Herpes Simplex Virus Type 1 Interactions with the Interferon System



Abstract
:1. Introduction
2. Herpes Simplex Virus
Entry, Replication, and Release of HSV-1
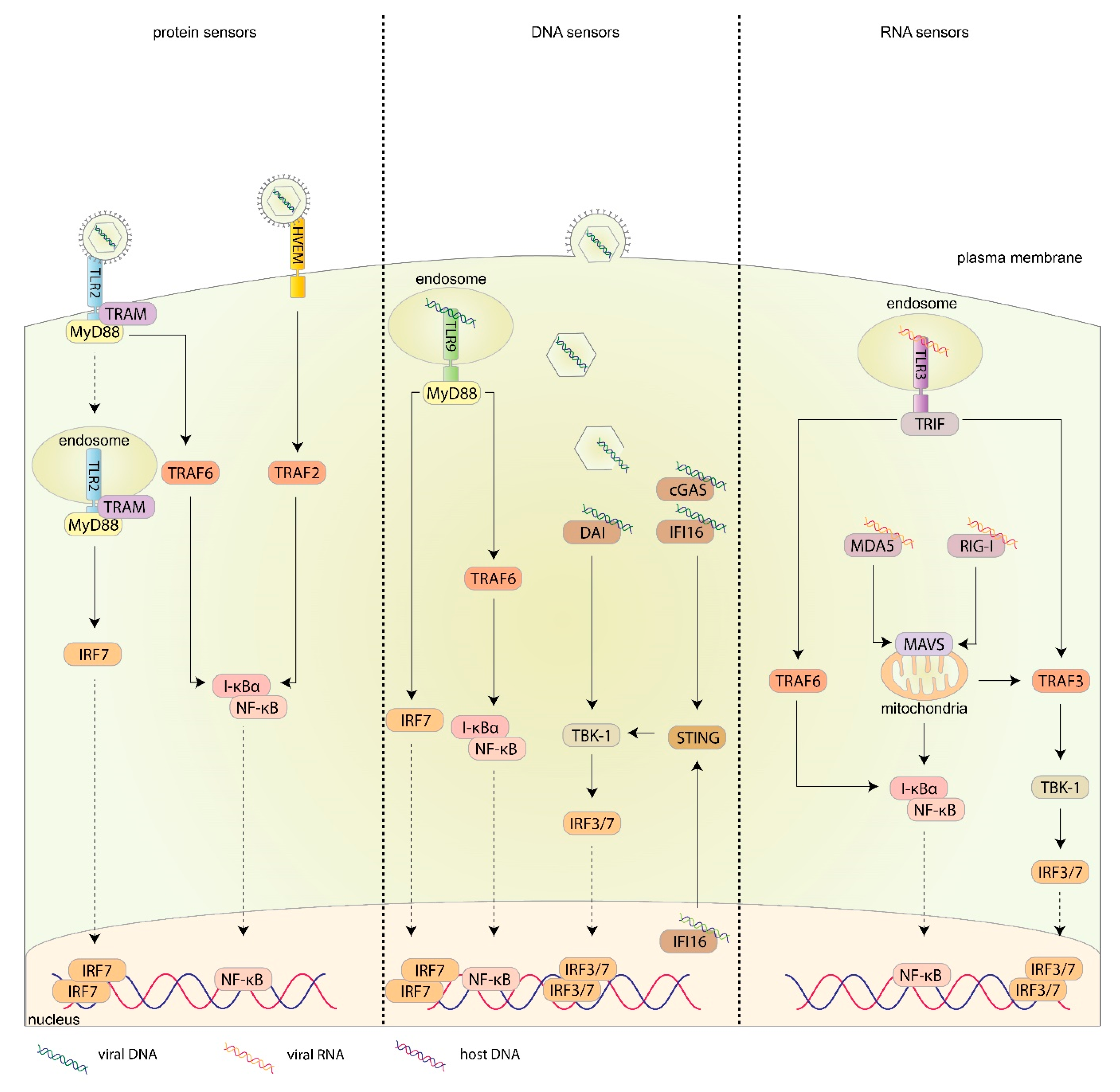
3. Recognition of HSV-1 by the Innate Immune System
3.1. Introduction
3.2. Sensing Viral Attachment and Fusion
3.3. Sensing Viral DNA
3.4. Sensing Viral RNA
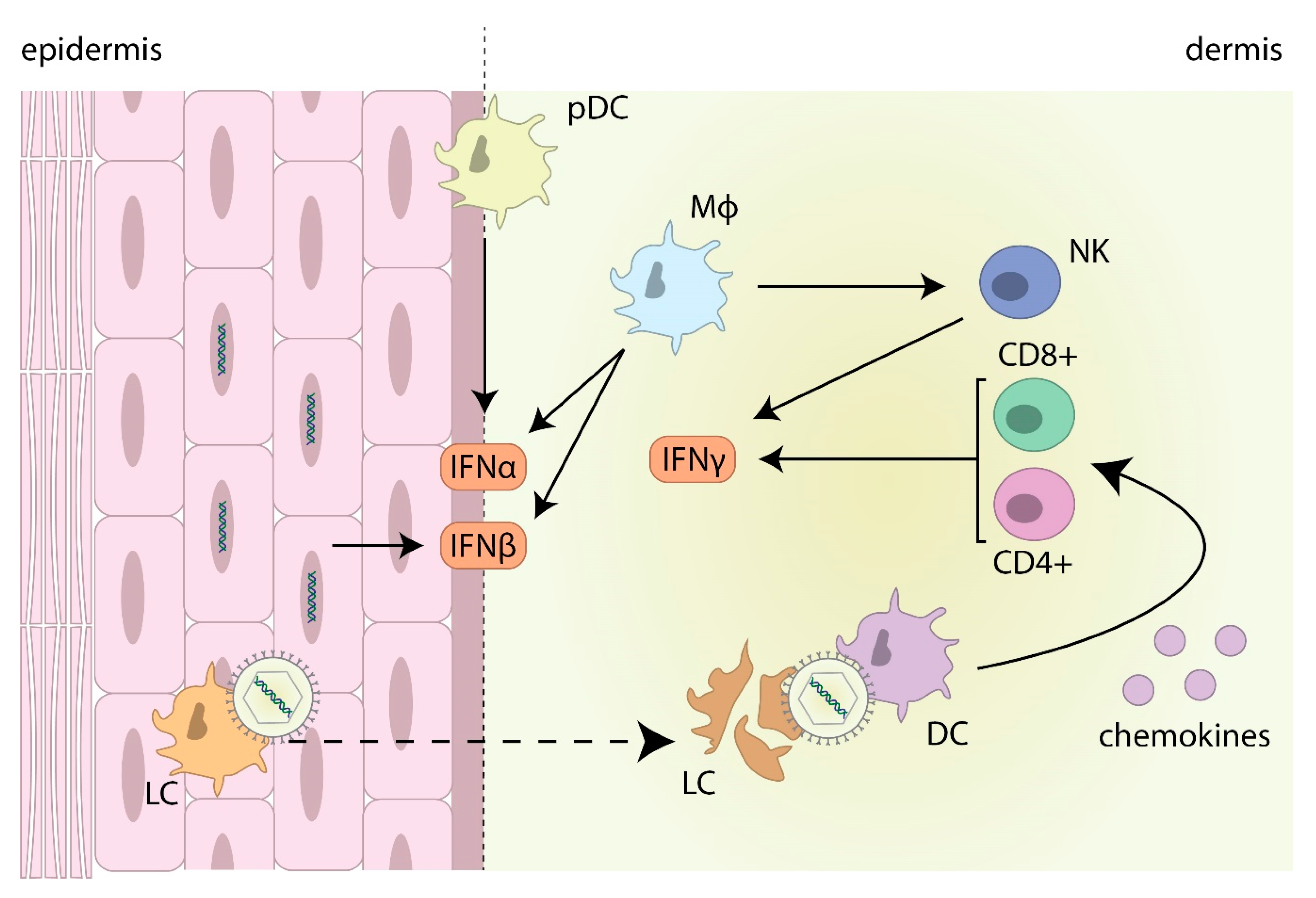
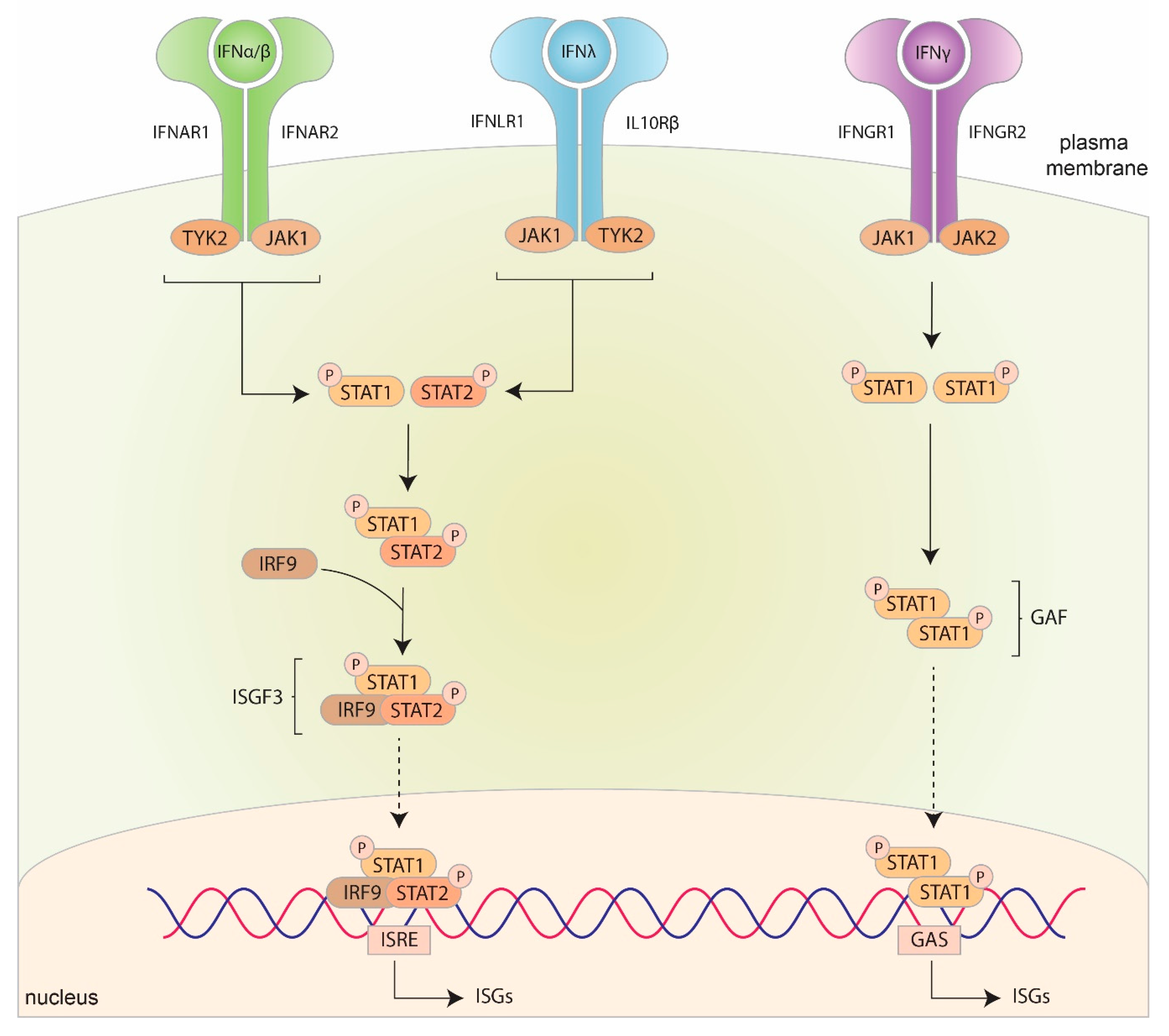
4. The Interferon Response against HSV
4.1. IFN Production in Herpes Lesions
4.2. Canonical vs. Non-Canonical IFN Pathways
4.3. ISG Induction in Response to HSV
4.4. Role of IFN in Controlling HSV-1 Infections
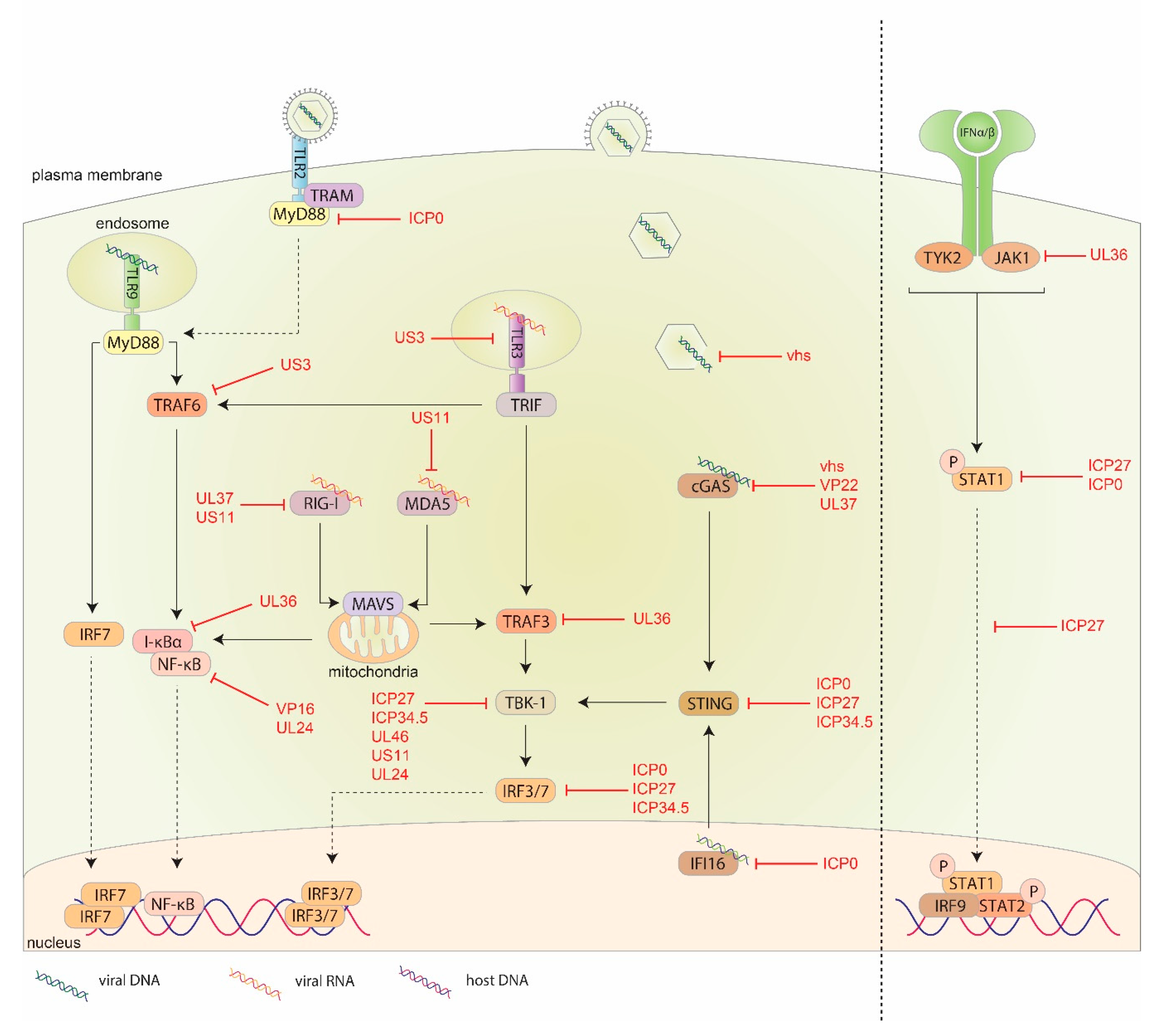
5. HSV-1 Evasion Response against the Innate Immune System
5.1. HSV-1 pUS3
5.2. HSV-1 Virion Host Shutoff Protein (vhs)
5.3. HSV-1 Viral Protein 16 (VP16)
5.4. HSV-1 ICP27
5.5. HSV-1 ICP0
5.6. HSV-1 ICP34.5
5.7. Other HSV-1 Proteins Involved in Suppressing IFN
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Bclaf1 | bcl-2-associated transcription factor |
| cGAS | Cyclic guanosine monophosphate-adenosine monophosphate synthase |
| CXCL10 | Interferon gamma induced protein 10 |
| DAI | DNA-dependent activator of IFN-regulatory factors |
| DAMP | Damage associated molecular pattern |
| DC | Dendritic cell |
| dsDNA | Double stranded DNA |
| dsRNA | Double stranded RNA |
| DRG | Dorsal root ganglia |
| GAF | Gamma interferon activation factor |
| GAS | Gamma interferon activated sites |
| gB | HSV-1 envelope glycoprotein B |
| gC | HSV-1 envelope glycoprotein C |
| gD | HSV-1 envelope glycoprotein D |
| gH/gL | HSV-1 envelope glycoprotein H/L heterodimer |
| HSE | Herpes encephalitis |
| HSV | Herpes simplex virus |
| HVEM | Herpes virus entry mediator |
| ICP0 | HSV-1 infected cell protein 0 |
| ICP27 | HSV-1 infected cell protein 27 |
| ICP34.5 | HSV-1 infected cell protein 34.5 |
| IFI16 | Interferon inducible protein 16 |
| IL | Interleukin |
| IFN | Interferon |
| IFNAR | Interferon alpha receptor |
| IFNGR | Interferon gamma receptor |
| IFNLR | Interferon lambda receptor |
| ISGF3 | Interferon-stimulated gene factor 3 |
| ISRE | Interferon-stimulated response element |
| IRF | Interferon regulatory factor |
| ISG | Interferon-stimulated gene |
| JAK | Janus-activated kinase |
| LAT | Latency associated transcript |
| MAVS | Mitochondria antiviral signaling protein |
| MDA5 | Melanoma differentiation-associated protein 5 |
| Mx | Myxovirus resistance protein |
| MyD88 | Myeloid differentiation factor 88 |
| NF-κB | Nuclear factor kappa B |
| NK | Natural killer |
| OAS | 2′,5′-oligoadenylate synthase |
| PAMP | Pathogen associated molecular pattern |
| pDC | Plasmacytoid dendritic cell |
| PRR | Pattern recognition receptor |
| RIG-I | Retinoic acid-inducible gene I |
| RNApol III | RNA polymerase III |
| ssRNA | Single stranded RNA |
| STAT | Signal transducer and activator of transcription |
| STING | Stimulator of interferon genes |
| TBK | TANK-binding kinase |
| TG | Trigeminal ganglia |
| TLR | Toll-like receptor |
| TNF | Tumor necrosis factor |
| TRAF | Tumour necrosis factor associated factor |
| TRAM | TRIF related adaptor molecule |
| TRIF | Toll/IL1 receptor domain containing adaptor inducing interferon beta |
| TYK | Tyrosine kinase |
| pUL13 | HSV-1 protein unique long 13 |
| pUL24 | HSV-1 protein unique long 24 |
| pUL36 | HSV-1 protein unique long 36 |
| pUL37 | HSV-1 protein unique long 37 |
| pUL42 | HSV-1 protein unique long 42 |
| pUL46 | HSV-1 tegument protein UL46 |
| pUS3 | HSV-1 unique short 3 |
| pUS11 | HSV-1 unique short 11 |
| USP | Ubiquitin carboxyl-terminal hydrolase |
| vhs | HSV-1 virion host shutoff protein |
| VP16 | HSV-1 viral protein 16 |
| VP22 | HSV-1 viral protein 22 |
| VZV | Varicella zoster virus |
References
- Sehrawat, S.; Kumar, D.; Rouse, B.T. Herpesviruses: Harmonious pathogens but relevant cofactors in other diseases? Front. Cell. Infect. Microbiol. 2018, 8, 177. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Mobeen, F.; Prakash, T. Comparative genomics of herpesviridae family to look for potential signatures of human infecting strains. Int. J. Genom. 2016, 2016, 9543274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denes, C.E.; Everett, R.D.; Diefenbach, R.J. Tour de herpes: Cycling through the life and biology of HSV-1. In Herpes Simplex Virus: Methods in Molecular Biology, 2nd ed.; Diefenbach, R.J., Fraefel, C., Eds.; Humana: New York, NY, USA, 2020; Volume 2060, pp. 1–30. [Google Scholar]
- Wu, W.; Newcomb, W.W.; Cheng, N.; Aksyuk, A.; Winkler, D.C.; Steven, A.C. Internal proteins of the procapsid and mature capsids of herpes simplex virus 1 mapped by bubblegram imaging. J. Virol. 2016, 90, 5176–5186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laine, R.F.; Albecka, A.; van de Linde, S.; Rees, E.J.; Crump, C.M.; Kaminski, C.F. Structural analysis of herpes simplex virus by optical super-resolution imaging. Nat. Commun. 2015, 6, 5980. [Google Scholar] [CrossRef] [Green Version]
- Miranda-Saksena, M.; Denes, C.E.; Diefenbach, R.J.; Cunningham, A.L. Infection and transport of herpes simplex virus type 1 in neurons: Role of the cytoskeleton. Viruse 2018, 10, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magdaleno-Tapial, J.; Hernandez-Bel, P.; Valenzuela-Onate, C.; Ortiz-Salvador, J.M.; Garcia-Legaz-Martinez, M.; Martinez-Domenech, A.; Perez-Pastor, G.; Esteve-Martinez, A.; Zaragoza-Ninet, V.; Sanchez-Carazo, J.L.; et al. Genital infection with herpes simplex virus type 1 and type 2 in Valencia, Spain: A retrospective observational study. Actas Dermosifiliogr. 2020, 111, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Durukan, D.; Fairley, C.K.; Bradshaw, C.S.; Read, T.R.H.; Druce, J.; Catton, M.; Caly, L.; Chow, E.P.F. Increasing proportion of herpes simplex virus type 1 among women and men diagnosed with first-episode anogenital herpes: A retrospective observational study over 14 years in Melbourne, Australia. Sex. Transm. Infect. 2019, 95, 307–313. [Google Scholar] [CrossRef]
- Ayoub, H.H.; Chemaitelly, H.; Abu-Raddad, L.J. Characterizing the transitioning epidemiology of herpes simplex virus type 1 in the USA: Model-based predictions. BMC. Med. 2019, 17, 57. [Google Scholar] [CrossRef] [PubMed]
- Belshe, R.B.; Leone, P.A.; Bernstein, D.I.; Wald, A.; Levin, M.J.; Stapleton, J.T.; Gorfinkel, I.; Morrow, R.L.; Ewell, M.G.; Stokes-Riner, A.; et al. Efficacy results of a trial of a herpes simplex vaccine. N. Engl. J. Med. 2012, 366, 34–43. [Google Scholar] [CrossRef] [Green Version]
- Johnson, H.M.; Ahmed, C.M. Noncanonical IFN signaling: Mechanistic linkage of genetic and epigenetic events. Mediat. Inflamm. 2016, 2016, 9564814. [Google Scholar] [CrossRef]
- Richter, E.R.; Dias, J.K.; Gilbert, J.E., II; Atherton, S.S. Distribution of herpes simplex virus type 1 and varicella zoster virus in ganglia of the human head and neck. J. Infect. Dis. 2009, 200, 1901–1906. [Google Scholar] [CrossRef] [Green Version]
- Cohen, C.; Corpet, A.; Roubille, S.; Maroui, M.A.; Poccardi, N.; Rousseau, A.; Kleijwegt, C.; Binda, O.; Texier, P.; Sawtell, N.; et al. Promyelocytic leukemia (PML) nuclear bodies (NBs) induce latent/quiescent HSV-1 genomes chromatinization through a PML NB/histone H3.3/H3.3 chaperone axis. PLoS Pathog. 2018, 14, e1007313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denes, C.E.; Miranda-Saksena, M.; Cunningham, A.L.; Diefenbach, R.J. Cytoskeletons in the closet: Subversion in alphaherpesvirus infections. Viruses-Basel 2018, 10, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tormanen, K.; Allen, S.; Mott, K.R.; Ghiasi, H. The latency-associated transcript inhibits apoptosis via downregulation of components of the type I interferon pathway during latent herpes simplex virus 1 ocular infection. J. Virol. 2019, 93, e00103-19. [Google Scholar] [CrossRef] [Green Version]
- Miranda-Saksena, M.; Armati, P.; Boadle, R.A.; Holland, D.J.; Cunningham, A.L. Anterograde transport of herpes simplex virus type 1 in cultured, dissociated human and rat dorsal root ganglion neurons. J. Virol. 2000, 74, 1827–1839. [Google Scholar] [CrossRef] [Green Version]
- Diefenbach, R.J.; Davis, A.; Miranda-Saksena, M.; Fernandez, M.A.; Kelly, B.J.; Jones, C.A.; LaVail, J.H.; Xue, J.; Lai, J.; Cunningham, A.L. The basic domain of herpes simplex virus 1 pUS9 recruits kinesin-1 to facilitate egress from neurons. J. Virol. 2016, 90, 2102–2111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miranda-Saksena, M.; Boadle, R.A.; Diefenbach, R.J.; Cunningham, A.L. Dual role of herpes simplex virus 1 pUS9 in virus anterograde axonal transport and final assembly in growth cones in distal axons. J. Virol. 2015, 90, 2653–2663. [Google Scholar] [CrossRef] [Green Version]
- Ramchandani, M.S.; Jing, L.; Russell, R.M.; Tran, T.; Laing, K.J.; Magaret, A.S.; Selke, S.; Cheng, A.; Huang, M.L.; Xie, H.; et al. Viral genetics modulate orolabial herpes simplex virus type 1 shedding in humans. J. Infect. Dis. 2019, 219, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Looker, K.J.; Magaret, A.S.; May, M.T.; Turner, K.M.E.; Vickerman, P.; Newman, L.M.; Gottlieb, S.L. First estimates of the global and regional incidence of neonatal herpes infection. Lancet Glob. Health 2017, 5, e300–e309. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.Y. Herpes simplex virus encephalitis of childhood: Inborn errors of central nervous system cell-intrinsic immunity. Hum. Genet. 2020. [Google Scholar] [CrossRef]
- Sharthiya, H.; Seng, C.; Van Kuppevelt, T.H.; Tiwari, V.; Fornaro, M. HSV-1 interaction to 3-O-sulfated heparan sulfate in mouse-derived DRG explant and profiles of inflammatory markers during virus infection. J. Neurovirol. 2017, 23, 483–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirtz, L.; Mockel, M.; Knebel-Morsdorf, D. Invasion of herpes simplex virus 1 into murine dermis: Role of nectin-1 and herpesvirus entry mediator as cellular receptors during aging. J. Virol. 2020, 94, e02046-19. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, R.I.; Warner, M.S.; Lum, B.J.; Spear, P.G. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell 1996, 87, 427–436. [Google Scholar] [CrossRef] [Green Version]
- Petermann, P.; Thier, K.; Rahn, E.; Rixon, F.J.; Bloch, W.; Ozcelik, S.; Krummenacher, C.; Barron, M.J.; Dixon, M.J.; Scheu, S.; et al. Entry mechanisms of herpes simplex virus 1 into murine epidermis: Involvement of nectin-1 and herpesvirus entry mediator as cellular receptors. J. Virol. 2015, 89, 262–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petermann, P.; Rahn, E.; Thier, K.; Hsu, M.J.; Rixon, F.J.; Kopp, S.J.; Knebel-Morsdorf, D. Role of nectin-1 and herpesvirus entry mediator as cellular receptors for herpes simplex virus 1 on primary murine dermal fibroblasts. J. Virol. 2015, 89, 9407–9416. [Google Scholar] [CrossRef] [Green Version]
- Simpson, S.A.; Manchak, M.D.; Hager, E.J.; Krummenacher, C.; Whitbeck, J.C.; Levin, M.J.; Freed, C.R.; Wilcox, C.L.; Cohen, G.H.; Eisenberg, R.J.; et al. Nectin-1/HveC mediates herpes simplex virus type 1 entry into primary human sensory neurons and fibroblasts. J. Neurovirol. 2005, 11, 208–218. [Google Scholar] [CrossRef]
- Sayers, C.L.; Elliott, G. Herpes simplex virus 1 enters human keratinocytes by a nectin-1-dependent, rapid plasma membrane fusion pathway that functions at low temperature. J. Virol. 2016, 90, 10379–10389. [Google Scholar] [CrossRef] [Green Version]
- Thier, K.; Petermann, P.; Rahn, E.; Rothamel, D.; Bloch, W.; Knebel-Morsdorf, D. Mechanical barriers restrict invasion of herpes simplex virus 1 into human oral mucosa. J. Virol. 2017, 91, e01295-17. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Tomaras, G.D.; Jegaskanda, S.; Moody, M.A.; Liao, H.X.; Goodman, K.N.; Berman, P.W.; Rerks-Ngarm, S.; Pitisuttithum, P.; Nitayapan, S.; et al. Monoclonal antibodies, derived from humans vaccinated with the RV144 HIV vaccine containing the HVEM binding domain of herpes simplex virus (HSV) glycoprotein D, neutralize HSV infection, mediate antibody-dependent cellular cytotoxicity, and protect mice from ocular challenge with HSV-1. J. Virol. 2017, 91, e00411-17. [Google Scholar] [CrossRef] [Green Version]
- Atanasiu, D.; Saw, W.T.; Lazear, E.; Whitbeck, J.C.; Cairns, T.M.; Lou, H.; Eisenberg, R.J.; Cohen, G.H. Using antibodies and mutants to localize the presumptive gH/gL binding site on herpes simplex virus gD. J. Virol. 2018, 92, e01694-18. [Google Scholar] [CrossRef] [Green Version]
- Nicola, A.V. Herpesvirus entry into host cells mediated by endosomal low pH. Traffic 2016, 17, 965–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicola, A.V.; Hou, J.; Major, E.O.; Straus, S.E. Herpes simplex virus type 1 enters human epidermal keratinocytes, but not neurons, via a pH-dependent endocytic pathway. J. Virol. 2005, 79, 7609–7616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurt-Jones, E.A.; Orzalli, M.H.; Knipe, D.M. Innate immune mechanisms and herpes simplex virus infection and disease. In Cell Biology of Herpes Virus; Osterrieder, K., Ed.; Springer International Publishing: Cham, Switzerland, 2017; Volume 2060, pp. 49–75. [Google Scholar]
- Lee, A.J.; Ashkar, A.A. The dual nature of type I and type II interferons. Front. Immunol. 2018, 9, 2061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moll, H.P.; Maier, T.; Zommer, A.; Lavoie, T.; Brostjan, C. The differential activity of interferon-alpha subtypes is consistent among distinct target genes and cell types. Cytokine 2011, 53, 52–59. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Li, S.; Zhou, Y.; Liu, J.; Francois, S.; Lu, M.; Yang, D.; Dittmer, U.; Sutter, K. Different antiviral effects of IFNalpha subtypes in a mouse model of HBV infection. Sci. Rep. 2017, 7, 334. [Google Scholar] [CrossRef] [Green Version]
- Stanifer, M.L.; Pervolaraki, K.; Boulant, S. Differential regulation of type I and type III interferon signaling. Int. J. Mol. Sci. 2019, 20, 1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Hamming, O.J.; Ank, N.; Paludan, S.R.; Nielsen, A.L.; Hartmann, R. Type III interferon (IFN) induces a type I IFN-like response in a restricted subset of cells through signaling pathways involving both the Jak-STAT pathway and the mitogen-activated protein kinases. J. Virol. 2007, 81, 7749–7758. [Google Scholar] [CrossRef] [Green Version]
- Holm, C.K.; Jensen, S.B.; Jakobsen, M.R.; Cheshenko, N.; Horan, K.A.; Moeller, H.B.; Gonzalez-Dosal, R.; Rasmussen, S.B.; Christensen, M.H.; Yarovinsky, T.O.; et al. Virus-cell fusion as a trigger of innate immunity dependent on the adaptor STING. Nat. Immunol. 2012, 13, 737–743. [Google Scholar] [CrossRef] [Green Version]
- Lucinda, N.; Figueiredo, M.M.; Pessoa, N.L.; Santos, B.S.; Lima, G.K.; Freitas, A.M.; Machado, A.M.; Kroon, E.G.; Antonelli, L.R.; Campos, M.A. Dendritic cells, macrophages, NK and CD8(+) T lymphocytes play pivotal roles in controlling HSV-1 in the trigeminal ganglia by producing IL1-beta, iNOS and granzyme B. Virol. J. 2017, 14, 37. [Google Scholar] [CrossRef] [Green Version]
- Kurt-Jones, E.A.; Chan, M.; Zhou, S.; Wang, J.; Reed, G.; Bronson, R.; Arnold, M.M.; Knipe, D.M.; Finberg, R.W. Herpes simplex virus 1 interaction with toll-like receptor 2 contributes to lethal encephalitis. Proc. Natl. Acad. Sci. USA 2004, 101, 1315–1320. [Google Scholar] [CrossRef] [Green Version]
- Kurt-Jones, E.A.; Belko, J.; Yu, C.; Newburger, P.E.; Wang, J.; Chan, M.; Knipe, D.M.; Finberg, R.W. The role of toll-like receptors in herpes simplex infection in neonates. J. Infect. Dis. 2005, 191, 746–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.P.; Bowen, G.N.; Zhou, S.; Cerny, A.; Zacharia, A.; Knipe, D.M.; Finberg, R.W.; Kurt-Jones, E.A. Role of specific innate immune responses in herpes simplex virus infection of the central nervous system. J. Virol. 2012, 86, 2273–2281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stack, J.; Doyle, S.L.; Connolly, D.J.; Reinert, L.S.; O’Keeffe, K.M.; McLoughlin, R.M.; Paludan, S.R.; Bowie, A.G. TRAM is required for TLR2 endosomal signaling to type I IFN induction. J. Immunol. 2014, 193, 6090–6102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gianni, T.; Leoni, V.; Campadelli-Fiume, G. Type I interferon and NF-kappaB activation elicited by herpes simplex virus gH/gL via alphavbeta3 integrin in epithelial and neuronal cell lines. J. Virol. 2013, 87, 13911–13916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leoni, V.; Gianni, T.; Salvioli, S.; Campadelli-Fiume, G. Herpes simplex virus glycoproteins gH/gL and gB bind Toll-like receptor 2, and soluble gH/gL is sufficient to activate NF-kappaB. J. Virol. 2012, 86, 6555–6562. [Google Scholar] [CrossRef] [Green Version]
- Cai, M.; Li, M.; Wang, K.; Wang, S.; Lu, Q.; Yan, J.; Mossman, K.L.; Lin, R.; Zheng, C. The herpes simplex virus 1-encoded envelope glycoprotein B activates NF-kappaB through the toll-like receptor 2 and MyD88/TRAF6-dependent signaling pathway. PLoS ONE 2013, 8, e54586. [Google Scholar] [CrossRef] [Green Version]
- Marino, A.; Pergolizzi, S.; Cimino, F.; Lauriano, E.R.; Speciale, A.; D’Angelo, V.; Sicurella, M.; Argnani, R.; Manservigi, R.; Marconi, P. Role of herpes simplex envelope glycoprotein B and toll-like receptor 2 in ocular inflammation: An ex vivo organotypic rabbit corneal model. Viruses 2019, 11, 819. [Google Scholar] [CrossRef] [Green Version]
- Brun, P.; Scarpa, M.; Marchiori, C.; Conti, J.; Kotsafti, A.; Porzionato, A.; De Caro, R.; Scarpa, M.; Calistri, A.; Castagliuolo, I. Herpes simplex virus type 1 engages toll like receptor 2 to recruit macrophages during infection of enteric neurons. Front. Microbiol. 2018, 9, 2148. [Google Scholar] [CrossRef]
- Guo, Y.J.; Luo, T.; Wu, F.; Mei, Y.W.; Peng, J.; Liu, H.; Li, H.R.; Zhang, S.L.; Dong, J.H.; Fang, Y.; et al. Involvement of TLR2 and TLR9 in the anti-inflammatory effects of chlorogenic acid in HSV-1-infected microglia. Life Sci. 2015, 127, 12–18. [Google Scholar] [CrossRef]
- Liu, H.; Chen, K.; Feng, W.; Wu, X.; Li, H. TLR4-MyD88/Mal-NF-kB axis is involved in infection of HSV-2 in human cervical epithelial cells. PLoS ONE 2013, 8, e80327. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Chen, K.; Feng, W.; Guo, J.; Li, H. HSV-2 increases TLR4-dependent phosphorylated IRFs and IFN-beta induction in cervical epithelial cells. PLoS ONE 2014, 9, e94806. [Google Scholar] [CrossRef]
- Lv, X.; Wang, H.; Su, A.; Xu, S.; Chu, Y. Herpes simplex virus type 2 infection triggers AP-1 transcription activity through TLR4 signaling in genital epithelial cells. Virol. J. 2018, 15, 173. [Google Scholar] [CrossRef] [PubMed]
- Boivin, N.; Sergerie, Y.; Rivest, S.; Boivin, G. Effect of pretreatment with toll-like receptor agonists in a mouse model of herpes simplex virus type 1 encephalitis. J. Infect. Dis. 2008, 198, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Sarangi, P.P.; Kim, B.; Kurt-Jones, E.; Rouse, B.T. Innate recognition network driving herpes simplex virus-induced corneal immunopathology: Role of the toll pathway in early inflammatory events in stromal keratitis. J. Virol. 2007, 81, 11128–11138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flacher, V.; Bouschbacher, M.; Verronese, E.; Massacrier, C.; Sisirak, V.; Berthier-Vergnes, O.; de Saint-Vis, B.; Caux, C.; Dezutter-Dambuyant, C.; Lebecque, S.; et al. Human Langerhans cells express a specific TLR profile and differentially respond to viruses and Gram-positive bacteria. J. Immunol. 2006, 177, 7959–7967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hornung, V.; Rothenfusser, S.; Britsch, S.; Krug, A.; Jahrsdorfer, B.; Giese, T.; Endres, S.; Hartmann, G. Quantitative expression of toll-like receptor 1-10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J. Immunol. 2002, 168, 4531–4537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sciortino, M.T.; Medici, M.A.; Marino-Merlo, F.; Zaccaria, D.; Giuffre-Cuculletto, M.; Venuti, A.; Grelli, S.; Mastino, A. Involvement of HVEM receptor in activation of nuclear factor kappaB by herpes simplex virus 1 glycoprotein D. Cell. Microbiol. 2008, 10, 2297–2311. [Google Scholar] [CrossRef]
- Edwards, R.G.; Longnecker, R. Herpesvirus entry mediator and ocular herpesvirus infection: More than meets the eye. J. Virol. 2017, 91, e00115-17. [Google Scholar] [CrossRef] [Green Version]
- Edwards, R.G.; Kopp, S.J.; Karaba, A.H.; Wilcox, D.R.; Longnecker, R. Herpesvirus entry mediator on radiation-resistant cell lineages promotes ocular herpes simplex virus 1 pathogenesis in an entry-independent manner. MBio 2015, 6, e01532-15. [Google Scholar] [CrossRef] [Green Version]
- Edwards, R.G.; Kopp, S.J.; Ifergan, I.; Shui, J.W.; Kronenberg, M.; Miller, S.D.; Longnecker, R. Murine corneal inflammation and nerve damage after infection with HSV-1 are promoted by HVEM and ameliorated by immune-modifying nanoparticle therapy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 282–291. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Rajasagi, N.K.; Veiga-Parga, T.; Rouse, B.T. Herpes virus entry mediator (HVEM) modulates proliferation and activation of regulatory T cells following HSV-1 infection. Microbes Infect. 2014, 16, 648–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, H.; Pang, K.; Wei, Y.; Yi, C.; Wu, X. Herpes virus entry mediator in human corneal epithelial cells modulates the production of inflammatory cytokines in response to HSV type 1 challenge. Ophthalmic. Res. 2015, 54, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Horan, K.A.; Hansen, K.; Jakobsen, M.R.; Holm, C.K.; Soby, S.; Unterholzner, L.; Thompson, M.; West, J.A.; Iversen, M.B.; Rasmussen, S.B.; et al. Proteasomal degradation of herpes simplex virus capsids in macrophages releases DNA to the cytosol for recognition by DNA sensors. J. Immunol. 2013, 190, 2311–2319. [Google Scholar] [CrossRef] [PubMed]
- Krug, A.; Luker, G.D.; Barchet, W.; Leib, D.A.; Akira, S.; Colonna, M. Herpes simplex virus type 1 activates murine natural interferon-producing cells through toll-like receptor 9. Blood 2004, 103, 1433–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasmussen, S.B.; Sorensen, L.N.; Malmgaard, L.; Ank, N.; Baines, J.D.; Chen, Z.J.; Paludan, S.R. Type I interferon production during herpes simplex virus infection is controlled by cell-type-specific viral recognition through Toll-like receptor 9, the mitochondrial antiviral signaling protein pathway, and novel recognition systems. J. Virol. 2007, 81, 13315–13324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honda, K.; Yanai, H.; Negishi, H.; Asagiri, M.; Sato, M.; Mizutani, T.; Shimada, N.; Ohba, Y.; Takaoka, A.; Yoshida, N.; et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 2005, 434, 772–777. [Google Scholar] [CrossRef]
- Iversen, M.B.; Ank, N.; Melchjorsen, J.; Paludan, S.R. Expression of type III interferon (IFN) in the vaginal mucosa is mediated primarily by dendritic cells and displays stronger dependence on NF-kappaB than type I IFNs. J. Virol. 2010, 84, 4579–4586. [Google Scholar] [CrossRef] [Green Version]
- Malmgaard, L.; Melchjorsen, J.; Bowie, A.G.; Mogensen, S.C.; Paludan, S.R. Viral activation of macrophages through TLR-dependent and -independent pathways. J. Immunol. 2004, 173, 6890–6898. [Google Scholar] [CrossRef] [Green Version]
- Miettinen, M.; Sareneva, T.; Julkunen, I.; Matikainen, S. IFNs activate toll-like receptor gene expression in viral infections. Genes Immun. 2001, 2, 349–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uyangaa, E.; Choi, J.Y.; Patil, A.M.; Hossain, F.M.A.; Park, S.O.; Kim, B.; Kim, K.; Eo, S.K. Dual TLR2/9 recognition of herpes simplex virus infection is required for recruitment and activation of monocytes and NK cells and restriction of viral dissemination to the central nervous system. Front. Immunol. 2018, 9, 905. [Google Scholar] [CrossRef]
- Sorensen, L.N.; Reinert, L.S.; Malmgaard, L.; Bartholdy, C.; Thomsen, A.R.; Paludan, S.R. TLR2 and TLR9 synergistically control herpes simplex virus infection in the brain. J. Immunol. 2008, 181, 8604–8612. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Brinkmann, M.M.; Paquet, M.E.; Ploegh, H.L. UNC93B1 delivers nucleotide-sensing toll-like receptors to endolysosomes. Nature 2008, 452, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, M.M.; Spooner, E.; Hoebe, K.; Beutler, B.; Ploegh, H.L.; Kim, Y.M. The interaction between the ER membrane protein UNC93B and TLR3, 7, and 9 is crucial for TLR signaling. J. Cell Biol. 2007, 177, 265–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casrouge, A.; Zhang, S.Y.; Eidenschenk, C.; Jouanguy, E.; Puel, A.; Yang, K.; Alcais, A.; Picard, C.; Mahfoufi, N.; Nicolas, N.; et al. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science 2006, 314, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [Green Version]
- Ablasser, A.; Goldeck, M.; Cavlar, T.; Deimling, T.; Witte, G.; Rohl, I.; Hopfner, K.P.; Ludwig, J.; Hornung, V. cGAS produces a 2'-5'-linked cyclic dinucleotide second messenger that activates STING. Nature 2013, 498, 380–384. [Google Scholar] [CrossRef] [Green Version]
- Abe, T.; Barber, G.N. Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-kappaB activation through TBK1. J. Virol. 2014, 88, 5328–5341. [Google Scholar] [CrossRef] [Green Version]
- You, H.; Lin, Y.; Lin, F.; Yang, M.; Li, J.; Zhang, R.; Huang, Z.; Shen, Q.; Tang, R.; Zheng, C. Beta-catenin is required for the cGAS/STING signaling pathway but antagonized by the herpes simplex virus 1 US3 protein. J. Virol. 2020, 94, e01847-19. [Google Scholar] [CrossRef]
- Orzalli, M.H.; Broekema, N.M.; Diner, B.A.; Hancks, D.C.; Elde, N.C.; Cristea, I.M.; Knipe, D.M. cGAS-mediated stabilization of IFI16 promotes innate signaling during herpes simplex virus infection. Proc. Natl. Acad. Sci. USA 2015, 112, E1773–E1781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diner, B.A.; Lum, K.K.; Toettcher, J.E.; Cristea, I.M. Viral DNA sensors IFI16 and Cyclic GMP-AMP synthase possess distinct functions in regulating viral gene expression, immune defenses, and apoptotic responses during herpesvirus infection. MBio 2016, 7, e01553-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bansode, Y.D.; Chattopadhyay, D.; Saha, B. Innate immune response in astrocytes infected with herpes simplex virus 1. Arch. Virol. 2019, 164, 1433–1439. [Google Scholar] [CrossRef] [PubMed]
- Latif, M.B.; Raja, R.; Kessler, P.M.; Sen, G.C. Relative contributions of the cGAS-STING and TLR3 signaling pathways to attenuation of herpes simplex virus 1 replication. J. Virol. 2020, 94, e01717-19. [Google Scholar] [CrossRef] [PubMed]
- Jeffries, A.M.; Nitika; Truman, A.W.; Marriott, I. The intracellular DNA sensors cGAS and IFI16 do not mediate effective antiviral immune responses to HSV-1 in human microglial cells. J. Neurovirol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Reinert, L.S.; Lopusna, K.; Winther, H.; Sun, C.; Thomsen, M.K.; Nandakumar, R.; Mogensen, T.H.; Meyer, M.; Vaegter, C.; Nyengaard, J.R.; et al. Sensing of HSV-1 by the cGAS-STING pathway in microglia orchestrates antiviral defence in the CNS. Nat. Commun. 2016, 7, 13348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Jiang, F.; Kong, L.; Li, B.; Yang, Y.; Zhang, L.; Liu, B.; Zheng, Y.; Gao, C. Cutting edge: USP27X deubiquitinates and stabilizes the DNA sensor cGAS to regulate cytosolic DNA-mediated signaling. J. Immunol. 2019, 203, 2049–2054. [Google Scholar] [CrossRef]
- Chen, X.; Chen, Y. Ubiquitination of cGAS by TRAF6 regulates anti-DNA viral innate immune responses. Biochem. Biophys. Res. Commun. 2019, 514, 659–664. [Google Scholar] [CrossRef]
- Seo, G.J.; Kim, C.; Shin, W.J.; Sklan, E.H.; Eoh, H.; Jung, J.U. TRIM56-mediated monoubiquitination of cGAS for cytosolic DNA sensing. Nat. Commun. 2018, 9, 613. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.Y.; Liao, B.W.; Xu, Z.S.; Ran, Y.; Wang, D.P.; Yang, Y.; Luo, W.W.; Wang, Y.Y. USP44 positively regulates innate immune response to DNA viruses through deubiquitinating MITA. PLoS Pathog. 2020, 16, e1008178. [Google Scholar] [CrossRef]
- Wang, Z.S.; Liu, Y.L.; Mi, N.; Duan, D.Y. Intracellular DNA sensing pathway of cGAS-cGAMP is decreased in human newborns and young children. Mol. Immunol. 2017, 87, 76–85. [Google Scholar] [CrossRef]
- Li, T.; Diner, B.A.; Chen, J.; Cristea, I.M. Acetylation modulates cellular distribution and DNA sensing ability of interferon-inducible protein IFI16. Proc. Natl. Acad. Sci. USA 2012, 109, 10558–10563. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, J.; Ansari, M.A.; Kumar, B.; Dutta, D.; Roy, A.; Chikoti, L.; Pisano, G.; Dutta, S.; Vahedi, S.; Veettil, M.V.; et al. Histone H2B-IFI16 recognition of nuclear herpesviral genome induces cytoplasmic interferon-beta responses. PLoS Pathog. 2016, 12, e1005967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orzalli, M.H.; DeLuca, N.A.; Knipe, D.M. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc. Natl. Acad. Sci. USA 2012, 109, E3008–E3017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conrady, C.D.; Zheng, M.; Fitzgerald, K.A.; Liu, C.; Carr, D.J. Resistance to HSV-1 infection in the epithelium resides with the novel innate sensor, IFI-16. Mucosal. Immunol. 2012, 5, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.R.; Sharma, S.; Atianand, M.; Jensen, S.B.; Carpenter, S.; Knipe, D.M.; Fitzgerald, K.A.; Kurt-Jones, E.A. Interferon gamma-inducible protein (IFI) 16 transcriptionally regulates type i interferons and other interferon-stimulated genes and controls the interferon response to both DNA and RNA viruses. J. Biol. Chem. 2014, 289, 23568–23581. [Google Scholar] [CrossRef] [Green Version]
- Unterholzner, L.; Keating, S.E.; Baran, M.; Horan, K.A.; Jensen, S.B.; Sharma, S.; Sirois, C.M.; Jin, T.; Latz, E.; Xiao, T.S.; et al. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 2010, 11, 997–1004. [Google Scholar] [CrossRef] [Green Version]
- Johnson, K.E.; Bottero, V.; Flaherty, S.; Dutta, S.; Singh, V.V.; Chandran, B. Correction: IFI16 restricts HSV-1 replication by accumulating on the HSV-1 genome, repressing HSV-1 gene expression, and directly or indirectly modulating histone modifications. PLoS Pathog. 2018, 14, e1007113. [Google Scholar] [CrossRef] [PubMed]
- Orzalli, M.H.; Conwell, S.E.; Berrios, C.; DeCaprio, J.A.; Knipe, D.M. Nuclear interferon-inducible protein 16 promotes silencing of herpesviral and transfected DNA. Proc. Natl. Acad. Sci. USA 2013, 110, E4492–E4501. [Google Scholar] [CrossRef] [Green Version]
- Merkl, P.E.; Knipe, D.M. Role for a filamentous nuclear assembly of IFI16, DNA, and host factors in restriction of herpesviral infection. MBio 2019, 10, e02621-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merkl, P.E.; Orzalli, M.H.; Knipe, D.M. Mechanisms of host IFI16, PML, and Daxx protein restriction of herpes simplex virus 1 replication. J. Virol. 2018, 92, e00057-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takaoka, A.; Wang, Z.; Choi, M.K.; Yanai, H.; Negishi, H.; Ban, T.; Lu, Y.; Miyagishi, M.; Kodama, T.; Honda, K.; et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 2007, 448, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Rebsamen, M.; Heinz, L.X.; Meylan, E.; Michallet, M.C.; Schroder, K.; Hofmann, K.; Vazquez, J.; Benedict, C.A.; Tschopp, J. DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-kappaB. EMBO Rep. 2009, 10, 916–922. [Google Scholar] [CrossRef] [PubMed]
- Furr, S.R.; Chauhan, V.S.; Moerdyk-Schauwecker, M.J.; Marriott, I. A role for DNA-dependent activator of interferon regulatory factor in the recognition of herpes simplex virus type 1 by glial cells. J. Neuroinflammation 2011, 8, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, T.H.; Kwon, K.M.; Kim, Y.E.; Kim, K.K.; Ahn, J.H. DNA sensing-independent inhibition of herpes simplex virus 1 replication by DAI/ZBP1. J. Virol. 2013, 87, 3076–3086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, K.J.; Kawagoe, T.; Koyama, S.; Matsui, K.; Kumar, H.; Kawai, T.; Uematsu, S.; Takeuchi, O.; Takeshita, F.; Coban, C.; et al. TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature 2008, 451, 725–729. [Google Scholar] [CrossRef]
- Crill, E.K.; Furr-Rogers, S.R.; Marriott, I. RIG-I is required for VSV-induced cytokine production by murine glia and acts in combination with DAI to initiate responses to HSV-1. Glia 2015, 63, 2168–2180. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.A.; Smith, B.R.C.; Tate, M.D.; Belz, G.T.; Barrios, M.H.; Elgass, K.D.; Weisman, A.S.; Baker, P.J.; Preston, S.P.; Whitehead, L.; et al. SIDT2 transports extracellular dsRNA into the cytoplasm for innate immune recognition. Immunity 2017, 47, 498–509 e496. [Google Scholar] [CrossRef] [Green Version]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-kappaB by toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef]
- Yamamoto, M.; Sato, S.; Hemmi, H.; Hoshino, K.; Kaisho, T.; Sanjo, H.; Takeuchi, O.; Sugiyama, M.; Okabe, M.; Takeda, K.; et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 2003, 301, 640–643. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Y.; Jouanguy, E.; Ugolini, S.; Smahi, A.; Elain, G.; Romero, P.; Segal, D.; Sancho-Shimizu, V.; Lorenzo, L.; Puel, A.; et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science 2007, 317, 1522–1527. [Google Scholar] [CrossRef]
- Guo, Y.; Audry, M.; Ciancanelli, M.; Alsina, L.; Azevedo, J.; Herman, M.; Anguiano, E.; Sancho-Shimizu, V.; Lorenzo, L.; Pauwels, E.; et al. Herpes simplex virus encephalitis in a patient with complete TLR3 deficiency: TLR3 is otherwise redundant in protective immunity. J. Exp. Med. 2011, 208, 2083–2098. [Google Scholar] [CrossRef]
- Tal, Y.; Ribak, Y.; Khalaila, A.; Shamriz, O.; Marcus, N.; Zinger, A.; Meiner, V.; Schuster, R.; Lewis, E.C.; Nahum, A. Toll-like receptor 3 (TLR3) variant and NLRP12 mutation confer susceptibility to a complex clinical presentation. Clin. Immunol. 2020, 212, 108249. [Google Scholar] [CrossRef] [PubMed]
- Lafaille, F.G.; Pessach, I.M.; Zhang, S.Y.; Ciancanelli, M.J.; Herman, M.; Abhyankar, A.; Ying, S.W.; Keros, S.; Goldstein, P.A.; Mostoslavsky, G.; et al. Impaired intrinsic immunity to HSV-1 in human iPSC-derived TLR3-deficient CNS cells. Nature 2012, 491, 769–773. [Google Scholar] [CrossRef] [PubMed]
- Reinert, L.S.; Harder, L.; Holm, C.K.; Iversen, M.B.; Horan, K.A.; Dagnaes-Hansen, F.; Ulhoi, B.P.; Holm, T.H.; Mogensen, T.H.; Owens, T.; et al. TLR3 deficiency renders astrocytes permissive to herpes simplex virus infection and facilitates establishment of CNS infection in mice. J. Clin. Investig. 2012, 122, 1368–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davey, G.M.; Wojtasiak, M.; Proietto, A.I.; Carbone, F.R.; Heath, W.R.; Bedoui, S. Cutting edge: Priming of CD8 T cell immunity to herpes simplex virus type 1 requires cognate TLR3 expression in vivo. J. Immunol. 2010, 184, 2243–2246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmer, B.; Ewaleifoh, O.; Harschnitz, O.; Lee, Y.S.; Peneau, C.; McAlpine, J.L.; Liu, B.; Tchieu, J.; Steinbeck, J.A.; Lafaille, F.; et al. Human iPSC-derived trigeminal neurons lack constitutive TLR3-dependent immunity that protects cortical neurons from HSV-1 infection. Proc. Natl. Acad. Sci. USA 2018, 115, E8775–E8782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelka, K.; Bertheloot, D.; Reimer, E.; Phulphagar, K.; Schmidt, S.V.; Christ, A.; Stahl, R.; Watson, N.; Miyake, K.; Hacohen, N.; et al. The chaperone UNC93B1 regulates toll-like receptor stability independently of endosomal TLR transport. Immunity 2018, 48, 911–922. [Google Scholar] [CrossRef]
- Schenten, D.; Medzhitov, R. The control of adaptive immune responses by the innate immune system. Adv. Immunol. 2011, 109, 87–124. [Google Scholar] [CrossRef]
- Odendall, C.; Dixit, E.; Stavru, F.; Bierne, H.; Franz, K.M.; Durbin, A.F.; Boulant, S.; Gehrke, L.; Cossart, P.; Kagan, J.C. Diverse intracellular pathogens activate type III interferon expression from peroxisomes. Nat. Immunol. 2014, 15, 717–726. [Google Scholar] [CrossRef]
- Melchjorsen, J.; Rintahaka, J.; Soby, S.; Horan, K.A.; Poltajainen, A.; Ostergaard, L.; Paludan, S.R.; Matikainen, S. Early innate recognition of herpes simplex virus in human primary macrophages is mediated via the MDA5/MAVS-dependent and MDA5/MAVS/RNA polymerase III-independent pathways. J. Virol. 2010, 84, 11350–11358. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, S.B.; Jensen, S.B.; Nielsen, C.; Quartin, E.; Kato, H.; Chen, Z.J.; Silverman, R.H.; Akira, S.; Paludan, S.R. Herpes simplex virus infection is sensed by both toll-like receptors and retinoic acid-inducible gene- like receptors, which synergize to induce type I interferon production. J. Gen. Virol. 2009, 90, 74–78. [Google Scholar] [CrossRef]
- Cotter, C.R.; Kim, W.K.; Nguyen, M.L.; Yount, J.S.; Lopez, C.B.; Blaho, J.A.; Moran, T.M. The virion host shutoff protein of herpes simplex virus 1 blocks the replication-independent activation of NF-kappaB in dendritic cells in the absence of type I interferon signaling. J. Virol. 2011, 85, 12662–12672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, M.K.; Wang, Z.; Ban, T.; Yanai, H.; Lu, Y.; Koshiba, R.; Nakaima, Y.; Hangai, S.; Savitsky, D.; Nakasato, M.; et al. A selective contribution of the RIG-I-like receptor pathway to type I interferon responses activated by cytosolic DNA. Proc. Natl. Acad. Sci. USA 2009, 106, 17870–17875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Ye, X.; Dunker, W.; Song, Y.; Karijolich, J. RIG-I like receptor sensing of host RNAs facilitates the cell-intrinsic immune response to KSHV infection. Nat. Commun. 2018, 9, 4841. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.H.; Macmillan, J.B.; Chen, Z.J. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 2009, 138, 576–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogunjimi, B.; Zhang, S.Y.; Sorensen, K.B.; Skipper, K.A.; Carter-Timofte, M.; Kerner, G.; Luecke, S.; Prabakaran, T.; Cai, Y.; Meester, J.; et al. Inborn errors in RNA polymerase III underlie severe varicella zoster virus infections. J. Clin. Investig. 2017, 127, 3543–3556. [Google Scholar] [CrossRef] [Green Version]
- Balachandran, S.; Roberts, P.C.; Brown, L.E.; Truong, H.; Pattnaik, A.K.; Archer, D.R.; Barber, G.N. Essential role for the dsRNA-dependent protein kinase PKR in innate immunity to viral infection. Immunity 2000, 13, 129–141. [Google Scholar] [CrossRef] [Green Version]
- Meurs, E.; Chong, K.; Galabru, J.; Thomas, N.S.; Kerr, I.M.; Williams, B.R.; Hovanessian, A.G. Molecular cloning and characterization of the human double-stranded RNA-activated protein kinase induced by interferon. Cell 1990, 62, 379–390. [Google Scholar] [CrossRef]
- Kumar, A.; Haque, J.; Lacoste, J.; Hiscott, J.; Williams, B.R. Double-stranded RNA-dependent protein kinase activates transcription factor NF-kappa B by phosphorylating I kappa B. Proc. Natl. Acad. Sci. USA 1994, 91, 6288–6292. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.; Zamanian-Daryoush, M.; Nie, H.; Silva, A.M.; Williams, B.R.; Li, X. Poly(I-C)-induced toll-like receptor 3 (TLR3)-mediated activation of NFkappa B and MAP kinase is through an interleukin-1 receptor-associated kinase (IRAK)-independent pathway employing the signaling components TLR3-TRAF6-TAK1-TAB2-PKR. J. Biol. Chem. 2003, 278, 16713–16719. [Google Scholar] [CrossRef] [Green Version]
- Horng, T.; Barton, G.M.; Medzhitov, R. TIRAP: An adapter molecule in the toll signaling pathway. Nat. Immunol. 2001, 2, 835–841. [Google Scholar] [CrossRef]
- Pham, A.M.; Santa Maria, F.G.; Lahiri, T.; Friedman, E.; Marie, I.J.; Levy, D.E. PKR transduces MDA5-dependent signals for Type I IFN induction. PLoS Pathog. 2016, 12, e1005489. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Gross, M.; Roizman, B. The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc. Natl. Acad. Sci. USA 1997, 94, 843–848. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Zhang, J.; Kumar, A.; Zheng, M.; Atherton, S.S.; Yu, F.S. Herpes simplex virus 1 infection induces the expression of proinflammatory cytokines, interferons and TLR7 in human corneal epithelial cells. Immunology 2006, 117, 167–176. [Google Scholar] [CrossRef]
- Torseth, J.W.; Nickoloff, B.J.; Basham, T.Y.; Merigan, T.C. Beta interferon produced by keratinocytes in human cutaneous infection with herpes simplex virus. J. Infect. Dis. 1987, 155, 641–648. [Google Scholar] [CrossRef]
- Siegal, F.P.; Kadowaki, N.; Shodell, M.; Fitzgerald-Bocarsly, P.A.; Shah, K.; Ho, S.; Antonenko, S.; Liu, Y.J. The nature of the principal type 1 interferon-producing cells in human blood. Science 1999, 284, 1835–1837. [Google Scholar] [CrossRef] [Green Version]
- Baranek, T.; Zucchini, N.; Dalod, M. Plasmacytoid dendritic cells and the control of herpesvirus infections. Viruses 2009, 1, 383–419. [Google Scholar] [CrossRef]
- Donaghy, H.; Bosnjak, L.; Harman, A.N.; Marsden, V.; Tyring, S.K.; Meng, T.C.; Cunningham, A.L. Role for plasmacytoid dendritic cells in the immune control of recurrent human herpes simplex virus infection. J. Virol. 2009, 83, 1952–1961. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Song, Y.; Zhu, L.; Wang, X.; Richers, B.; Leung, D.Y.M.; Bin, L. Interferon kappa is important for keratinocyte host defense against herpes simplex virus-1. J. Immunol. Res. 2020, 2020, 5084682. [Google Scholar] [CrossRef]
- Gill, N.; Chenoweth, M.J.; Verdu, E.F.; Ashkar, A.A. NK cells require type I IFN receptor for antiviral responses during genital HSV-2 infection. Cell. Immunol. 2011, 269, 29–37. [Google Scholar] [CrossRef]
- Dhanushkodi, N.R.; Srivastava, R.; Prakash, S.; Roy, S.; Coulon, P.A.; Vahed, H.; Nguyen, A.M.; Salazar, S.; Nguyen, L.; Amezquita, C.; et al. High frequency of gamma interferon-producing PLZF(lo)RORgammat(lo) invariant natural killer 1 cells infiltrating herpes simplex virus 1-infected corneas is associated with asymptomatic ocular herpesvirus infection. J. Virol. 2020, 94, e00140-20. [Google Scholar] [CrossRef]
- Koelle, D.M.; Posavad, C.M.; Barnum, G.R.; Johnson, M.L.; Frank, J.M.; Corey, L. Clearance of HSV-2 from recurrent genital lesions correlates with infiltration of HSV-specific cytotoxic T lymphocytes. J. Clin. Investig. 1998, 101, 1500–1508. [Google Scholar] [CrossRef]
- Posavad, C.M.; Zhao, L.; Mueller, D.E.; Stevens, C.E.; Huang, M.L.; Wald, A.; Corey, L. Persistence of mucosal T-cell responses to herpes simplex virus type 2 in the female genital tract. Mucosal. Immunol. 2015, 8, 115–126. [Google Scholar] [CrossRef] [Green Version]
- Koelle, D.M.; Liu, Z.; McClurkan, C.M.; Topp, M.S.; Riddell, S.R.; Pamer, E.G.; Johnson, A.S.; Wald, A.; Corey, L. Expression of cutaneous lymphocyte-associated antigen by CD8(+) T cells specific for a skin-tropic virus. J. Clin. Investig. 2002, 110, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Truong, N.R.; James, V.; Bosnjak, L.; Sandgren, K.J.; Harman, A.N.; Nasr, N.; Bertram, K.M.; Olbourne, N.; Sawleshwarkar, S.; et al. Relay of herpes simplex virus between Langerhans cells and dermal dendritic cells in human skin. PLoS Pathog. 2015, 11, e1004812. [Google Scholar] [CrossRef] [PubMed]
- Harpur, C.M.; Kato, Y.; Dewi, S.T.; Stankovic, S.; Johnson, D.N.; Bedoui, S.; Whitney, P.G.; Lahoud, M.H.; Caminschi, I.; Heath, W.R.; et al. Classical type 1 dendritic cells dominate priming of Th1 responses to herpes simplex virus type 1 skin infection. J. Immunol. 2019, 202, 653–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Koelle, D.M.; Cao, J.; Vazquez, J.; Huang, M.L.; Hladik, F.; Wald, A.; Corey, L. Virus-specific CD8+ T cells accumulate near sensory nerve endings in genital skin during subclinical HSV-2 reactivation. J. Exp. Med. 2007, 204, 595–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Hladik, F.; Woodward, A.; Klock, A.; Peng, T.; Johnston, C.; Remington, M.; Magaret, A.; Koelle, D.M.; Wald, A.; et al. Persistence of HIV-1 receptor-positive cells after HSV-2 reactivation is a potential mechanism for increased HIV-1 acquisition. Nat. Med. 2009, 15, 886–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellermann-Eriksen, S. Macrophages and cytokines in the early defence against herpes simplex virus. Virol. J. 2005, 2, 59. [Google Scholar] [CrossRef] [Green Version]
- Sainz, B., Jr.; Halford, W.P. Alpha/Beta interferon and gamma interferon synergize to inhibit the replication of herpes simplex virus type 1. J. Virol. 2002, 76, 11541–11550. [Google Scholar] [CrossRef] [Green Version]
- Mikloska, Z.; Cunningham, A.L. Alpha and gamma interferons inhibit herpes simplex virus type 1 infection and spread in epidermal cells after axonal transmission. J. Virol. 2001, 75, 11821–11826. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Gamero, A.M.; Jensen, L.E. IL-36 promotes anti-viral immunity by boosting sensitivity to IFN-alpha/beta in IRF1 dependent and independent manners. Nat. Commun. 2019, 10, 4700. [Google Scholar] [CrossRef] [PubMed]
- Deschamps, T.; Kalamvoki, M. Extracellular vesicles released by herpes simplex virus 1-infected cells block virus replication in recipient cells in a STING-dependent manner. J. Virol. 2018, 92, e01102-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierangeli, A.; Statzu, M.; Nenna, R.; Santinelli, L.; Petrarca, L.; Frassanito, A.; Gentile, M.; Antonelli, G.; Midulla, F.; Scagnolari, C. Interferon lambda receptor 1 (IFNL1R) transcript is highly expressed in rhinovirus bronchiolitis and correlates with disease severity. J. Clin. Virol. 2018, 102, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Hemann, E.A.; Gale, M., Jr.; Savan, R. Interferon lambda genetics and biology in regulation of viral control. Front. Immunol. 2017, 8, 1707. [Google Scholar] [CrossRef]
- Yanai, H.; Chiba, S.; Hangai, S.; Kometani, K.; Inoue, A.; Kimura, Y.; Abe, T.; Kiyonari, H.; Nishio, J.; Taguchi-Atarashi, N.; et al. Revisiting the role of IRF3 in inflammation and immunity by conditional and specifically targeted gene ablation in mice. Proc. Natl. Acad. Sci. USA 2018, 115, 5253–5258. [Google Scholar] [CrossRef] [Green Version]
- Silvennoinen, O.; Ihle, J.N.; Schlessinger, J.; Levy, D.E. Interferon-induced nuclear signalling by Jak protein tyrosine kinases. Nature 1993, 366, 583–585. [Google Scholar] [CrossRef]
- Platanias, L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef]
- Donnelly, R.P.; Kotenko, S.V. Interferon-lambda: A new addition to an old family. J. Interferon. Cytokine Res. 2010, 30, 555–564. [Google Scholar] [CrossRef] [Green Version]
- Qin, C.; Zhang, R.; Lang, Y.; Shao, A.; Xu, A.; Feng, W.; Han, J.; Wang, M.; He, W.; Yu, C.; et al. Bclaf1 critically regulates the type I interferon response and is degraded by alphaherpesvirus US3. PLoS Pathog. 2019, 15, e1007559. [Google Scholar] [CrossRef] [Green Version]
- Green, D.S.; Young, H.A.; Valencia, J.C. Current prospects of type II interferon gamma signaling and autoimmunity. J. Biol. Chem. 2017, 292, 13925–13933. [Google Scholar] [CrossRef] [Green Version]
- Lavender, K.J.; Gibbert, K.; Peterson, K.E.; Van Dis, E.; Francois, S.; Woods, T.; Messer, R.J.; Gawanbacht, A.; Muller, J.A.; Munch, J.; et al. Interferon alpha subtype-specific suppression of HIV-1 infection in vivo. J. Virol. 2016, 90, 6001–6013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pervolaraki, K.; Rastgou Talemi, S.; Albrecht, D.; Bormann, F.; Bamford, C.; Mendoza, J.L.; Garcia, K.C.; McLauchlan, J.; Hofer, T.; Stanifer, M.L.; et al. Differential induction of interferon stimulated genes between type I and type III interferons is independent of interferon receptor abundance. PLoS Pathog. 2018, 14, e1007420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhushal, S.; Wolfsmuller, M.; Selvakumar, T.A.; Kemper, L.; Wirth, D.; Hornef, M.W.; Hauser, H.; Koster, M. Cell polarization and epigenetic status shape the heterogeneous response to type III interferons in intestinal epithelial cells. Front. Immunol. 2017, 8, 671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavoie, T.B.; Kalie, E.; Crisafulli-Cabatu, S.; Abramovich, R.; DiGioia, G.; Moolchan, K.; Pestka, S.; Schreiber, G. Binding and activity of all human alpha interferon subtypes. Cytokine 2011, 56, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; O’Neal, A.J.; Gonzalez, C.; Wittling, M.; Gjinaj, E.; Parsons, L.M.; Panda, D.; Khalenkov, A.; Scott, D.; Misra, S.; et al. S27 of IFNalpha1 contributes to its low affinity for IFNAR2 and weak antiviral activity. J. Interferon. Cytokine Res. 2019, 39, 283–292. [Google Scholar] [CrossRef]
- Lamken, P.; Lata, S.; Gavutis, M.; Piehler, J. Ligand-induced assembling of the type I interferon receptor on supported lipid bilayers. J. Mol. Biol. 2004, 341, 303–318. [Google Scholar] [CrossRef]
- Majoros, A.; Platanitis, E.; Kernbauer-Holzl, E.; Rosebrock, F.; Muller, M.; Decker, T. Canonical and non-canonical aspects of JAK-STAT signaling: Lessons from interferons for cytokine responses. Front. Immunol. 2017, 8, 29. [Google Scholar] [CrossRef] [Green Version]
- Mohr, A.; Chatain, N.; Domoszlai, T.; Rinis, N.; Sommerauer, M.; Vogt, M.; Muller-Newen, G. Dynamics and non-canonical aspects of JAK/STAT signalling. Eur. J. Cell Biol. 2012, 91, 524–532. [Google Scholar] [CrossRef]
- Siren, J.; Pirhonen, J.; Julkunen, I.; Matikainen, S. IFN-alpha regulates TLR-dependent gene expression of IFN-alpha, IFN-beta, IL-28, and IL-29. J. Immunol. 2005, 174, 1932–1937. [Google Scholar] [CrossRef] [Green Version]
- Ma, F.; Li, B.; Liu, S.Y.; Iyer, S.S.; Yu, Y.; Wu, A.; Cheng, G. Positive feedback regulation of type I IFN production by the IFN-inducible DNA sensor cGAS. J. Immunol. 2015, 194, 1545–1554. [Google Scholar] [CrossRef]
- Veeranki, S.; Duan, X.; Panchanathan, R.; Liu, H.; Choubey, D. IFI16 protein mediates the anti-inflammatory actions of the type-I interferons through suppression of activation of caspase-1 by inflammasomes. PLoS ONE 2011, 6, e27040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, D.C.; Gopalkrishnan, R.V.; Lin, L.; Randolph, A.; Valerie, K.; Pestka, S.; Fisher, P.B. Expression analysis and genomic characterization of human melanoma differentiation associated gene-5, mda-5: A novel type I interferon-responsive apoptosis-inducing gene. Oncogene 2004, 23, 1789–1800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Cao, T.; Shao, S.; Li, B.; Jin, L.; Lei, J.; Qiao, H.; Wang, G. Up-regulation of Interferon-inducible protein 16 contributes to psoriasis by modulating chemokine production in keratinocytes. Sci. Rep. 2016, 6, 25381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ku, C.C.; Che, X.B.; Reichelt, M.; Rajamani, J.; Schaap-Nutt, A.; Huang, K.J.; Sommer, M.H.; Chen, Y.S.; Chen, Y.Y.; Arvin, A.M. Herpes simplex virus-1 induces expression of a novel MxA isoform that enhances viral replication. Immunol. Cell Biol. 2011, 89, 173–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crameri, M.; Bauer, M.; Caduff, N.; Walker, R.; Steiner, F.; Franzoso, F.D.; Gujer, C.; Boucke, K.; Kucera, T.; Zbinden, A.; et al. MxB is an interferon-induced restriction factor of human herpesviruses. Nat. Commun. 2018, 9, 1980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schilling, M.; Bulli, L.; Weigang, S.; Graf, L.; Naumann, S.; Patzina, C.; Wagner, V.; Bauersfeld, L.; Goujon, C.; Hengel, H.; et al. Human MxB protein is a pan-herpesvirus restriction factor. J. Virol. 2018, 92, e01056-18. [Google Scholar] [CrossRef] [Green Version]
- Zenner, H.L.; Mauricio, R.; Banting, G.; Crump, C.M. Herpes simplex virus 1 counteracts tetherin restriction via its virion host shutoff activity. J. Virol. 2013, 87, 13115–13123. [Google Scholar] [CrossRef] [Green Version]
- Royer, D.J.; Carr, D.J. A STING-dependent innate-sensing pathway mediates resistance to corneal HSV-1 infection via upregulation of the antiviral effector tetherin. Mucosal. Immunol. 2016, 9, 1065–1075. [Google Scholar] [CrossRef] [Green Version]
- Blondeau, C.; Pelchen-Matthews, A.; Mlcochova, P.; Marsh, M.; Milne, R.S.; Towers, G.J. Tetherin restricts herpes simplex virus 1 and is antagonized by glycoprotein M. J. Virol. 2013, 87, 13124–13133. [Google Scholar] [CrossRef] [Green Version]
- Lenschow, D.J.; Lai, C.; Frias-Staheli, N.; Giannakopoulos, N.V.; Lutz, A.; Wolff, T.; Osiak, A.; Levine, B.; Schmidt, R.E.; Garcia-Sastre, A.; et al. IFN-stimulated gene 15 functions as a critical antiviral molecule against influenza, herpes, and Sindbis viruses. Proc. Natl. Acad. Sci. USA 2007, 104, 1371–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katzenell, S.; Leib, D.A. Herpes simplex virus and interferon signaling induce novel autophagic clusters in sensory neurons. J. Virol. 2016, 90, 4706–4719. [Google Scholar] [CrossRef] [Green Version]
- Al-khatib, K.; Williams, B.R.; Silverman, R.H.; Halford, W.; Carr, D.J. The murine double-stranded RNA-dependent protein kinase PKR and the murine 2′,5′-oligoadenylate synthetase-dependent RNase L are required for IFN-beta-mediated resistance against herpes simplex virus type 1 in primary trigeminal ganglion culture. Virology 2003, 313, 126–135. [Google Scholar] [CrossRef] [Green Version]
- Thapa, M.; Welner, R.S.; Pelayo, R.; Carr, D.J. CXCL9 and CXCL10 expression are critical for control of genital herpes simplex virus type 2 infection through mobilization of HSV-specific CTL and NK cells to the nervous system. J. Immunol. 2008, 180, 1098–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, R.; Khan, A.A.; Chilukuri, S.; Syed, S.A.; Tran, T.T.; Furness, J.; Bahraoui, E.; BenMohamed, L. CXCL10/CXCR3-dependent mobilization of herpes simplex virus-specific CD8(+) TEM and CD8(+) TRM cells within infected tissues allows efficient protection against recurrent herpesvirus infection and disease. J. Virol. 2017, 91, e00278-17. [Google Scholar] [CrossRef] [Green Version]
- Wilcox, D.R.; Folmsbee, S.S.; Muller, W.J.; Longnecker, R. The type I interferon response determines differences in choroid plexus susceptibility between newborns and adults in herpes simplex virus encephalitis. MBio 2016, 7, e00437-16. [Google Scholar] [CrossRef] [Green Version]
- Zawatzky, R.; Gresser, I.; DeMaeyer, E.; Kirchner, H. The role of interferon in the resistance of C57BL/6 mice to various doses of herpes simplex virus type 1. J. Infect. Dis. 1982, 146, 405–410. [Google Scholar] [CrossRef]
- Luker, G.D.; Prior, J.L.; Song, J.; Pica, C.M.; Leib, D.A. Bioluminescence imaging reveals systemic dissemination of herpes simplex virus type 1 in the absence of interferon receptors. J. Virol. 2003, 77, 11082–11093. [Google Scholar] [CrossRef] [Green Version]
- Conrady, C.D.; Halford, W.P.; Carr, D.J. Loss of the type I interferon pathway increases vulnerability of mice to genital herpes simplex virus 2 infection. J. Virol. 2011, 85, 1625–1633. [Google Scholar] [CrossRef] [Green Version]
- Cantin, E.; Tanamachi, B.; Openshaw, H. Role for gamma interferon in control of herpes simplex virus type 1 reactivation. J. Virol. 1999, 73, 3418–3423. [Google Scholar] [CrossRef] [Green Version]
- Andersen, L.L.; Mork, N.; Reinert, L.S.; Kofod-Olsen, E.; Narita, R.; Jorgensen, S.E.; Skipper, K.A.; Honing, K.; Gad, H.H.; Ostergaard, L.; et al. Functional IRF3 deficiency in a patient with herpes simplex encephalitis. J. Exp. Med. 2015, 212, 1371–1379. [Google Scholar] [CrossRef]
- Canivet, C.; Rheaume, C.; Lebel, M.; Piret, J.; Gosselin, J.; Boivin, G. Both IRF3 and especially IRF7 play a key role to orchestrate an effective cerebral inflammatory response in a mouse model of herpes simplex virus encephalitis. J. Neurovirol. 2018, 24, 761–768. [Google Scholar] [CrossRef]
- Song, R.; Koyuncu, O.O.; Greco, T.M.; Diner, B.A.; Cristea, I.M.; Enquist, L.W. Two modes of the axonal interferon response limit alphaherpesvirus neuroinvasion. MBio 2016, 7, e02145-15. [Google Scholar] [CrossRef] [Green Version]
- Rosato, P.C.; Leib, D.A. Neuronal interferon signaling is required for protection against herpes simplex virus replication and pathogenesis. PLoS Pathog. 2015, 11, e1005028. [Google Scholar] [CrossRef]
- Li, J.; Hu, S.; Zhou, L.; Ye, L.; Wang, X.; Ho, J.; Ho, W. Interferon lambda inhibits herpes simplex virus type I infection of human astrocytes and neurons. Glia 2011, 59, 58–67. [Google Scholar] [CrossRef] [Green Version]
- Linderman, J.A.; Kobayashi, M.; Rayannavar, V.; Fak, J.J.; Darnell, R.B.; Chao, M.V.; Wilson, A.C.; Mohr, I. Immune escape via a transient gene expression program enables productive replication of a latent pathogen. Cell. Rep. 2017, 18, 1312–1323. [Google Scholar] [CrossRef]
- van Velzen, M.; Jing, L.; Osterhaus, A.D.; Sette, A.; Koelle, D.M.; Verjans, G.M. Local CD4 and CD8 T-cell reactivity to HSV-1 antigens documents broad viral protein expression and immune competence in latently infected human trigeminal ganglia. PLoS Pathog. 2013, 9, e1003547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knickelbein, J.E.; Khanna, K.M.; Yee, M.B.; Baty, C.J.; Kinchington, P.R.; Hendricks, R.L. Noncytotoxic lytic granule-mediated CD8+ T cell inhibition of HSV-1 reactivation from neuronal latency. Science 2008, 322, 268–271. [Google Scholar] [CrossRef] [Green Version]
- Peri, P.; Mattila, R.K.; Kantola, H.; Broberg, E.; Karttunen, H.S.; Waris, M.; Vuorinen, T.; Hukkanen, V. Herpes simplex virus type 1 Us3 gene deletion influences toll-like receptor responses in cultured monocytic cells. Virol. J. 2008, 5, 140. [Google Scholar] [CrossRef] [Green Version]
- Sen, J.; Liu, X.; Roller, R.; Knipe, D.M. Herpes simplex virus US3 tegument protein inhibits toll-like receptor 2 signaling at or before TRAF6 ubiquitination. Virology 2013, 439, 65–73. [Google Scholar] [CrossRef] [Green Version]
- Liang, L.; Roizman, B. Expression of gamma interferon-dependent genes is blocked independently by virion host shutoff RNase and by US3 protein kinase. J. Virol. 2008, 82, 4688–4696. [Google Scholar] [CrossRef] [Green Version]
- Barzilai, A.; Zivony-Elbom, I.; Sarid, R.; Noah, E.; Frenkel, N. The herpes simplex virus type 1 vhs-UL41 gene secures viral replication by temporarily evading apoptotic cellular response to infection: Vhs-UL41 activity might require interactions with elements of cellular mRNA degradation machinery. J. Virol. 2006, 80, 505–513. [Google Scholar] [CrossRef] [Green Version]
- Taddeo, B.; Roizman, B. The virion host shutoff protein (UL41) of herpes simplex virus 1 is an endoribonuclease with a substrate specificity similar to that of RNase, A. J. Virol. 2006, 80, 9341–9345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, G.; Wang, K.; Wang, S.; Cai, M.; Li, M.L.; Zheng, C. Herpes simplex virus 1 counteracts viperin via its virion host shutoff protein UL41. J. Virol. 2014, 88, 12163–12166. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Liao, Z.; Xu, Z.; Zou, X.; Wang, Y.; Peng, H.; Li, Y.; Ou, X.; Deng, Y.; Guo, Y.; et al. The interaction mechanism between herpes simplex virus 1 glycoprotein D and host antiviral protein viperin. Front. Immunol. 2019, 10, 2810. [Google Scholar] [CrossRef] [Green Version]
- Dauber, B.; Saffran, H.A.; Smiley, J.R. The herpes simplex virus host shutoff (vhs) RNase limits accumulation of double stranded RNA in infected cells: Evidence for accelerated decay of duplex RNA. PLoS Pathog. 2019, 15, e1008111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasieka, T.J.; Lu, B.; Crosby, S.D.; Wylie, K.M.; Morrison, L.A.; Alexander, D.E.; Menachery, V.D.; Leib, D.A. Herpes simplex virus virion host shutoff attenuates establishment of the antiviral state. J. Virol. 2008, 82, 5527–5535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, C.; Zheng, C. Herpes simplex virus 1 abrogates the cGAS/STING-mediated cytosolic DNA-sensing pathway via its virion host shutoff protein, UL41. J. Virol. 2017, 91, e02414-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, J.; Ni, L.; Wang, S.; Wang, K.; Lin, R.; Zheng, C. Herpes simplex virus 1-encoded tegument protein VP16 abrogates the production of beta interferon (IFN) by inhibiting NF-kappaB activation and blocking IFN regulatory factor 3 to recruit its coactivator CBP. J. Virol. 2013, 87, 9788–9801. [Google Scholar] [CrossRef] [Green Version]
- Zheng, C.; Su, C. Herpes simplex virus 1 infection dampens the immediate early antiviral innate immunity signaling from peroxisomes by tegument protein VP16. Virol. J. 2017, 14, 35. [Google Scholar] [CrossRef] [Green Version]
- Johnson, K.E.; Song, B.; Knipe, D.M. Role for herpes simplex virus 1 ICP27 in the inhibition of type I interferon signaling. Virology 2008, 374, 487–494. [Google Scholar] [CrossRef] [Green Version]
- Johnson, K.E.; Knipe, D.M. Herpes simplex virus-1 infection causes the secretion of a type I interferon-antagonizing protein and inhibits signaling at or before JAK-1 activation. Virology 2010, 396, 21–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christensen, M.H.; Jensen, S.B.; Miettinen, J.J.; Luecke, S.; Prabakaran, T.; Reinert, L.S.; Mettenleiter, T.; Chen, Z.J.; Knipe, D.M.; Sandri-Goldin, R.M.; et al. HSV-1 ICP27 targets the TBK1-activated STING signalsome to inhibit virus-induced type I IFN expression. EMBO J. 2016, 35, 1385–1399. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Yeh, K.C.; Liu, J.; Knipe, D.M. Herpes simplex virus gene products required for viral inhibition of expression of G1-phase functions. Virology 2001, 290, 320–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paladino, P.; Collins, S.E.; Mossman, K.L. Cellular localization of the herpes simplex virus ICP0 protein dictates its ability to block IRF3-mediated innate immune responses. PLoS ONE 2010, 5, e10428. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Noyce, R.S.; Collins, S.E.; Everett, R.D.; Mossman, K.L. The herpes simplex virus ICP0 RING finger domain inhibits IRF3- and IRF7-mediated activation of interferon-stimulated genes. J. Virol. 2004, 78, 1675–1684. [Google Scholar] [CrossRef] [Green Version]
- Diner, B.A.; Lum, K.K.; Javitt, A.; Cristea, I.M. Interactions of the antiviral factor interferon gamma-inducible protein 16 (IFI16) mediate immune signaling and herpes simplex virus-1 immunosuppression. Mol. Cell. Proteom. 2015, 14, 2341–2356. [Google Scholar] [CrossRef] [Green Version]
- Orzalli, M.H.; Broekema, N.M.; Knipe, D.M. Relative contributions of herpes simplex virus 1 ICP0 and vhs to loss of cellular IFI16 vary in different human cell types. J. Virol. 2016, 90, 8351–8359. [Google Scholar] [CrossRef] [Green Version]
- Deschamps, T.; Kalamvoki, M. Impaired STING pathway in human osteosarcoma U2OS cells contributes to the growth of ICP0-null mutant herpes simplex virus. J. Virol. 2017, 91, e00006-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Lint, A.L.; Murawski, M.R.; Goodbody, R.E.; Severa, M.; Fitzgerald, K.A.; Finberg, R.W.; Knipe, D.M.; Kurt-Jones, E.A. Herpes simplex virus immediate-early ICP0 protein inhibits toll-like receptor 2-dependent inflammatory responses and NF-kappaB signaling. J. Virol. 2010, 84, 10802–10811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verpooten, D.; Ma, Y.; Hou, S.; Yan, Z.; He, B. Control of TANK-binding kinase 1-mediated signaling by the gamma(1)34.5 protein of herpes simplex virus 1. J. Biol. Chem. 2009, 284, 1097–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manivanh, R.; Mehrbach, J.; Knipe, D.M.; Leib, D.A. Role of herpes simplex virus 1 gamma34.5 in the regulation of IRF3 signaling. J. Virol. 2017, 91, e01156-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, S.; Liu, X.; Ma, Y.; Cao, Y.; He, B. Herpes simplex virus 1 gamma134.5 protein inhibits STING activation that restricts viral replication. J. Virol. 2018, 92, e01015-18. [Google Scholar] [CrossRef] [Green Version]
- Sato, Y.; Koshizuka, T.; Ishibashi, K.; Hashimoto, K.; Ishioka, K.; Ikuta, K.; Yokota, S.I.; Fujii, N.; Suzutani, T. Involvement of herpes simplex virus type 1 UL13 protein kinase in induction of SOCS genes, the negative regulators of cytokine signaling. Microbiol. Immunol. 2017, 61, 159–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapon, M.; Parvatiyar, K.; Aliyari, S.R.; Zhao, J.S.; Cheng, G. Comprehensive mutagenesis of herpes simplex virus 1 genome identifies UL42 as an inhibitor of type I interferon induction. J. Virol. 2019, 93, e01446-19. [Google Scholar] [CrossRef]
- Xu, H.; Su, C.; Pearson, A.; Mody, C.H.; Zheng, C. Herpes simplex virus 1 UL24 abrogates the DNA sensing signal pathway by inhibiting NF-kappaB activation. J. Virol. 2017, 91, e00025-17. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Su, C.; Zheng, C. Herpes simplex virus 1 serine protease VP24 blocks the DNA-sensing signal pathway by abrogating activation of interferon regulatory factor 3. J. Virol. 2016, 90, 5824–5829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, H.; Zheng, S.; Huang, Z.; Lin, Y.; Shen, Q.; Zheng, C. Herpes simplex virus 1 tegument protein UL46 inhibits TANK-binding kinase 1-mediated signaling. MBio 2019, 10, e00919-19. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; You, H.; Su, C.; Li, Y.; Chen, S.; Zheng, C. Herpes simplex virus 1 tegument protein VP22 abrogates cGAS/STING-mediated antiviral innate immunity. J. Virol. 2018, 92, e00841-18. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Wang, K.; Li, J.; Zheng, C. Herpes simplex virus 1 ubiquitin-specific protease UL36 inhibits beta interferon production by deubiquitinating TRAF3. J. Virol. 2013, 87, 11851–11860. [Google Scholar] [CrossRef] [Green Version]
- Ye, R.; Su, C.; Xu, H.; Zheng, C. Herpes simplex virus 1 ubiquitin-specific protease UL36 abrogates NF-kappaB activation in DNA sensing signal pathway. J. Virol. 2017, 91, e02417-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, H.; You, J.; You, H.; Zheng, C. Herpes simplex virus 1 UL36USP antagonizes type I interferon-mediated antiviral innate immunity. J. Virol. 2018, 92, e01161-18. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Zeng, Y.; Xu, S.; Chen, J.; Shen, G.; Yu, C.; Knipe, D.; Yuan, W.; Peng, J.; Xu, W.; et al. A viral deamidase targets the helicase domain of RIG-I to block RNA-induced activation. Cell Host Microbe 2016, 20, 770–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Zhao, J.; Xu, S.; Li, J.; He, S.; Zeng, Y.; Xie, L.; Xie, N.; Liu, T.; Lee, K.; et al. Species-specific deamidation of cGAS by herpes simplex virus UL37 protein facilitates viral replication. Cell Host Microbe 2018, 24, 234–248 e235. [Google Scholar] [CrossRef] [Green Version]
- Xing, J.; Wang, S.; Lin, R.; Mossman, K.L.; Zheng, C. Herpes simplex virus 1 tegument protein US11 downmodulates the RLR signaling pathway via direct interaction with RIG-I and MDA-5. J. Virol. 2012, 86, 3528–3540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Main, D.; Ma, Y.; He, B. Herpes simplex virus 1 inhibits TANK-binding kinase 1 through formation of the Us11-Hsp90 complex. J. Virol. 2018, 92, e00402-18. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ISG | Function | Effect on HSV-1 | Reference |
|---|---|---|---|
| MxA | GPTase protein which limits genome replication and viral capsid transport | MxA induced by IFNα reduces HSV-1 replication in human fibroblasts. pDCs infiltrating into the dermis of herpes lesions may be involved in stimulating the production of MxA by surrounding cells | [139,177] |
| MxB | GPTase protein which blocks viral DNA entering the nucleus | Inhibits HSV-1 replication in several human epithelial and neuronal cell lines | [178,179] |
| Tetherin | Membrane glycoprotein that ‘tethers’ viral particles to the cellular membrane, preventing release | Reduced HSV-1 spread in human epithelial cells, monkey fibroblasts and mouse cornea following upregulation of tetherin. This reduction is enhanced in virion host shutoff factor (vhs) null HSV-1 | [180,181,182] |
| ISG15 | Ubiquitin-like protein which conjugates to proteins (>100 known) to induce post-translational modifications | ISG15 knockout mice are unable to control HSV-1 infections and are associated with the formation of autophagic clusters of TG neurons | [183,184] |
| PKR | Phosphorylates eIF-2α upon binding viral dsRNA to limit mRNA translation | Mouse TG neurons with PKR or RNase L knockouts have increased HSV-1 replication compared to wild type TG neurons in the presence of IFNβ | [185] |
| OAS/RNase L | OAS synthesizes 2′,5′-oligoadenylate that binds to RNase L, which then cleaves mRNA | ||
| CXCL10 | IFNγ induced protein involved in recruiting NK and CD8+ T cells to infection sites | CXCL10 null mice infected with HSV-1 and 2 have reduced numbers of NK and CD8+ T cells at the site of infection and increased viral replication | [186,187] |
| Viral Protein | Role in Suppressing the Innate Immune Response | Reference |
|---|---|---|
| pUS3 | Inhibits TLR3 expression | [201] |
| Induces the degradation of bclaf2 | [163] | |
| Inhibits the ubiquitination of TRAF6 | [204] | |
| Inhibits the phosphorylation of the IFNGR1 subunit | [205] | |
| Inhibits the nuclear translocation of β-catenin | [84] | |
| vhs | Degrades host mRNA to inhibit ISG translation | [206,207] |
| Prevents accumulation of viral DNA in cell cytoplasm | [210] | |
| Inhibits the phosphorylation of eIF2α | [211] | |
| Downregulates cGAS production | [212] | |
| VP16 | Binds to IRF3 and prevents its interaction with CBP | [213] |
| Blocks NF-κB binding to promoter region | [213] | |
| Inhibits peroxisomal MAVS activation | [214] | |
| ICP27 | Inhibits the phosphorylation and nuclear translocation of STAT1 | [215] |
| Inhibits TBK1 and STING downstream signaling | [217] | |
| Degrades host mRNA to inhibit ISG translation | [218] | |
| ICP0 | Inhibits IRF3 and IRF7 activation | [219,220] |
| Degrades IFI16 | [98,221,222] | |
| Inhibits STING activation | [223] | |
| Inhibits the phosphorylation of STAT1 | [215] | |
| Reduces levels of MyD88 downstream from TLR2 activation | [224] | |
| ICP34.5 | Inhibits the phosphorylation of eIF2α | [138] |
| Indirectly binds to and inhibits TBK1 | [225,226] | |
| Inhibits STING activation | [227] |
| HSV-1 Proteins | Role in Suppressing the Innate Immune Response | Reference |
|---|---|---|
| pUL13 | Induces the expression of suppressor of cytokine signaling 1 and 3, which act to suppress IFN production | [226] |
| pUL42 | Inhibits the phosphorylation of IRF3 | [227] |
| pUL24 | Prevent the nuclear translocation of NF-κB subunits | [228] |
| Inhibits the interaction between TBK1 and IRF3, preventing the dimerization of IRF3 and its translocation to the nucleus | [229] | |
| pUL46 | Inhibits TBK1 activation and IRF3 translocation to the nucleus | [230] |
| VP22 | Inhibits cGAS from synthesizing cGAMP | [231] |
| pUL36 | Deubiquitinates TRAF3 impairing downstream signaling, inhibiting IRF3 dimerization | [232] |
| Deubiquitinates I-κBα, inhibiting its degradation. This results in sequestering NF-κB in the cytoplasm and preventing its translocation to the nucleus | [233] | |
| Binds to IFNAR2 and prevents the phosphorylation of JAK1 | [234] | |
| pUL37 | Deamidates RIG-I and cGAS inhibiting the ability to sense dsRNA and dsDNA respectively | [235,236] |
| pUS11 | Interacts with both MDA5 and RIG-I blocking their interaction with MAVS | [237] |
| Inhibits TBK1 | [238] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Danastas, K.; Miranda-Saksena, M.; Cunningham, A.L. Herpes Simplex Virus Type 1 Interactions with the Interferon System. Int. J. Mol. Sci. 2020, 21, 5150. https://doi.org/10.3390/ijms21145150
Danastas K, Miranda-Saksena M, Cunningham AL. Herpes Simplex Virus Type 1 Interactions with the Interferon System. International Journal of Molecular Sciences. 2020; 21(14):5150. https://doi.org/10.3390/ijms21145150
Chicago/Turabian StyleDanastas, Kevin, Monica Miranda-Saksena, and Anthony L. Cunningham. 2020. "Herpes Simplex Virus Type 1 Interactions with the Interferon System" International Journal of Molecular Sciences 21, no. 14: 5150. https://doi.org/10.3390/ijms21145150
APA StyleDanastas, K., Miranda-Saksena, M., & Cunningham, A. L. (2020). Herpes Simplex Virus Type 1 Interactions with the Interferon System. International Journal of Molecular Sciences, 21(14), 5150. https://doi.org/10.3390/ijms21145150