribund (from Original to 3rd Passage) |
|---|---|---|---|---|---|---|
| 1711 | 75, male | IDH1wt | 163 | 107 | 118 | 28% |
| 1768 | 51, male | IDH1wt | 131 | 130 | 102 | 22% |
| 1783 | 75, male | N/A | 138 | 93 | 72 | 48% |
| 1786 | 67, male | IDH1wt | 93 | 60 | 53 | 43% |
| 1892 | 63, female | N/A | 126 | 143 | 109 | 13% |
| 1914 | N/A | 112 | 158 | 100 | 11% | |
| 1918 | 63, female | IDH1wt | 90 | 152 | 50 | 44% |
| 1919 | 53, male | IDH1wt, ATRXwt | 85 | 119 | 135 | - |
| 1949 | N/A | IDH1wt | 149 | 122 | 109 | 27% |
| 1951 | N/A | N/A | 148 | 86 | 102 | 31% |
| 1953 | N/A | IDH1wt | 166 | 142 | 109 | 34% |
| 1959 | N/A | N/A | 94 | 49 | 34 | 64% |
| 1962 | N/A | IDH1wt | 154 | 51 | 86 | 44% |
| 1963 | N/A | N/A | 154 | 89 | 109 | 30% |
| 1997 | N/A | N/A | 90 | 107 | 96 | - |
| 2014 | N/A | N/A | 88 | 115 | 112 | - |
| 2025 | N/A | N/A | 156 | 120 | 85 | 45% |
| 2033 | N/A | N/A | 151 | 131 | 106 | 30% |
| 2070 | N/A | IDH1wt | 148 | 125 | 92 | 38% |
| 2072 | N/A | IDH1wt | 167 | 113 | 63 | 62% |
| 2078 | N/A | IDH1wt | 161 | 113 | 108 | 33% |
| 2091 | N/A | IDH1wt | 166 | 110 | 95 | 43% |
| 2095 | N/A | N/A | 154 | 79 | 89 | 42% |
| 2096 | N/A | N/A | 135 | 16 | 106 | 22% |
| 2114 | N/A | IDH1wt | 112 | 110 | 100 | - |
| 2142 | 36, female | IDH1mut, ATRXmut | 154 | 74 | 95 | 38% |
| 2144 | N/A | IDH1wt, ATRXwt | 139 | 113 | 92 | 34% |
| 2152 | 79, male | IDH1wt, ATRXwt | 152 | 118 | 82 | 46% |
| 2188 | N/A | IDH1wt | 115 | 90 | 92 | 20% |
| 2187 | 35, male | N/A | 65 | 97 | 82 | - |
| 2214 | N/A | IDH1wt, ATRXwt | 83 | 97 | 82 | - |
| 2216 | 71, male | IDH1wt, ATRXwt | 155 | 102 | 36 | 77% |
| 2273 | 59, female | IDH1wt, ATRXwt | 42 | 93 | 78 | - |
| 2284 | 64, female | IDH1wt, ATRXwt | 40 | 17 | 99 | - |
| 2302 | 60, male | IDH1wt | 127 | 71 | 85 | 33% |
| 2381 | 67, male | IDH1wt, ATRXwt | 92 | 101 | 71 | 23% |
| 2409 | 57, male | IDH1wt, ATRXwt | 78 | 72 | 71 | 9% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kerstetter-Fogle, A.E.; Harris, P.L.R.; Brady-Kalnay, S.M.; Sloan, A.E. Generation of Glioblastoma Patient-Derived Intracranial Xenografts for Preclinical Studies. Int. J. Mol. Sci. 2020, 21, 5113. https://doi.org/10.3390/ijms21145113
Kerstetter-Fogle AE, Harris PLR, Brady-Kalnay SM, Sloan AE. Generation of Glioblastoma Patient-Derived Intracranial Xenografts for Preclinical Studies. International Journal of Molecular Sciences. 2020; 21(14):5113. https://doi.org/10.3390/ijms21145113
Chicago/Turabian StyleKerstetter-Fogle, Amber E., Peggy L. R. Harris, Susann M. Brady-Kalnay, and Andrew E. Sloan. 2020. "Generation of Glioblastoma Patient-Derived Intracranial Xenografts for Preclinical Studies" International Journal of Molecular Sciences 21, no. 14: 5113. https://doi.org/10.3390/ijms21145113
APA StyleKerstetter-Fogle, A. E., Harris, P. L. R., Brady-Kalnay, S. M., & Sloan, A. E. (2020). Generation of Glioblastoma Patient-Derived Intracranial Xenografts for Preclinical Studies. International Journal of Molecular Sciences, 21(14), 5113. https://doi.org/10.3390/ijms21145113