Tumor-Associated Macrophages in Osteosarcoma: From Mechanisms to Therapy

 , ,
, , 
 and
and 
Abstract
:
1. Introduction
2. Osteosarcoma and Macrophage Function
2.1. Osteosarcoma: Clinical and Molecular Features
2.2. Macrophages and Bone Homeostasis
2.3. Macrophages: Origin and Plasticity
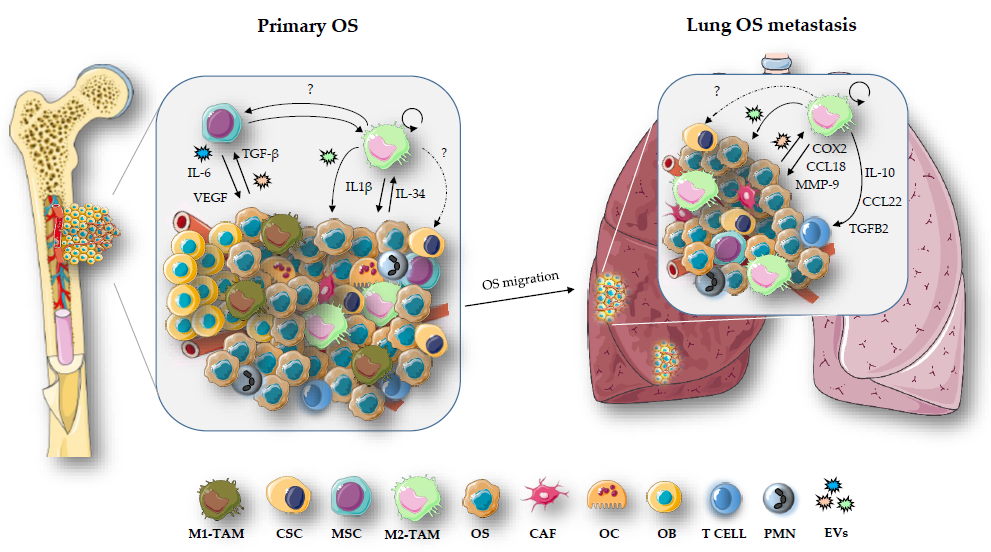
3. TAMs and TME in OS
3.1. TAMs and OS Metastasis
3.2. Crosstalk between TAMs, CSCs and MSCs in OS
3.3. Extracellular Vesicles in OS
4. TAM and OS Therapy
TAMs as a Target in OS Therapy
5. Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Nie, Z.; Peng, H. Osteosarcoma in patients below 25 years of age: An observational study of incidence, metastasis, treatment and outcomes. Oncol. Lett. 2018, 16, 6502–6514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Zhang, Y.; Li, R.; Li, J.; Lu, X.; Zhang, Y. The efficacy and safety comparison of first-line chemotherapeutic agents (high-dose methotrexate, doxorubicin, cisplatin, and ifosfamide) for osteosarcoma: A network meta-analysis. J. Orthop. Surg. Res. 2020, 15, 51. [Google Scholar] [CrossRef] [Green Version]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Buddingh, E.P.; Kuijjer, M.L.; Duim, R.A.; Bürger, H.; Agelopoulos, K.; Myklebost, O.; Serra, M.; Mertens, F.; Hogendoorn, P.C.; Lankester, A.C.; et al. Tumor-infiltrating macrophages are associated with metastasis suppression in high-grade osteosarcoma: A rationale for treatment with macrophage activating agents. Clin. Cancer Res. 2011, 17, 2110–2119. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Guo, W.; Ren, T.; Huang, Y.; Wang, S.; Liu, K.; Zheng, B.; Yang, K.; Zhang, H.; Liang, X. Tumor-associated macrophages promote lung metastasis and induce epithelial-mesenchymal transition in osteosarcoma by activating the COX-2/STAT3 axis. Cancer Lett. 2019, 440–441, 116–125. [Google Scholar] [CrossRef]
- Dumars, C.; Ngyuen, J.M.; Gaultier, A.; Lanel, R.; Corradini, N.; Gouin, F.; Heymann, D.; Heymann, M.F. Dysregulation of macrophage polarization is associated with the metastatic process in osteosarcoma. Oncotarget 2016, 7, 78343–78354. [Google Scholar] [CrossRef] [Green Version]
- Ségaliny, A.I.; Mohamadia, A.; Dizier, B.; Lokajczyk, A.; Brion, R.; Lanel, R.; Amiaud, J.; Charrier, C.; Boisson-Vidal, C.; Heymann, D. Interleukin-34 promotes tumor progression and metastatic process in osteosarcoma through induction of angiogenesis and macrophage recruitment. Int. J. Cancer 2015, 137, 73–85. [Google Scholar] [CrossRef]
- Zhou, Q.; Xian, M.; Xiang, S.; Xiang, D.; Shao, X.; Wang, J.; Cao, J.; Yang, X.; Yang, B.; Ying, M.; et al. All-Trans Retinoic Acid Prevents Osteosarcoma Metastasis by Inhibiting M2 Polarization of Tumor-Associated Macrophages. Cancer Immunol. Res. 2017, 5, 547–559. [Google Scholar] [CrossRef] [Green Version]
- Ottaviani, G.; Jaffe, N. The epidemiology of osteosarcoma. Cancer Treat. Res. 2009, 152, 3–13. [Google Scholar] [CrossRef]
- Gonzalez, A.L.; Cates, J.M.M. Osteosarcoma: Differential Diagnostic Considerations. Surg. Pathol. Clin. 2012, 5, 117–146. [Google Scholar] [CrossRef] [PubMed]
- Nouri, H.; Maitigue, M.B.; Abid, L.; Nouri, N.; Abdelkader, A.; Bouaziz, M.; Mestiri, M. Surface osteosarcoma: Clinical features and therapeutic implications. J. Bone Oncol. 2015, 4, 115–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelini, A.; Mavrogenis, A.F.; Trovarelli, G.; Ferrari, S.; Picci, P.; Ruggieri, P. Telangiectatic osteosarcoma: A review of 87 cases. J. Cancer Res. Clin. Oncol. 2016, 142, 2197–2207. [Google Scholar] [CrossRef] [PubMed]
- Laschi, M.; Bernardini, G.; Geminiani, M.; Ghezzi, L.; Amato, L.; Braconi, D.; Millucci, L.; Frediani, B.; Spreafico, A.; Franchi, A.; et al. Establishment of Four New Human Primary Cell Cultures from Chemo-Naïve Italian Osteosarcoma Patients. J. Cell Physiol. 2015, 230, 2718–2727. [Google Scholar] [CrossRef] [PubMed]
- Rickel, K.; Fang, F.; Tao, J. Molecular genetics of osteosarcoma. Bone 2017, 102, 69–79. [Google Scholar] [CrossRef]
- Gianferante, D.M.; Mirabello, L.; Savage, S.A. Germline and somatic genetics of osteosarcoma—Connecting aetiology, biology and therapy. Nat. Rev. Endocrinol. 2017, 13, 480–491. [Google Scholar] [CrossRef]
- Mirabello, L.; Zhu, B.; Koster, R.; Karlins, E.; Dean, M.; Yeager, M.; Gianferante, M.; Spector, L.G.; Morton, L.M.; Karyadi, D.; et al. Frequency of Pathogenic Germline Variants in Cancer-Susceptibility Genes in Patients with Osteosarcoma. JAMA Oncol. 2020. [Google Scholar] [CrossRef]
- Perry, J.A.; Kiezun, A.; Tonzi, P.; Van Allen, E.M.; Carter, S.L.; Baca, S.C.; Cowley, G.S.; Bhatt, A.S.; Rheinbay, E.; Pedamallu, C.S.; et al. Complementary genomic approaches highlight the PI3K/mTOR pathway as a common vulnerability in osteosarcoma. Proc. Natl. Acad. Sci. USA 2014, 111, E5564–E5573. [Google Scholar] [CrossRef] [Green Version]
- Kovac, M.; Blattmann, C.; Ribi, S.; Smida, J.; Mueller, N.S.; Engert, F.; Castro-Giner, F.; Weischenfeldt, J.; Kovacova, M.; Krieg, A.; et al. Exome sequencing of osteosarcoma reveals mutation signatures reminiscent of BRCA deficiency. Nat. Commun. 2015, 6, 8940. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Velletri, T.; Xie, N.; Wang, Y.; Huang, Y.; Yang, Q.; Chen, X.; Chen, Q.; Shou, P.; Gan, Y.; Cao, G.; et al. P53 functional abnormality in mesenchymal stem cells promotes osteosarcoma development. Cell Death Dis. 2016, 7, e2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franceschini, N.; Lam, S.W.; Cleton-Jansen, A.M.; Bovée, J.V.M.G. What’s new in bone forming tumours of the skeleton? Virchows Arch. 2020, 476, 147–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, W.; Li, Y.; Zhang, J.; Li, J.; Gao, J. Combined analysis of DNA methylation and gene expression profiles of osteosarcoma identified several prognosis signatures. Gene 2018, 650, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Liu, W.; Pan, Y.; Ni, P.; Ji, J.; Guo, L.; Zhang, J.; Wu, J.; Jiang, J.; Chen, X.; et al. Homeobox gene IRX1 is a tumor suppressor gene in gastric carcinoma. Oncogene 2010, 29, 3908–3920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriarity, B.S.; Otto, G.M.; Rahmann, E.P.; Rathe, S.K.; Wolf, N.K.; Weg, M.T.; Manlove, L.A.; LaRue, R.S.; Temiz, N.A.; Molyneux, S.D.; et al. A Sleeping Beauty forward genetic screen identifies new genes and pathways driving osteosarcoma development and metastasis. Nat. Genet. 2015, 47, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, J.M.; Wijetunga, N.A.; Fazzari, M.J.; Krailo, M.; Barkauskas, D.A.; Gorlick, R.; Greally, J.M. Predictive properties of DNA methylation patterns in primary tumor samples for osteosarcoma relapse status. Epigenetics 2015, 10, 31–39. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Hao, M.; Du, X.; Chen, K.; Wang, G.; Yang, J. Advances in targeted therapy for osteosarcoma. Discov. Med. 2014, 17, 301–307. [Google Scholar]
- Zhang, J.; Yu, X.H.; Yan, Y.G.; Wang, C.; Wang, W.J. PI3K/Akt signaling in osteosarcoma. Clin. Chim. Acta 2015, 444, 182–192. [Google Scholar] [CrossRef]
- Chen, C.; Zhao, M.; Tian, A.; Zhang, X.; Yao, Z.; Ma, X. Aberrant activation of Wnt/β-catenin signaling drives proliferation of bone sarcoma cells. Oncotarget 2015, 6, 17570–17583. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.; Cao, Y.; Luo, Y.; Hu, H.; Ling, H. Signalling mechanism(s) of epithelial-mesenchymal transition and cancer stem cells in tumour therapeutic resistance. Clin. Chim. Acta 2018, 483, 156–163. [Google Scholar] [CrossRef]
- Iwaya, K.; Ogawa, H.; Kuroda, M.; Izumi, M.; Ishida, T.; Mukai, K. Cytoplasmic and/or nuclear staining of beta-catenin is associated with lung metastasis. Clin. Exp. Metastasis 2003, 20, 525–529. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Kurenbekova, L.; Gao, Y.; Roos, A.; Creighton, C.J.; Rao, P.; Hicks, J.; Man, T.K.; Lau, C.; Brown, A.M.; et al. NKD2, a negative regulator of Wnt signaling, suppresses tumor growth and metastasis in osteosarcoma. Oncogene 2015, 34, 5069–5079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luke, J.J.; Bao, R.; Sweis, R.F.; Spranger, S.; Gajewski, T.F. WNT/β-catenin Pathway Activation Correlates with Immune Exclusion across Human Cancers. Clin. Cancer Res. 2019, 25, 3074–3083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirotsu, M.; Setoguchi, T.; Sasaki, H.; Matsunoshita, Y.; Gao, H.; Nagao, H.; Kunigou, O.; Komiya, S. Smoothened as a new therapeutic target for human osteosarcoma. Mol. Cancer 2010, 9, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, W.W.; Pinnaduwage, D.; Gokgoz, N.; Wunder, J.S.; Andrulis, I.L. Aberrant hedgehog signaling and clinical outcome in osteosarcoma. Sarcoma 2014, 2014, 261804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardini, G.; Geminiani, M.; Gambassi, S.; Orlandini, M.; Petricci, E.; Marzocchi, B.; Laschi, M.; Taddei, M.; Manetti, F.; Santucci, A. Novel smoothened antagonists as anti-neoplastic agents for the treatment of osteosarcoma. J. Cell. Physiol. 2018, 233, 4961–4971. [Google Scholar] [CrossRef] [PubMed]
- Lo, W.W.; Wunder, J.S.; Dickson, B.C.; Campbell, V.; McGovern, K.; Alman, B.A.; Andrulis, I.L. Involvement and targeted intervention of dysregulated Hedgehog signaling in osteosarcoma. Cancer 2014, 120, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Righi, A.; Gambarotti, M.; Sbaraglia, M.; Sisto, A.; Ferrari, S.; Dei Tos, A.P.; Picci, P. P16 Expression as a Prognostic and Predictive Marker in High-Grade Localized Osteosarcoma of the Extremities: An Analysis of 357 Cases. Hum. Pathol. 2016, 58, 15–23. [Google Scholar] [CrossRef]
- Abdou, A.G.; Kandil, M.; Asaad, N.Y.; Dawoud, M.M.; Shahin, A.A.; Abd Eldayem, A.F. The prognostic role of Ezrin and HER2/neu expression in osteosarcoma. Appl. Immunohistochem. Mol. Morphol. 2016, 24, 355–363. [Google Scholar] [CrossRef]
- Xie, X.; Li, Y.; Zhu, H.; Kuang, Z.; Chen, D.; Fan, T. Prognostic Significance of β-Catenin Expression in Osteosarcoma: A Meta-Analysis. Front. Oncol. 2020, 10, 402. [Google Scholar] [CrossRef]
- Corre, I.; Verrecchia, F.; Crenn, V.; Redini, F.; Trichet, V. The Osteosarcoma Microenvironment: A Complex but Targetable Ecosystem. Cells 2020, 9, 976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giurisato, E.; Xu, Q.; Lonardi, S.; Telfer, B.; Russo, I.; Pearson, A.; Finegan, K.G.; Wang, W.; Wang, J.; Gray, N.S.; et al. Myeloid ERK5 deficiency suppresses tumor growth by blocking protumor macrophage polarization via STAT3 inhibition. Proc. Natl. Acad. Sci. USA 2018, 115, E2801–E2810. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.; Plüddemann, A.; Martinez Estrada, F. Macrophage heterogeneity in tissues: Phenotypic diversity and functions. Immunol. Rev. 2014, 262, 36–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollard, J.W. Trophic macrophages in development and disease. Nat. Rev. Immunol. 2009, 9, 259–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhopadhyay, S.; Plüddemann, A.; Gordon, S. Macrophage pattern recognition receptors in immunity, homeostasis and self tolerance. Adv. Exp. Med. Biol. 2009, 653, 1–14. [Google Scholar] [CrossRef]
- Aderem, A. Phagocytosis and the inflammatory response. J. Infect. Dis. 2003, 187 (Suppl. 2), S340–S345. [Google Scholar] [CrossRef]
- Tsukasaki, M.; Takayanagi, H. Osteoimmunology: Evolving concepts in bone-immune interactions in health and disease. Nat. Rev. Immunol. 2019, 19, 626–642. [Google Scholar] [CrossRef]
- Chang, M.K.; Raggatt, L.J.; Alexander, K.A.; Kuliwaba, J.S.; Fazzalari, N.L.; Schroder, K.; Maylin, E.R.; Ripoll, V.M.; Hume, D.A.; Pettit, A.R. Osteal tissue macrophages are intercalated throughout human and mouse bone lining tissues and regulate osteoblast function in vitro and in vivo. J. Immunol. 2008, 181, 1232–1244. [Google Scholar] [CrossRef] [Green Version]
- Champagne, C.M.; Takebe, J.; Offenbacher, S.; Cooper, L.F. Macrophage cell lines produce osteoinductive signals that include bone morphogenetic protein-2. Bone 2002, 30, 26–31. [Google Scholar] [CrossRef]
- Nicolaidou, V.; Wong, M.M.; Redpath, A.N.; Ersek, A.; Baban, D.F.; Williams, L.M.; Cope, A.P.; Horwood, N.J. Monocytes induce STAT3 activation in human mesenchymal stem cells to promote osteoblast formation. PLoS ONE 2012, 7, e39871. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, T.J.; Hodge, J.M.; Singh, P.P.; Eeles, D.G.; Collier, F.M.; Holten, I.; Ebeling, P.R.; Nicholson, G.C.; Quinn, J.M. Cord blood-derived macrophage-lineage cells rapidly stimulate osteoblastic maturation in mesenchymal stem cells in a glycoprotein-130 dependent manner. PLoS ONE 2013, 8, e73266. [Google Scholar] [CrossRef] [PubMed]
- Guihard, P.; Danger, Y.; Brounais, B.; David, E.; Brion, R.; Delecrin, J.; Richards, C.D.; Chevalier, S.; Rédini, F.; Heymann, D.; et al. Induction of osteogenesis in mesenchymal stem cells by activated monocytes/macrophages depends on oncostatin M signaling. Stem Cells 2012, 30, 762–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnett, S.H.; Beus, B.J.; Avdiushko, R.; Qualls, J.; Kaplan, A.M.; Cohen, D.A. Development of peritoneal adhesions in macrophage depleted mice. J. Surg. Res. 2006, 131, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Burnett, S.H.; Kershen, E.J.; Zhang, J.; Zeng, L.; Straley, S.C.; Kaplan, A.M.; Cohen, D.A. Conditional macrophage ablation in transgenic mice expressing a Fas-based suicide gene. J. Leukocyte Biol. 2004, 75, 612–623. [Google Scholar] [CrossRef]
- Vi, L.; Baht, G.S.; Whetstone, H.; Ng, A.; Wei, Q.; Poon, R.; Mylvaganam, S.; Grynpas, M.; Alman, B.A. Macrophages promote osteoblastic differentiation in-vivo: Implications in fracture repair and bone homeostasis. J. Bone Miner. Res. 2015, 30, 1090–1102. [Google Scholar] [CrossRef]
- Cho, S.W.; Soki, F.N.; Koh, A.J.; Eber, M.R.; Entezami, P.; Park, S.I.; van Rooijen, N.; McCauley, L.K. Osteal macrophages support physiologic skeletal remodeling and anabolic actions of parathyroid hormone in bone. PNAS 2014, 111, 1545–1550. [Google Scholar] [CrossRef] [Green Version]
- Winkler, I.G.; Sims, N.A.; Pettit, A.R.; Barbier, V.; Nowlan, B.; Helwani, F.; Poulton, I.J.; van Rooijen, N.; Alexander, K.A.; Raggatt, L.J.; et al. Bone marrow macrophages maintain hematopoietic stem cell (HSC) niches and their depletion mobilizes HSCs. Blood 2010, 116, 4815–4828. [Google Scholar] [CrossRef] [Green Version]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Martinez, F.O. Regulators of macrophage activation. Eur. J. Immunol. 2011, 41, 1531–1534. [Google Scholar] [CrossRef]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Poon, I.K.; Lucas, C.D.; Rossi, A.G.; Ravichandran, K.S. Apoptotic cell clearance: Basic biology and therapeutic potential. Nat. Rev. Immunol. 2014, 14, 166–180. [Google Scholar] [CrossRef] [Green Version]
- Van Furth, R.; Cohn, Z.A. The origin and kinetics of mononuclear phagocytes. J. Exp. Med. 1968, 128, 415–435. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, R.T.; Strausbauch, P.H.; Volkman, A. Resident macrophage proliferation in mice depleted of blood monocytes by strontium-89. Lab. Investig. 1982, 46, 165–170. [Google Scholar]
- Schulz, C.; Gomez Perdiguero, E.; Chorro, L.; Szabo-Rogers, H.; Cagnard, N.; Kierdorf, K.; Prinz, M.; Wu, B.; Jacobsen, S.E.; Pollard, J.W.; et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 2012, 336, 86–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yona, S.; Kim, K.W.; Wolf, Y.; Mildner, A.; Varol, D.; Breker, M.; Strauss-Ayali, D.; Viukov, S.; Guilliams, M.; Misharin, A.; et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 2013, 38, 79–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, D.; Chow, A.; Noizat, C.; Teo, P.; Beasley, M.B.; Leboeuf, M.; Becker, C.D.; See, P.; Price, J.; Lucas, D.; et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 2013, 38, 792–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guilliams, M.; De Kleer, I.; Henri, S.; Post, S.; Vanhoutte, L.; De Prijck, S.; Deswarte, K.; Malissen, B.; Hammad, H.; Lambrecht, B.N. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J. Exp. Med. 2013, 210, 1977–1992. [Google Scholar] [CrossRef] [Green Version]
- Jakubzick, C.; Gautier, E.L.; Gibbings, S.L.; Sojka, D.K.; Schlitzer, A.; Johnson, T.E.; Ivanov, S.; Duan, Q.; Bala, S.; Condon, T.; et al. Minimal differentiation of classical monocytes as they survey steady-state tissues and transport antigen to lymph nodes. Immunity 2013, 39, 599–610. [Google Scholar] [CrossRef] [Green Version]
- Epelman, S.; Lavine, K.J.; Beaudin, A.E.; Sojka, D.K.; Carrero, J.A.; Calderon, B.; Brija, T.; Gautier, E.L.; Ivanov, S.; Satpathy, A.T.; et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 2014, 40, 91–104. [Google Scholar] [CrossRef] [Green Version]
- Perdiguero, E.G.; Geissmann, F. The development and maintenance of resident macrophages. Nat. Immunol. 2016, 17, 2–8. [Google Scholar] [CrossRef] [Green Version]
- Lavin, Y.; Mortha, A.; Rahman, A.; Merad, M. Regulation of macrophage development and function in peripheral tissues. Nat. Rev. Immunol. 2015, 15, 731–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginhoux, F.; Jung, S. Monocytes and macrophages: Developmental pathways and tissue homeostasis. Nat. Rev. Immunol. 2014, 14, 392–404. [Google Scholar] [CrossRef] [PubMed]
- Mass, E.; Ballesteros, I.; Farlik, M.; Halbritter, F.; Günther, P.; Crozet, L.; Jacome-Galarza, C.E.; Händler, K.; Klughammer, J.; Kobayashi, Y.; et al. Specification of tissue-resident macrophages during organogenesis. Science 2016, 353, aaf4238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoeffel, G.; Chen, J.; Lavin, Y.; Low, D.; Almeida, F.F.; See, P.; Beaudin, A.E.; Lum, J.; Low, I.; Forsberg, E.C.; et al. C-Myb(+) erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity 2015, 42, 665–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cecchini, M.G.; Dominguez, M.G.; Mocci, S.; Wetterwald, A.; Felix, R.; Fleisch, H.; Chisholm, O.; Hofstetter, W.; Pollard, J.W.; Stanley, E.R. Role of colony stimulating factor-1 in the establishment and regulation of tissue macrophages during postnatal development of the mouse. Development 1994, 120, 1357–1372. [Google Scholar] [PubMed]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [Green Version]
- Novak, M.L.; Koh, T.J. Macrophage phenotypes during tissue repair. J. Leukoc. Biol. 2013, 93, 875–881. [Google Scholar] [CrossRef] [Green Version]
- Martinez, F.O.; Sica, A.; Mantovani, A.; Locati, M. Macrophage activation and polarization. Front. Biosci. 2008, 13, 453–461. [Google Scholar] [CrossRef] [Green Version]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Gordon, S. Alternative activation of macrophages. Nat. Rev. Immunol. 2003, 3, 23–35. [Google Scholar] [CrossRef]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Duluc, D.; Delneste, Y.; Tan, F.; Moles, M.P.; Grimaud, L.; Lenoir, J.; Preisser, L.; Anegon, I.; Catala, L.; Ifrah, N.; et al. Tumor-associated leukemia inhibitory factor and IL-6 skew monocyte differentiation into tumor-associated macrophage-like cells. Blood 2007, 110, 4319–4330. [Google Scholar] [CrossRef] [PubMed]
- Bingle, L.; Brown, N.J.; Lewis, C.E. The role of tumour-associated macrophages in tumour progression: Implications for new anticancer therapies. J. Pathol. 2002, 196, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Franklin, R.A.; Liao, W.; Sarkar, A.; Kim, M.V.; Bivona, M.R.; Liu, K.; Pamer, E.G.; Li, M.O. The cellular and molecular origin of tumor-associated macrophages. Science 2014, 344, 921–925. [Google Scholar] [CrossRef] [Green Version]
- Bottazzi, B.; Erba, E.; Nobili, N.; Fazioli, F.; Rambaldi, A.; Mantovani, A. A paracrine circuit in the regulation of the proliferation of macrophages infiltrating murine sarcomas. J. Immunol. 1990, 144, 2409–2412. [Google Scholar]
- Joyce, J.A.; Pollard, J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer 2009, 9, 239–252. [Google Scholar] [CrossRef]
- Wyckoff, J.B.; Wang, Y.; Lin, E.Y.; Li, J.F.; Goswami, S.; Stanley, E.R.; Segall, J.E.; Pollard, J.W.; Condeelis, J. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res. 2007, 67, 2649–2656. [Google Scholar] [CrossRef] [Green Version]
- Condeelis, J.; Pollard, J.W. Macrophages: Obligate partners for tumor cell migration, invasion, and metastasis. Cell 2006, 124, 263–266. [Google Scholar] [CrossRef] [Green Version]
- Harney, A.S.; Arwert, E.N.; Entenberg, D.; Wang, Y.; Guo, P.; Qian, B.Z.; Oktay, M.H.; Pollard, J.W.; Jones, J.G.; Condeelis, J.S. Real-Time Imaging Reveals Local, Transient Vascular Permeability, and Tumor Cell Intravasation Stimulated by TIE2hi Macrophage-Derived VEGFA. Cancer Discov. 2015, 5, 932–943. [Google Scholar] [CrossRef] [Green Version]
- Frankenberger, C.; Rabe, D.; Bainer, R.; Sankarasharma, D.; Chada, K.; Krausz, T.; Gilad, Y.; Becker, L.; Rosner, M.R. Metastasis Suppressors Regulate the Tumor Microenvironment by Blocking Recruitment of Prometastatic Tumor-Associated Macrophages. Cancer Res. 2015, 75, 4063–4073. [Google Scholar] [CrossRef] [Green Version]
- Terabe, M.; Matsui, S.; Park, J.M.; Mamura, M.; Noben-Trauth, N.; Donaldson, D.D.; Chen, W.; Wahl, S.M.; Ledbetter, S.; Pratt, B.; et al. Transforming growth factor-beta production and myeloid cells are an effector mechanism through which CD1d-restricted T cells block cytotoxic T lymphocyte-mediated tumor immunosurveillance: Abrogation prevents tumor recurrence. J. Exp. Med. 2003, 198, 1741–1752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zea, A.H.; Rodriguez, P.C.; Atkins, M.B.; Hernandez, C.; Signoretti, S.; Zabaleta, J.; McDermott, D.; Quiceno, D.; Youmans, A.; O’Neill, A.; et al. Arginase-producing myeloid suppressor cells in renal cell carcinoma patients: A mechanism of tumor evasion. Cancer Res. 2005, 65, 3044–3048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sica, A.; Bronte, V. Altered macrophage differentiation and immune dysfunction in tumor development. J. Clin. Investig. 2007, 117, 1155–1166. [Google Scholar] [CrossRef] [PubMed]
- Munder, M.; Eichmann, K.; Modolell, M. Alternative metabolic states in murine macrophages reflected by the nitric oxide synthase/arginase balance: Competitive regulation by CD4+ T cells correlates with Th1/Th2 phenotype. J. Immunol. 1998, 160, 5347–5354. [Google Scholar] [PubMed]
- Chen, L.; Li, J.; Wang, F.; Dai, C.; Wu, F.; Liu, X.; Li, T.; Glauben, R.; Zhang, Y.; Nie, G.; et al. Tie2 Expression on Macrophages Is Required for Blood Vessel Reconstruction and Tumor Relapse after Chemotherapy. Cancer Res. 2016, 76, 6828–6838. [Google Scholar] [CrossRef] [Green Version]
- Pucci, F.; Venneri, M.A.; Biziato, D.; Nonis, A.; Moi, D.; Sica, A.; Di Serio, C.; Naldini, L.; Di Palma, M. A distinguishing gene signature shared by tumor-infiltrating Tie2-expressing monocytes, blood “resident” monocytes, and embryonic macrophages suggests common functions and developmental relationships. Blood 2009, 114, 901–914. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Guo, W.; Ren, T.; Huang, Y.; Sun, K.; Zhang, H.; Yu, Y.; Wang, W.; Niu, J. Macrophages reduce the sensitivity of osteosarcoma to neoadjuvant chemotherapy drugs by secreting Interleukin-1 beta. Cancer Lett. 2020, 480, 4–14. [Google Scholar] [CrossRef]
- Han, Q.; Shi, H.; Liu, F. CD163(+) M2-type tumor-associated macrophage support the suppression of tumor-infiltrating T cells in osteosarcoma. Int. Immunopharmacol. 2016, 34, 101–106. [Google Scholar] [CrossRef]
- Su, Y.; Zhou, Y.; Sun, Y.J.; Wang, Y.L.; Yin, J.Y.; Huang, Y.J.; Zhang, J.J.; He, A.N.; Han, K.; Zhang, H.Z.; et al. Macrophage-derived CCL18 promotes osteosarcoma proliferation and migration by upregulating the expression of UCA1. J. Mol. Med. 2019, 97, 49–61. [Google Scholar] [CrossRef]
- Shao, X.J.; Xiang, S.F.; Chen, Y.Q.; Zhang, N.; Cao, J.; Zhu, H.; Yang, B.; Zhou, Q.; Ying, M.D.; He, Q.J. Inhibition of M2-like macrophages by all-trans retinoic acid prevents cancer initiation and stemness in osteosarcoma cells. Acta Pharmacol. Sin. 2019, 40, 1343–1350. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.H.; Feng, G.W.; Wang, Z.L.; Du, Y.; Shen, C.; Hui, H.; Peng, D.; Li, Z.J.; Kong, D.L.; Tian, J. Activation of mesenchymal stem cells by macrophages promotes tumor progression through immune suppressive effects. Oncotarget 2016, 7, 20934–20944. [Google Scholar] [CrossRef] [PubMed]
- Wolf-Dennen, K.; Gordon, N.; Kleinerman, E.S. Exosomal communication by metastatic osteosarcoma cells modulates alveolar macrophages to an M2 tumor-promoting phenotype and inhibits tumoricidal functions. Oncoimmunology 2020, 9, 1747677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Punzo, F.; Bellini, G.; Tortora, C.; Pinto, D.D.; Argenziano, M.; Pota, E.; Paola, A.D.; Martino, M.D.; Rossi, F. Mifamurtide and TAM-like macrophages: Effect on proliferation, migration and differentiation of osteosarcoma cells. Oncotarget 2020, 11, 687–698. [Google Scholar] [CrossRef] [Green Version]
- Najafi, M.; Farhood, B.; Mortezaee, K. Cancer stem cells (CSCs) in cancer progression and therapy. J. Cell Physiol. 2019, 234, 8381–8395. [Google Scholar] [CrossRef] [PubMed]
- Posthuma DeBoer, J.; van Royen, B.J.; Helder, M.N. Mechanisms of therapy resistance in osteosarcoma: A review. Oncol. Discov. 2013, 1, 8. [Google Scholar] [CrossRef] [Green Version]
- Honoki, K.; Fujii, H.; Kubo, A.; Kido, A.; Mori, T.; Tanaka, Y.; Tsujiuchi, T. Possible involvement of stem-like populations with elevated ALDH1 in sarcomas for chemotherapeutic drug resistance. Oncol. Rep. 2010, 24, 501–505. [Google Scholar] [CrossRef]
- Tirino, V.; Desiderio, V.; Paino, F.; De Rosa, A.; Papaccio, F.; Fazioli, F.; Pirozzi, G.; Papaccio, G. Human primary bone sarcomas contain CD133+ cancer stem cells displaying high tumorigenicity in vivo. FASEB J. 2011, 25, 2022–2030. [Google Scholar] [CrossRef]
- Adhikari, A.S.; Agarwal, N.; Wood, B.M.; Porretta, C.; Ruiz, B. CD117 and Stro-1 identify osteosarcoma tumor-initiating cells associated with metastasis and drug resistance. Cancer Res. 2010, 70, 4602–4612. [Google Scholar] [CrossRef] [Green Version]
- Abarrategi, A.; Tornin, J.; Martinez-Cruzado, L.; Hamilton, A.; Martinez-Campos, E.; Rodrigo, J.P.; González, M.V.; Baldini, N.; Garcia-Castro, J.; Rodriguez, R. Osteosarcoma: Cells-of-Origin, Cancer Stem Cells, and Targeted Therapies. Stem Cells Int. 2016, 2016, 3631764. [Google Scholar] [CrossRef] [Green Version]
- Cortini, M.; Massa, A.; Avnet, S.; Bonuccelli, G.; Baldini, N. Tumor-Activated Mesenchymal Stromal Cells Promote Osteosarcoma Stemness and Migratory Potential via IL-6 Secretion. PLoS ONE 2016, 11, e0166500. [Google Scholar] [CrossRef] [Green Version]
- Brune, J.C.; Tormin, A.; Johansson, M.C.; Rissler, P.; Brosjö, O.; Löfvenberg, R.; von Steyern, F.V.; Mertens, F.; Rydholm, A.; Scheding, S. Mesenchymal stromal cells from primary osteosarcoma are non-malignant and strikingly similar to their bone marrow counterparts. Int. J. Cancer 2011, 129, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Mohseny, A.B.; Hogendoorn, P.C.; Cleton-Jansen, A.M. Mesenchymal stem cell transformation and sarcoma genesis. Clin. Sarcoma Res. 2013, 3, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Nail, L.R.; Brennan, M.; Rosset, P.; Deschaseaux, F.; Piloquet, P.; Pichon, O.; Le Caignec, C.; Crenn, V.; Layrolle, P.; Hérault, O.; et al. Comparison of Tumor- and Bone Marrow-Derived Mesenchymal Stromal/Stem Cells from Patients with High-Grade Osteosarcoma. Int. J. Mol. Sci. 2018, 19, 707. [Google Scholar] [CrossRef] [Green Version]
- Ridge, S.M.; Sullivan, F.J.; Glynn, S.A. Mesenchymal stem cells: Key players in cancer progression. Mol. Cancer 2017, 16, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.T.; Bian, Z.Y.; Fan, Q.M.; Li, G.; Tang, T.T. Human mesenchymal stem cells (hMSCs) target osteosarcoma and promote its growth and pulmonary metastasis. Cancer Lett. 2009, 281, 32–41. [Google Scholar] [CrossRef]
- Bergfeld, S.A.; DeClerck, Y.A. Bone marrow-derived mesenchymal stem cells and the tumor microenvironment. Cancer Metastasis Rev. 2010, 29, 249–261. [Google Scholar] [CrossRef]
- Zhang, P.; Dong, L.; Yan, K.; Long, H.; Yang, T.T.; Dong, M.Q.; Zhou, Y.; Fan, Q.Y.; Ma, B.A. CXCR4-mediated osteosarcoma growth and pulmonary metastasis is promoted by mesenchymal stem cells through VEGF. Oncol. Rep. 2013, 30, 1753–1761. [Google Scholar] [CrossRef]
- Spaeth, E.L.; Dembinski, J.L.; Sasser, A.K.; Watson, K.; Klopp, A.; Hall, B.; Andreeff, M.; Marini, F. Mesenchymal stem cell transition to tumor-associated fibroblasts contributes to fibrovascular network expansion and tumor progression. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Tu, B.; Zhu, J.; Liu, S.; Wang, L.; Fan, Q.; Hao, Y.; Fan, C.; Tang, T.T. Mesenchymal stem cells promote osteosarcoma cell survival and drug resistance through activation of STAT3. Oncotarget 2016, 7, 48296–48308. [Google Scholar] [CrossRef] [Green Version]
- Sainz, B., Jr.; Carron, E.; Vallespinós, M.; Machado, H.L. Cancer Stem Cells and Macrophages: Implications in Tumor Biology and Therapeutic Strategies. Mediat. Inflamm. 2016, 2016, 9012369. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Liao, D.; Chen, C.; Liu, Y.; Chuang, T.H.; Xiang, R.; Markowitz, D.; Reisfeld, R.A.; Luo, Y. Tumor-associated macrophages regulate murine breast cancer stem cells through a novel paracrine EGFR/Stat3/Sox-2 signaling pathway. Stem Cells 2013, 31, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Dong, Y.; Li, Y.; Wang, D.; Liu, S.; Wang, D.; Gao, Q.; Ji, S.; Chen, X.; Lei, Q.; et al. IL-10 derived from M2 macrophage promotes cancer stemness via JAK1/STAT1/NF-κB/Notch1 pathway in non-small cell lung cancer. Int. J. Cancer 2019, 145, 1099–1110. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.M.; Jing, Y.Y.; Yu, G.F.; Kou, X.R.; Ye, F.; Gao, L.; Li, R.; Zhao, Q.D.; Yang, Y.; Lu, Z.H.; et al. Tumor-associated macrophages promote cancer stem cell-like properties via transforming growth factor-beta1-induced epithelial-mesenchymal transition in hepatocellular carcinoma. Cancer Lett. 2014, 352, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Mitchem, J.B.; Brennan, D.J.; Knolhoff, B.L.; Belt, B.A.; Zhu, Y.; Sanford, D.E.; Belaygorod, L.; Carpenter, D.; Collins, L.; Piwnica-Worms, D.; et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013, 73, 1128–1141. [Google Scholar] [CrossRef]
- Asimakopoulos, F.; Kim, J.; Denu, R.A.; Hope, C.; Jensen, J.L.; Ollar, S.J.; Hebron, E.; Flanagan, C.; Callander, N.; Hematti, P. Macrophages in multiple myeloma: Emerging concepts and therapeutic implications. Leuk. Lymphoma 2013, 54, 2112–2121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaturvedi, P.; Gilkes, D.M.; Takano, N.; Semenza, G.L. Hypoxia-inducible factor-dependent signaling between triple-negative breast cancer cells and mesenchymal stem cells promotes macrophage recruitment. Proc. Natl. Acad. Sci. USA 2014, 111, E2120–E2129. [Google Scholar] [CrossRef] [Green Version]
- Perut, F.; Roncuzzi, L.; Baldini, N. The Emerging Roles of Extracellular Vesicles in Osteosarcoma. Front. Oncol. 2019, 9, 1342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell. Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [Green Version]
- Garimella, R.; Washington, L.; Isaacson, J.; Vallejo, J.; Spence, M.; Tawfik, O.; Rowe, P.; Brotto, M.; Perez, R. Extracellular Membrane Vesicles Derived from 143B Osteosarcoma Cells Contain Pro-Osteoclastogenic Cargo: A Novel Communication Mechanism in Osteosarcoma Bone Microenvironment. Transl. Oncol. 2014, 7, 331–340. [Google Scholar] [CrossRef] [Green Version]
- Troyer, R.M.; Ruby, C.E.; Goodall, C.P.; Yang, L.; Maier, C.S.; Albarqi, H.A.; Brady, J.V.; Bathke, K.; Taratula, O.; Mourich, D.; et al. Exosomes from Osteosarcoma and normal osteoblast differ in proteomic cargo and immunomodulatory effects on T cells. Exp. Cell. Res. 2017, 358, 369–376. [Google Scholar] [CrossRef]
- Raimondi, L.; De Luca, A.; Gallo, A.; Costa, V.; Russelli, G.; Cuscino, N.; Manno, M.; Raccosta, S.; Carina, V.; Bellavia, D.; et al. Osteosarcoma cell-derived exosomes affect tumor microenvironment by specific packaging of microRNAs. Carcinogenesis 2019, bgz130. [Google Scholar] [CrossRef] [PubMed]
- Torreggiani, E.; Roncuzzi, L.; Perut, F.; Zini, N.; Baldini, N. Multimodal transfer of MDR by exosomes in human osteosarcoma. Int. J. Oncol. 2016, 49, 189–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, B.; Liu, Q.; Liu, G.; Huang, X.; Zhu, G.; Gao, L.; Xu, Y. Macrophage-derived exosomes mediate osteosarcoma cell behavior by activating AKT signaling. RSC Adv. 2020, 10, 5032–5039. [Google Scholar] [CrossRef] [Green Version]
- Tu, B.; Peng, Z.X.; Fan, Q.M.; Du, L.; Yan, W.; Tang, T.T. Osteosarcoma cells promote the production of pro-tumor cytokines in mesenchymal stem cells by inhibiting their osteogenic differentiation through the TGF-β/Smad2/3 pathway. Exp. Cell. Res. 2014, 320, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Sousa, S.; Määttä, J. The role of tumour-associated macrophages in bone metastasis. J. Bone Oncol. 2016, 5, 135–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giurisato, E.; Lonardi, S.; Telfer, B.; Lussoso, S.; Risa-Ebri, B.; Zhang, J.; Russo, I.; Wang, J.; Santucci, A.; Finegan, K.; et al. Extracellular-regulated protein kinase 5-mediated control of p21 expression promotes macrophage proliferation associated with tumor growth and metastasis. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Bishop, M.W.; Janeway, K.A.; Gorlick, R. Future directions in the treatment of osteosarcoma. Curr. Opin. Pediatr. 2016, 28, 26–33. [Google Scholar] [CrossRef] [Green Version]
- Heymann, M.F.; Brown, H.K.; Heymann, D. Drugs in early clinical development for the treatment of osteosarcoma. Expert Opin. Investig. Drugs 2016, 25, 1265–1280. [Google Scholar] [CrossRef]
- Liu, B.; Huang, Y.; Sun, Y.; Zhang, J.; Yao, Y.; Shen, Z.; Xiang, D.; He, A. Prognostic value of inflammation-based scores in patients with osteosarcoma. Sci. Rep. 2016, 6, 39862. [Google Scholar] [CrossRef] [Green Version]
- Endo-Munoz, L.; Evdokiou, A.; Saunders, N.A. The role of osteoclasts and tumour-associated macrophages in osteosarcoma metastasis. Biochim. Biophys. Acta 2012, 1826, 434–442. [Google Scholar] [CrossRef]
- Meyers, P.A.; Schwartz, C.L.; Krailo, M.; Kleinerman, E.S.; Betcher, D.; Bernstein, M.L.; Conrad, E.; Ferguson, W.; Gebhardt, M.; Goorin, A.M.; et al. Osteosarcoma: A randomized, prospective trial of the addition of ifosfamide and/or muramyl tripeptide to cisplatin, doxorubicin, and high-dose methotrexate. J. Clin. Oncol. 2005, 23, 2004–2011. [Google Scholar] [CrossRef] [PubMed]
- Meyers, P.A.; Schwartz, C.L.; Krailo, M.D.; Healey, J.H.; Bernstein, M.L.; Betcher, D.; Ferguson, W.S.; Gebhardt, M.C.; Goorin, A.M.; Harris, M.; et al. Osteosarcoma: The addition of muramyl tripeptide to chemotherapy improves overall survival--a report from the Children’s Oncology Group. J. Clin. Oncol. 2008, 26, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Chou, A.J.; Kleinerman, E.S.; Krailo, M.D.; Chen, Z.; Betcher, D.L.; Healey, J.H.; Conrad, E.U., 3rd; Nieder, M.L.; Weiner, M.A.; Wells, R.J.; et al. Addition of muramyl tripeptide to chemotherapy for patients with newly diagnosed metastatic osteosarcoma: A report from the Children’s Oncology Group. Cancer 2009, 115, 5339–5348. [Google Scholar] [CrossRef] [PubMed]
- Kleinerman, E.S.; Raymond, A.K.; Bucana, C.D.; Jaffe, N.; Harris, M.B.; Krakoff, I.H.; Benjamin, R.; Fidler, I.J. Unique histological changes in lung metastases of osteosarcoma patients following therapy with liposomal muramyl tripeptide (CGP 19835A lipid). Cancer Immunol. Immunother. 1992, 34, 211–220. [Google Scholar] [CrossRef]
- Kleinerman, E.S.; Gano, J.B.; Johnston, D.A.; Benjamin, R.S.; Jaffe, N. Efficacy of liposomal muramyl tripeptide (CGP 19835A) in the treatment of relapsed osteosarcoma. Am. J. Clin. Oncol. 1995, 18, 93–99. [Google Scholar] [CrossRef]
- Pahl, J.H.; Kwappenberg, K.M.; Varypataki, E.M.; Santos, S.J.; Kuijjer, M.L.; Mohamed, S.; Wijnen, J.T.; van Tol, M.J.; Cleton-Jansen, A.M.; Egeler, R.M.; et al. Macrophages inhibit human osteosarcoma cell growth after activation with the bacterial cell wall derivative liposomal muramyl tripeptide in combination with interferon-γ. J. Exp. Clin. Cancer Res. 2014, 33, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ando, K.; Mori, K.; Corradini, N.; Redini, F.; Heymann, D. Mifamurtide for the treatment of nonmetastatic osteosarcoma. Expert. Opin. Pharmacother. 2011, 12, 285–292. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Chen, Y.; Liu, X.; Zhang, J.; He, X.; Teng, G.; Yu, D. Tim3/Gal9 interactions between T cells and monocytes result in an immunosuppressive feedback loop that inhibits Th1 responses in osteosarcoma patients. Int. Immunopharmacol. 2017, 44, 153–159. [Google Scholar] [CrossRef]
- Oldridge, E.E.; Walker, H.F.; Stower, M.J.; Simms, M.S.; Mann, V.M.; Collins, A.T.; Pellacani, D.; Maitland, N.J. Retinoic acid represses invasion and stem cell phenotype by induction of the metastasis suppressors RARRES1 and LXN. Oncogenesis 2013, 2, e45. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Qi, J.; Li, X.T.; Zhou, K.; Xu, J.H.; Zhou, Y.; Zhang, G.Q.; Xu, J.P.; Zhou, R.J. ATRA and Genistein synergistically inhibit the metastatic potential of human lung adenocarcinoma cells. Int. J. Clin. Exp. Med. 2015, 8, 4220–4227. [Google Scholar]
- Kimura, Y.; Sumiyoshi, M. Antitumor and antimetastatic actions of dihydroxycoumarins (esculetin or fraxetin) through the inhibition of M2 macrophage differentiation in tumor-associated macrophages and/or G1 arrest in tumor cells. Eur. J. Pharmacol. 2015, 746, 115–125. [Google Scholar] [CrossRef]
- Tsagozis, P.; Eriksson, F.; Pisa, P. Zoledronic acid modulates antitumoral responses of prostate cancer-tumor associated macrophages. Cancer Immunol. Immunother. 2008, 57, 1451–1459. [Google Scholar] [CrossRef]
- Junankar, S.; Shay, G.; Jurczyluk, J.; Ali, N.; Down, J.; Pocock, N.; Parker, A.; Nguyen, A.; Sun, S.; Kashemirov, B.; et al. Real-time intravital imaging establishes tumor-associated macrophages as the extraskeletal target of bisphosphonate action in cancer. Cancer Discov. 2015, 5, 35–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dass, C.R.; Choong, P.F. Zoledronic acid inhibits osteosarcoma growth in an orthotopic model. Mol. Cancer Ther. 2007, 6, 3263–3270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labrinidis, A.; Hay, S.; Liapis, V.; Ponomarev, V.; Findlay, D.M.; Evdokiou, A. Zoledronic acid inhibits both the osteolytic and osteoblastic components of osteosarcoma lesions in a mouse model. Clin. Cancer Res. 2009, 15, 3451–3461. [Google Scholar] [CrossRef] [Green Version]
- Ory, B.; Heymann, M.F.; Kamijo, A.; Gouin, F.; Heymann, D.; Redini, F. Zoledronic acid suppresses lung metastases and prolongs overall survival of osteosarcoma-bearing mice. Cancer 2005, 104, 2522–2529. [Google Scholar] [CrossRef] [PubMed]
- Van Rooijen, N.; Sanders, A. Elimination, blocking, and activation of macrophages: Three of a kind? J. Leukoc. Biol. 1997, 62, 702–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Zhang, X.H.; Massagué, J. Macrophage binding to receptor VCAM-1 transmits survival signals in breast cancer cells that invade the lungs. Cancer Cell 2011, 20, 538–549. [Google Scholar] [CrossRef] [Green Version]
- Dhupkar, P.; Gordon, N.; Stewart, J.; Kleinerman, E.S. Anti-PD-1 therapy redirects macrophages from an M2 to an M1 phenotype inducing regression of OS lung metastases. Cancer Med. 2018, 7, 2654–2664. [Google Scholar] [CrossRef]
- Pinto, N.; Park, J.R.; Murphy, E.; Yearley, J.; McClanahan, T.; Annamalai, L.; Hawkins, D.S.; Rudzinski, E.R. Patterns of PD-1, PD-L1, and PD-L2 expression in pediatric solid tumors. Pediatr. Blood Cancer 2017, 64. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.K.; Cote, G.M.; Choy, E.; Yang, P.; Harmon, D.; Schwab, J.; Nielsen, G.P.; Chebib, I.; Ferrone, S.; Wang, X.; et al. Programmed cell death ligand 1 expression in osteosarcoma. Cancer Immunol. Res. 2014, 2, 690–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundara, Y.T.; Kostine, M.; Cleven, A.H.; Bovée, J.V.; Schilham, M.W.; Cleton-Jansen, A.M. Increased PD-L1 and T-cell infiltration in the presence of HLA class I expression in metastatic high-grade osteosarcoma: A rationale for T-cell-based immunotherapy. Cancer Immunol. Immunother. 2017, 66, 119–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Erp, A.E.M.; Versleijen-Jonkers, Y.M.H.; Hillebrandt-Roeffen, M.H.S.; van Houdt, L.; Gorris, M.A.J.; van Dam, L.S.; Mentzel, T.; Weidema, M.E.; Savci-Heijink, C.D.; Desar, I.M.E.; et al. Expression and clinical association of programmed cell death-1, programmed death-ligand-1 and CD8+ lymphocytes in primary sarcomas is subtype dependent. Oncotarget 2017, 8, 71371–71384. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Jin, Z.; Zhang, M.; Tang, Y.; Yang, G.; Yuan, X.; Yao, J.; Sun, D. Prognostic value of programmed death-ligand 1 in sarcoma: A meta-analysis. Oncotarget 2017, 8, 59570–59580. [Google Scholar] [CrossRef] [Green Version]
- Lussier, D.M.; O’Neill, L.; Nieves, L.M.; McAfee, M.S.; Holechek, S.A.; Collins, A.W.; Dickman, P.; Jacobsen, P.J.; Hingorani, P.; Blattman, J.N. Enhanced T-cell immunity to osteosarcoma through antibody blockade of PD-1/PD-L1 interactions. J. Immunother. 2015, 38, 96–106. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, Q.; Chen, W.; Shan, B.; Ding, Y.; Zhang, G.; Cao, N.; Liu, L.; Zhang, Y. B7-H3 is overexpressed in patients suffering osteosarcoma and associated with tumor aggressiveness and metastasis. PLoS ONE 2013, 8, e70689. [Google Scholar] [CrossRef]
- Kaneda, M.M.; Messer, K.S.; Ralainirina, N.; Li, H.; Leem, C.J.; Gorjestani, S.; Woo, G.; Nguyen, A.V.; Figueiredo, C.C.; Foubert, P.; et al. PI3Kγ is a molecular switch that controls immune suppression. Nature 2016, 539, 437–442. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Tong, Z.; Ding, C.; Luo, F.; Wu, S.; Wu, C.; Albeituni, S.; He, L.; Hu, X.; Tieri, D.; et al. Transcription factor c-Maf is a checkpoint that programs macrophages in lung cancer. J. Clin. Investig. 2020, 130, 2081–2096. [Google Scholar] [CrossRef] [Green Version]
- Zanganeh, S.; Hutter, G.; Spitler, R.; Lenkov, O.; Mahmoudi, M.; Shaw, A.; Pajarinen, J.S.; Nejadnik, H.; Goodman, S.; Moseley, M.; et al. Iron oxide nanoparticles inhibit tumour growth by inducing pro-inflammatory macrophage polarization in tumour tissues. Nat. Nanotechnol. 2016, 11, 986–994. [Google Scholar] [CrossRef]
- Boraschi, D.; Italiani, P.; Palomba, R.; Decuzzi, P.; Duschl, A.; Fadeel, B.; Moghimi, S.M. Nanoparticles and innate immunity: New perspectives on host defence. Semin. Immunol. 2017, 34, 33–51. [Google Scholar] [CrossRef]
- Li, K.; Lu, L.; Xue, C.; Liu, J.; He, Y.; Zhou, J.; Xia, Z.; Dai, L.; Luo, Z.; Mao, Y.; et al. Polarization of tumor-associated macrophage phenotype via porous hollow iron nanoparticles for tumor immunotherapy in vivo. Nanoscale 2020, 12, 130–144. [Google Scholar] [CrossRef] [PubMed]
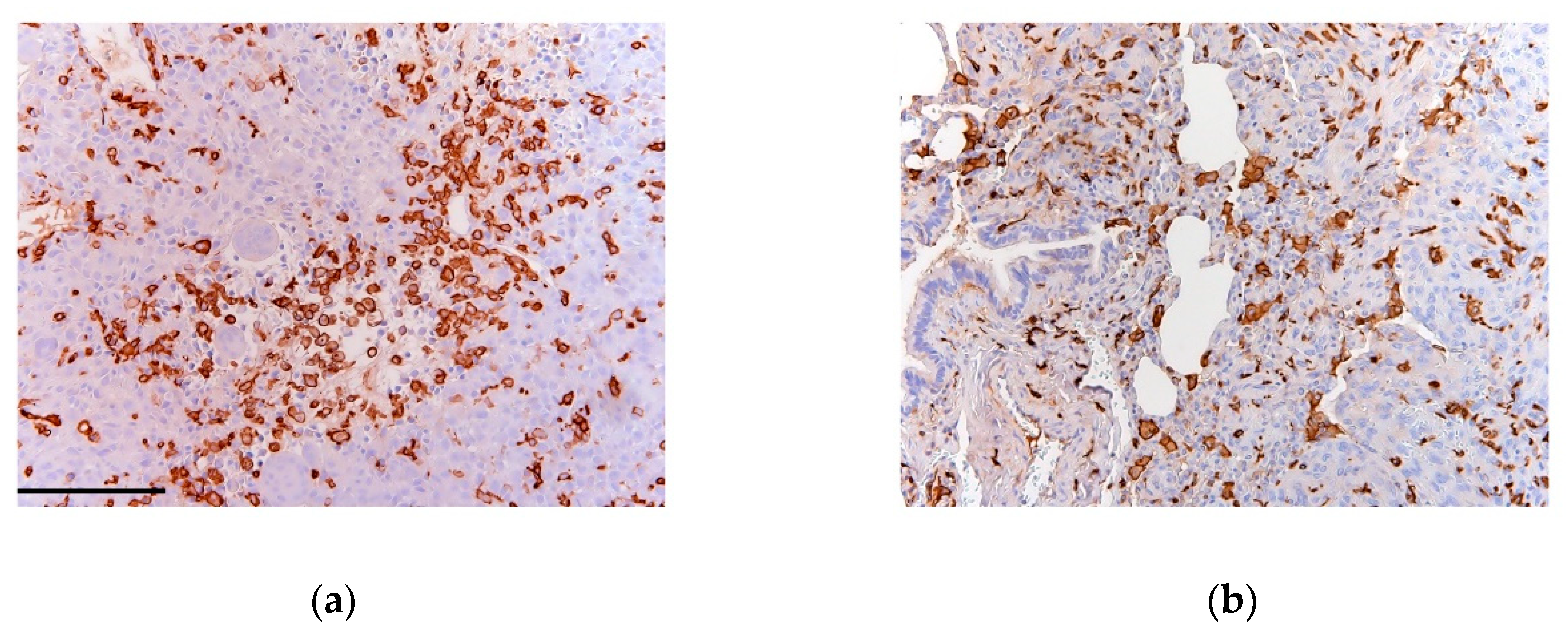
- Gomez-Brouchet, A.; Illac, C.; Gilhodes, J.; Bouvier, C.; Aubert, S.; Guinebretiere, J.M.; Marie, B.; Larousserie, F.; Entz-Werle, N.; de Pinieux, G.; et al. CD163-positive tumor-associated macrophages and CD8-positive cytotoxic lymphocytes are powerful diagnostic markers for the therapeutic stratification of osteosarcoma patients: An immunohistochemical analysis of the biopsies fromthe French OS2006 phase 3 trial. Oncoimmunology 2017, 6, e1331193. [Google Scholar] [PubMed]
- Fritzsching, B.; Fellenberg, J.; Moskovszky, L.; Sapi, Z.; Krenacs, T.; Machado, I.; Poeschl, J.; Lehner, B.; Szendroi, M.; Bosch, A.L.; et al. CD8(+)/FOXP3(+)-ratio in osteosarcoma microenvironment separates survivors from non-survivors: A multicenter validated retrospective study. Oncoimmunology 2015, 4, e990800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human chimeric antigen receptor macrophages for cancer immunotherapy Nat. Biotechnol. 2020. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Biomarkers | Function | Ref. |
|---|---|---|
| MMP-9 | Matrix metalloproteinase | [5] |
| COX2 | Proinflammatory enzyme | [5] |
| STAT3 | Transcription factor | [5] |
| CD163 | Scavenger receptor hemoglobulin | [6] |
| CCL18 | Chemokine | [98] |
| CD209 | Leptin receptor | [99] |
| IL-6 | Interleukin | [100] |
| CCL22 | Chemokine | [101] |
| IL-10 | Interleukin | [101] |
| TGFB2 | Cytokine | [101] |
| CD206 | Mannose receptor | [102] |
| Agent | Mechanism | Phase | Ref. |
|---|---|---|---|
| All trans retinoic acid | Reduces polarization of M2-like | Pre-clinical | [8] |
| Mifamurtide Esculetin | Induces M1-like activation Inhibits TAMs differentiation | 3 Pre-clinical | [145] [150] |
| Zoledronate | Polarizes TAMs to M1-like | 3 | [151] |
| Natalizumab | Interferes cross-talk between cancer cells and TAMs | NCT03811886 | [157] |
| Nivolumab | mAbs anti-PD-1 | NCT02304458 | [158] |
| Pembrolizumab | mAbs anti-PD-1 | NCT02301039 | [158] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cersosimo, F.; Lonardi, S.; Bernardini, G.; Telfer, B.; Mandelli, G.E.; Santucci, A.; Vermi, W.; Giurisato, E. Tumor-Associated Macrophages in Osteosarcoma: From Mechanisms to Therapy. Int. J. Mol. Sci. 2020, 21, 5207. https://doi.org/10.3390/ijms21155207
Cersosimo F, Lonardi S, Bernardini G, Telfer B, Mandelli GE, Santucci A, Vermi W, Giurisato E. Tumor-Associated Macrophages in Osteosarcoma: From Mechanisms to Therapy. International Journal of Molecular Sciences. 2020; 21(15):5207. https://doi.org/10.3390/ijms21155207
Chicago/Turabian StyleCersosimo, Francesca, Silvia Lonardi, Giulia Bernardini, Brian Telfer, Giulio Eugenio Mandelli, Annalisa Santucci, William Vermi, and Emanuele Giurisato. 2020. "Tumor-Associated Macrophages in Osteosarcoma: From Mechanisms to Therapy" International Journal of Molecular Sciences 21, no. 15: 5207. https://doi.org/10.3390/ijms21155207
APA StyleCersosimo, F., Lonardi, S., Bernardini, G., Telfer, B., Mandelli, G. E., Santucci, A., Vermi, W., & Giurisato, E. (2020). Tumor-Associated Macrophages in Osteosarcoma: From Mechanisms to Therapy. International Journal of Molecular Sciences, 21(15), 5207. https://doi.org/10.3390/ijms21155207