Atherosclerotic Pre-Conditioning Affects the Paracrine Role of Circulating Angiogenic Cells Ex-Vivo

 , , , , and
, , , , and
Abstract
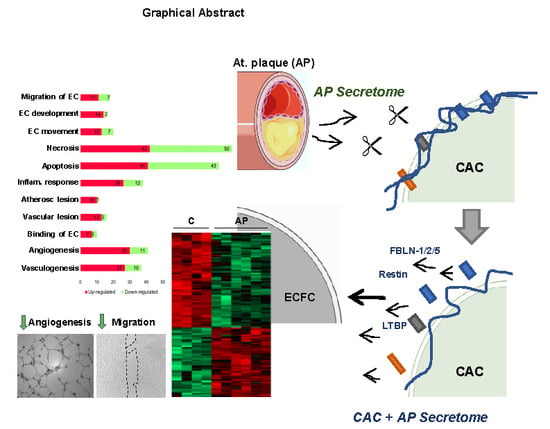
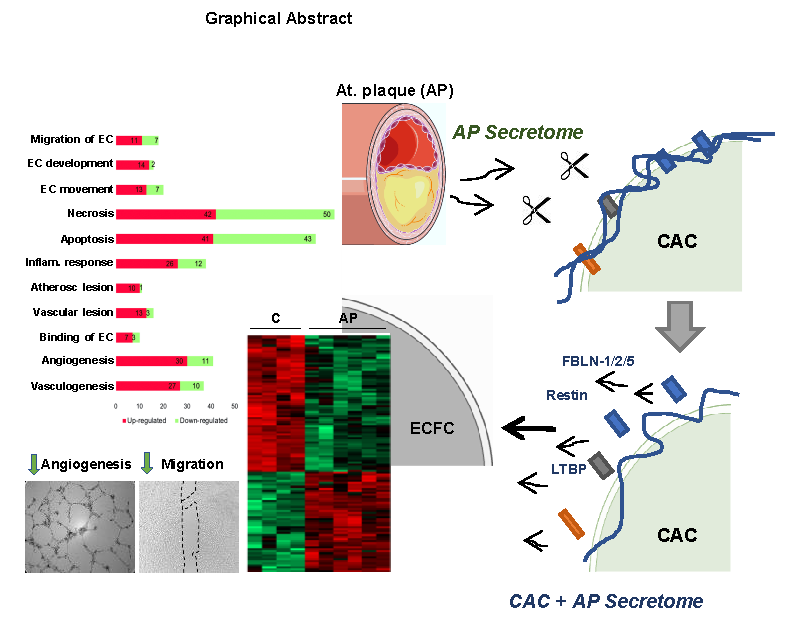
:
1. Introduction
2. Results
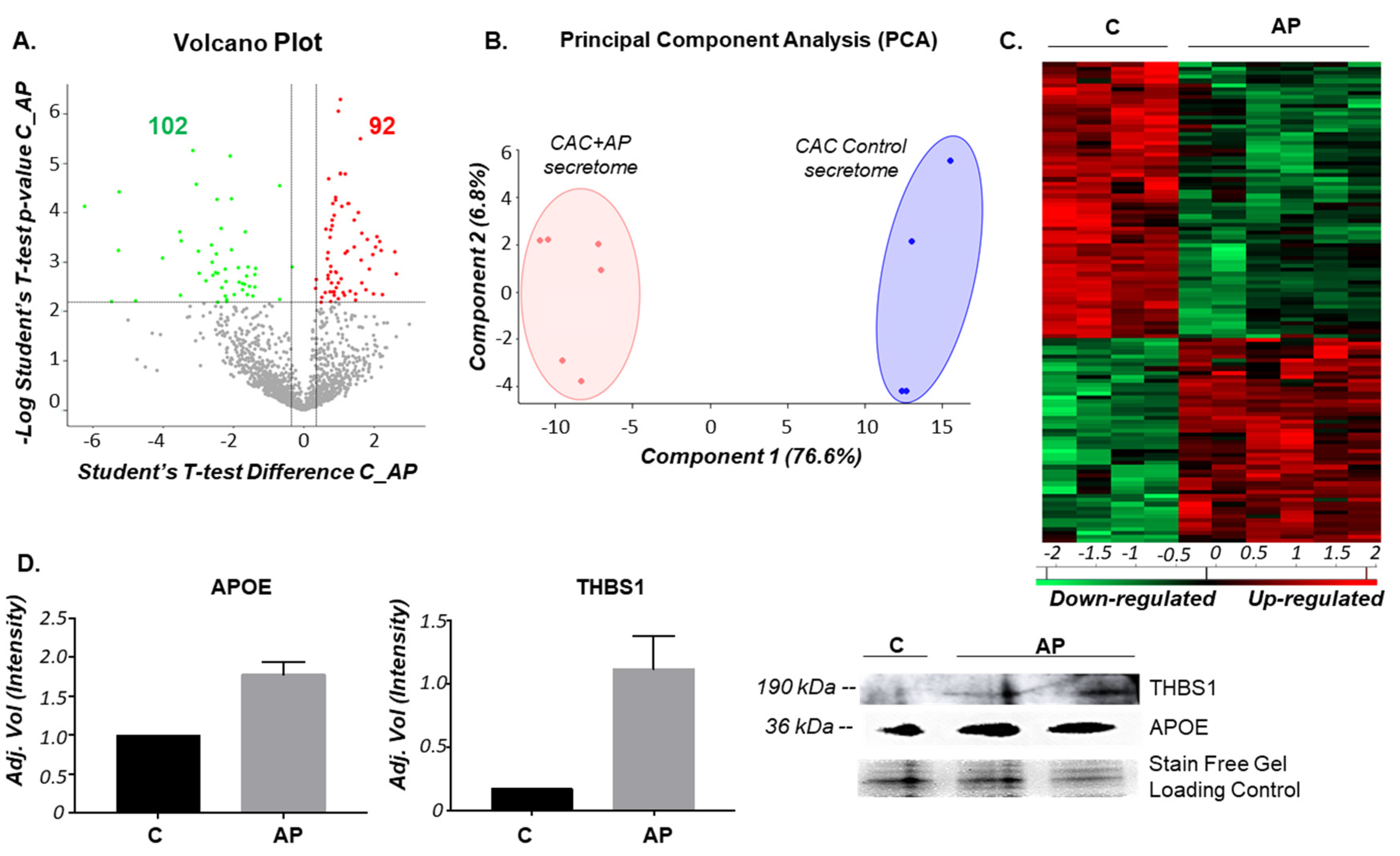
2.1. The Secretion Protein Profile of CAC Is Affected by the Incubation Ex-Vivo with Atherosclerotic Factors
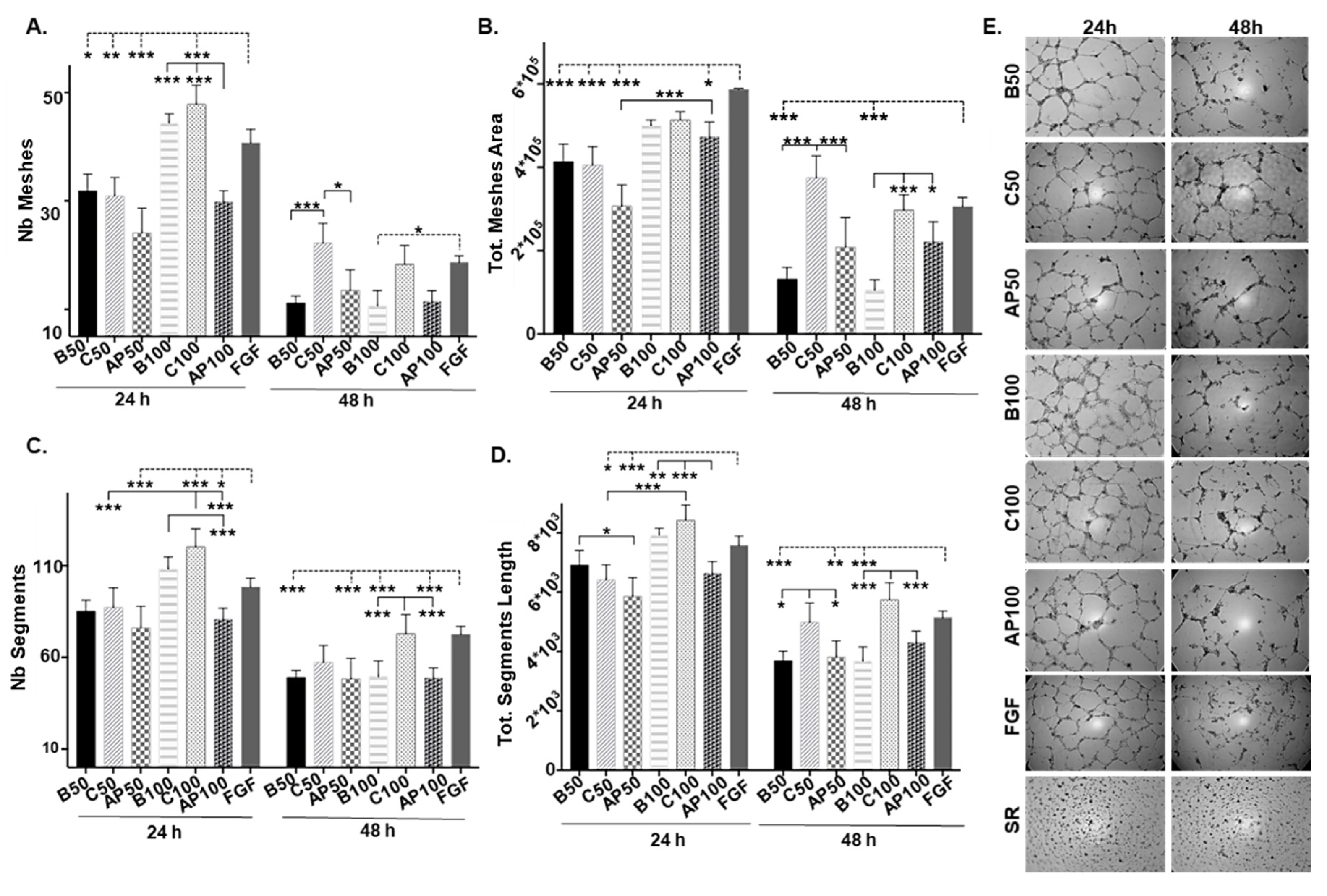
2.2. CAC Secretomes in Presence/Absence of Atherosclerotic Factors Alter Angiogenesis Potential of ECFC “Ex Vivo”
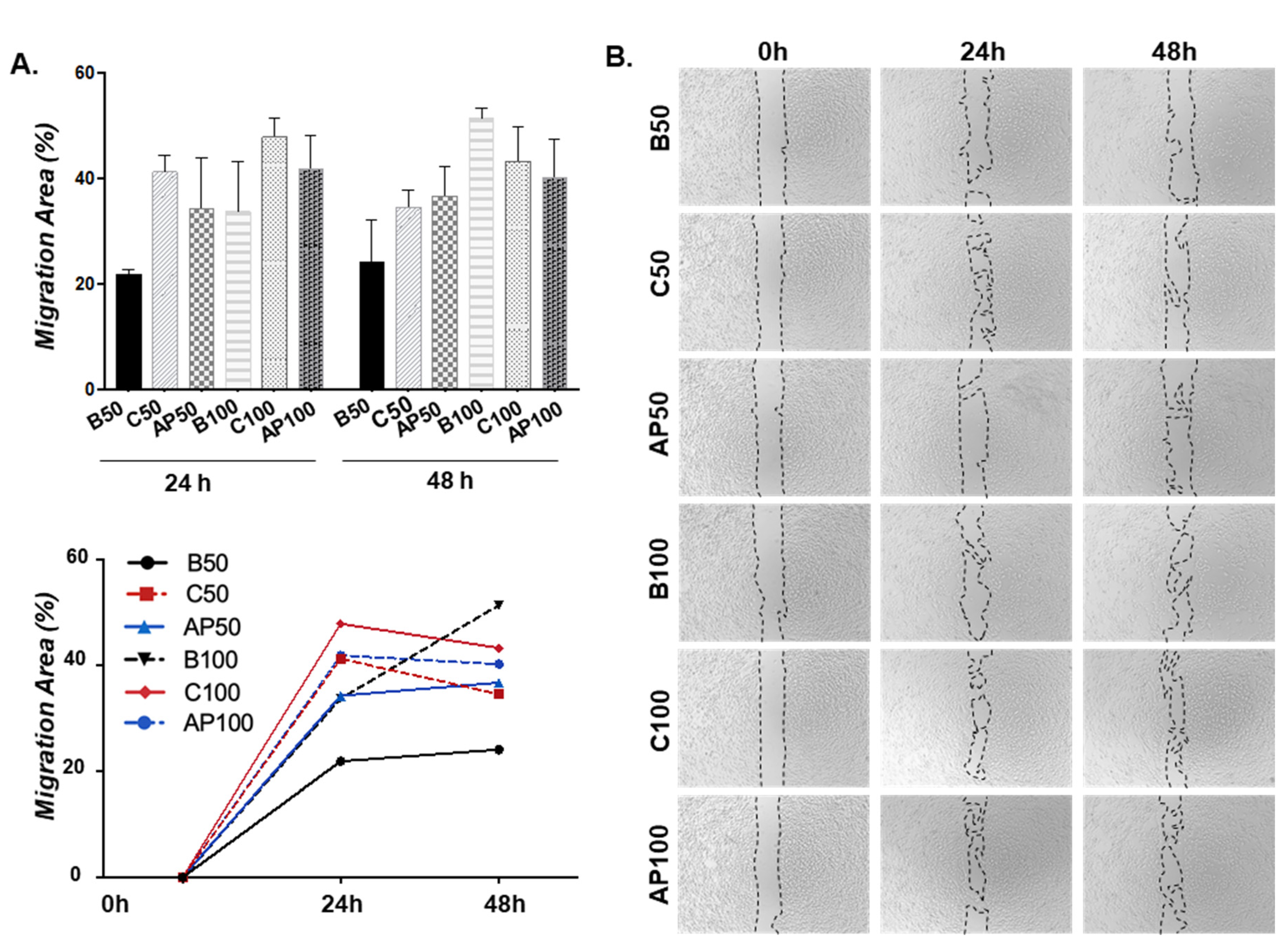
2.3. CAC Secretomes in Presence/Absence of Atherosclerotic Factors Effect on ECFC Migration “Ex Vivo”
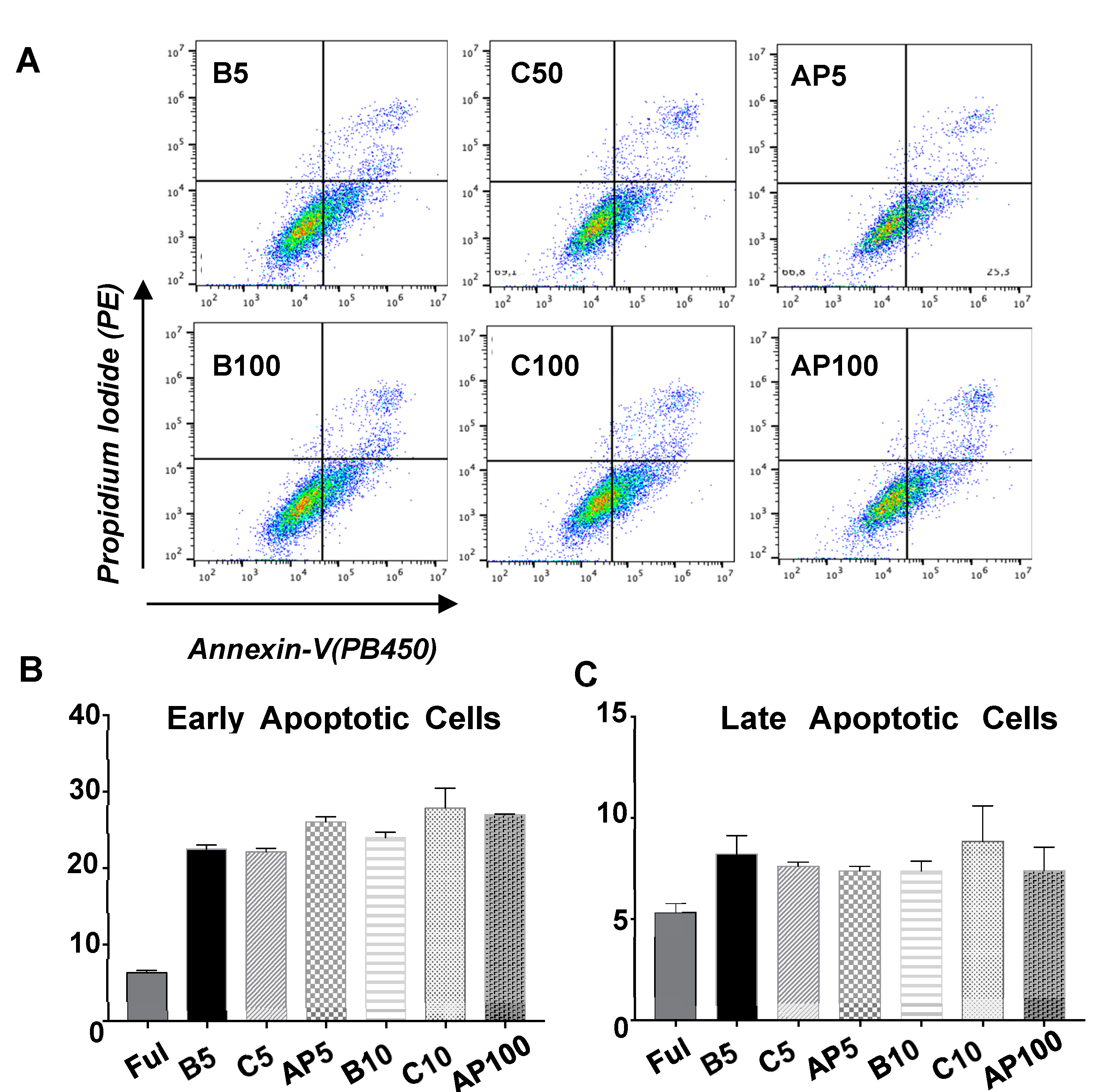
2.4. CAC Secretomes Effect over ECFC Apoptosis “Ex Vivo”
3. Discussion
4. Materials and Methods
4.1. Sample Acquisition
4.2. CAC Isolation and Culture
4.3. Atheroma Plaque Isolation and Culture
4.4. Characterization of CAC
4.5. CAC Incubation Ex Vivo with Atheroma Plaque Secretome
4.6. Proteomic Analysis
4.7. Validation of Protein Expression Changes
4.8. ECFC Isolation and Culture
4.9. Characterization of ECFC
4.10. Angiogenesis Assay
4.11. Wound Migration Assay
4.12. Apoptosis Assay
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AP | Atheroma plaque |
| APO | Apo-lipoproteins |
| APOE | Apolipoprotein-E |
| B | Basal |
| C | Control |
| CAC | Circulating angiogenic cells |
| Cb-ECFC | umbilical cord blood endothelial colony forming cell |
| CID | Collision induced dissociation |
| COL15A1 | Restin |
| COL18A1 | Endostatin |
| CVD | Cardiovascular diseases |
| EBM-2 | Endothelial Basal Medium-2 |
| EC | Endothelial cells |
| ECFC | Endothelial colony-forming cells |
| ECM | Extracellular matrix |
| EPC | Endothelial progenitor cells |
| eEPC | early EPC |
| FA | Formic acid |
| FBLN | Fibulin |
| FDR | False Discovery Rate |
| FGF | Fibroblast Growth Factor |
| FN1 | Fibronectin-1 |
| GC-SF | Granulocyte colony-stimulating factor |
| HMOX1 | Heme Oxygenase-1 |
| HTRA1 | HtrA Serine Peptidase-1 |
| HSP90 | Heat shock protein 90 |
| HSPG2 | Heparan Sulfate Proteoglycan-2 |
| HUVEC | Human umbilical vascular endothelial cell |
| IPA | Ingenuity Pathway Analysis |
| KGF | Keratinocyte growth factor |
| LC-MS/MS | Liquid Chromatography- Tandem Mass spectrometry |
| LFQ | Label Free Quantitation |
| LTBP | Latent TGF-beta binding proteins |
| MAC | Myeloid angiogenic cells |
| MMP9 | Matrix metallopeptidase-9 |
| MS | Mass spectrometry |
| PBS | Phosphate buffer saline |
| PCA | Principal Component Analysis |
| PDGF | Platelet-derived growth factor |
| P/S | Penicillin/streptomycin |
| SR | Sulforaphane |
| THBS | Thrombospondin |
| THBS1 | Thrombospondin-1 |
| TFA | Trifluoroacetic acid |
| TGFβ | Transforming Growth Factor-beta |
| TNC | Tenascin |
| VEGF | Vascular Endothelial growth factor |
References
- Chong, M.S.; Ng, W.K.; Chan, J.K. Concise Review: Endothelial Progenitor Cells in Regenerative Medicine: Applications and Challenges. Stem Cells Transl. Med. 2016, 5, 530–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basile, D.P.; Yoder, M.C. Circulating and tissue resident endothelial progenitor cells. J. Cell. Physiol. 2014, 229, 10–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoder, M.C. Endothelial progenitor cell: A blood cell by many other names may serve similar functions. J. Mol. Med. 2013, 91, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Zhou, B.; Gu, D.; Zhang, L.; Han, Z. Endothelial progenitor cell therapy in atherosclerosis: A double-edged sword? Ageing Res. Rev. 2009, 8, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Vega, F.M.; Gautier, V.; Fernandez-Ponce, C.M.; Extremera, M.J.; Altelaar, A.F.M.; Millan, J.; Tellez, J.C.; Hernandez-Campos, J.A.; Conejero, R.; Bolivar, J.; et al. The atheroma plaque secretome stimulates the mobilization of endothelial progenitor cells ex vivo. J. Mol. Cell. Cardiol. 2017, 105, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Hristov, M.; Weber, C. Ambivalence of progenitor cells in vascular repair and plaque stability. Curr. Opin. Lipidol. 2008, 19, 491–497. [Google Scholar] [CrossRef]
- Asahara, T.; Murohara, T.; Sullivan, A.; Silver, M.; van der Zee, R.; Li, T.; Witzenbichler, B.; Schatteman, G.; Isner, J.M. Isolation of putative progenitor endothelial cells for angiogenesis. Science 1997, 275, 964–967. [Google Scholar] [CrossRef]
- Medina, R.J.; Barber, C.L.; Sabatier, F.; Dignat-George, F.; Melero-Martin, J.M.; Khosrotehrani, K.; Ohneda, O.; Randi, A.M.; Chan, J.K.Y.; Yamaguchi, T.; et al. Endothelial Progenitors: A Consensus Statement on Nomenclature. Stem Cells Transl. Med. 2017, 6, 1316–1320. [Google Scholar] [CrossRef]
- Goligorsky, M.S.; Salven, P. Concise review: Endothelial stem and progenitor cells and their habitats. Stem Cells Transl. Med. 2013, 2, 499–504. [Google Scholar] [CrossRef]
- Banno, K.; Yoder, M.C. Tissue regeneration using endothelial colony-forming cells: Promising cells for vascular repair. Pediatr. Res. 2018, 83, 283–290. [Google Scholar] [CrossRef] [Green Version]
- Medina, R.J.; O’Neill, C.L.; O’Doherty, T.M.; Knott, H.; Guduric-Fuchs, J.; Gardiner, T.A.; Stitt, A.W. Myeloid angiogenic cells act as alternative M2 macrophages and modulate angiogenesis through interleukin-8. Mol. Med. 2011, 17, 1045–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beltran-Camacho, L.; Jimenez-Palomares, M.; Torres-Rojas, M.; Perez-Segura, M.C.; Serrano, A.; Sanchez-Gomar, I.; Rosal-Vela, A.; Antequera-González, B.; Alonso-Piñero, J.A.; Gonzalez-Rovira, A.; et al. Identification of the initial molecular changes detected in response to Circulating Angiogenic Cells-mediated therapy in critical limb ischemia. Stem Cell Res. Ther. 2020, 11, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, C.H.; Hur, J.; Park, K.W.; Kim, J.H.; Lee, C.S.; Oh, I.Y.; Kim, T.Y.; Cho, H.J.; Kang, H.J.; Chae, I.H.; et al. Synergistic neovascularization by mixed transplantation of early endothelial progenitor cells and late outgrowth endothelial cells: The role of angiogenic cytokines and matrix metalloproteinases. Circulation 2005, 112, 1618–1627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, Y.; Kawamoto, A. Stem cell-based peripheral vascular regeneration. Adv. Drug Deliv. Rev. 2017, 120, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Rigato, M.; Monami, M.; Fadini, G.P. Autologous Cell Therapy for Peripheral Arterial Disease: Systematic Review and Meta-Analysis of Randomized, Nonrandomized, and Noncontrolled Studies. Circ. Res. 2017, 120, 1326–1340. [Google Scholar] [CrossRef]
- Kalka, C.; Masuda, H.; Takahashi, T.; Kalka-Moll, W.M.; Silver, M.; Kearney, M.; Li, T.; Isner, J.M.; Asahara, T. Transplantation of ex vivo expanded endothelial progenitor cells for therapeutic neovascularization. Proc. Natl. Acad. Sci. USA 2000, 97, 3422–3427. [Google Scholar] [CrossRef]
- Lee, P.S.; Poh, K.K. Endothelial progenitor cells in cardiovascular diseases. World J. Stem Cells 2014, 6, 355–366. [Google Scholar] [CrossRef]
- Yang, J.X.; Pan, Y.Y.; Wang, X.X.; Qiu, Y.G.; Mao, W. Endothelial progenitor cells in age-related vascular remodeling. Cell Transpl. 2018, 27, 786–795. [Google Scholar] [CrossRef]
- Guven, H.; Shepherd, R.M.; Bach, R.G.; Capoccia, B.J.; Link, D.C. The number of endothelial progenitor cell colonies in the blood is increased in patients with angiographically significant coronary artery disease. J. Am. Coll. Cardiol. 2006, 48, 1579–1587. [Google Scholar] [CrossRef] [Green Version]
- Fadini, G.P.; Sartore, S.; Albiero, M.; Baesso, I.; Murphy, E.; Menegolo, M.; Grego, F.; Vigili de Kreutzenberg, S.; Tiengo, A.; Agostini, C.; et al. Number and function of endothelial progenitor cells as a marker of severity for diabetic vasculopathy. Arter. Thromb. Vasc. Biol. 2006, 26, 2140–2146. [Google Scholar] [CrossRef] [Green Version]
- Vasa, M.; Fichtlscherer, S.; Aicher, A.; Adler, K.; Urbich, C.; Martin, H.; Zeiher, A.M.; Dimmeler, S. Number and migratory activity of circulating endothelial progenitor cells inversely correlate with risk factors for coronary artery disease. Circ. Res. 2001, 89, e1–e7. [Google Scholar] [CrossRef] [Green Version]
- Lau, K.K.; Chan, Y.H.; Yiu, K.H.; Li, S.W.; Tam, S.; Lau, C.P.; Kwong, Y.L.; Tse, H.F. Burden of carotid atherosclerosis in patients with stroke: Relationships with circulating endothelial progenitor cells and hypertension. J. Hum. Hypertens. 2007, 21, 445–451. [Google Scholar] [CrossRef]
- Du, F.; Zhou, J.; Gong, R.; Huang, X.; Pansuria, M.; Virtue, A.; Li, X.; Wang, H.; Yang, X.F. Endothelial progenitor cells in atherosclerosis. Front. Biosci. 2012, 17, 2327–2349. [Google Scholar] [CrossRef] [Green Version]
- Mena, H.A.; Zubiry, P.R.; Dizier, B.; Schattner, M.; Boisson-Vidal, C.; Negrotto, S. Acidic preconditioning of endothelial colony-forming cells (ECFC) promote vasculogenesis under proinflammatory and high glucose conditions in vitro and in vivo. Stem Cell Res. Ther. 2018, 9, 120. [Google Scholar] [CrossRef]
- Kang, H.; Ma, X.; Liu, J.; Fan, Y.; Deng, X. High glucose-induced endothelial progenitor cell dysfunction. Diabetes Vasc. Dis. Res. 2017, 14, 381–394. [Google Scholar] [CrossRef] [Green Version]
- Ingram, D.A.; Krier, T.R.; Mead, L.E.; McGuire, C.; Prater, D.N.; Bhavsar, J.; Saadatzadeh, M.R.; Bijangi-Vishehsaraei, K.; Li, F.; Yoder, M.C.; et al. Clonogenic endothelial progenitor cells are sensitive to oxidative stress. Stem Cells 2007, 25, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Kalka, C.; Masuda, H.; Chen, D.; Silver, M.; Kearney, M.; Magner, M.; Isner, J.M.; Asahara, T. Ischemia- and cytokine-induced mobilization of bone marrow-derived endothelial progenitor cells for neovascularization. Nat. Med. 1999, 5, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Sprengers, R.W.; Lips, D.J.; Moll, F.L.; Verhaar, M.C. Progenitor cell therapy in patients with critical limb ischemia without surgical options. Ann. Surg. 2008, 247, 411–420. [Google Scholar] [CrossRef]
- Tanaka, R.; Masuda, H.; Kato, S.; Imagawa, K.; Kanabuchi, K.; Nakashioya, C.; Yoshiba, F.; Fukui, T.; Ito, R.; Kobori, M.; et al. Autologous G-CSF-mobilized peripheral blood CD34+ cell therapy for diabetic patients with chronic nonhealing ulcer. Cell Transpl. 2014, 23, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Sukmawati, D.; Tanaka, R. Introduction to next generation of endothelial progenitor cell therapy: A promise in vascular medicine. Am. J. Transl. Res. 2015, 7, 411–421. [Google Scholar]
- Alev, C.; Ii, M.; Asahara, T. Endothelial progenitor cells: A novel tool for the therapy of ischemic diseases. Antioxid. Redox Signal. 2011, 15, 949–965. [Google Scholar] [CrossRef]
- Kim, J.Y.; Song, S.H.; Kim, K.L.; Ko, J.J.; Im, J.E.; Yie, S.W.; Ahn, Y.K.; Kim, D.K.; Suh, W. Human cord blood-derived endothelial progenitor cells and their conditioned media exhibit therapeutic equivalence for diabetic wound healing. Cell Transpl. 2010, 19, 1635–1644. [Google Scholar] [CrossRef] [Green Version]
- Rehman, J.; Li, J.; Orschell, C.M.; March, K.L. Peripheral blood “endothelial progenitor cells” are derived from monocyte/macrophages and secrete angiogenic growth factors. Circulation 2003, 107, 1164–1169. [Google Scholar] [CrossRef] [Green Version]
- Urbich, C.; De Souza, A.I.; Rossig, L.; Yin, X.; Xing, Q.; Prokopi, M.; Drozdov, I.; Steiner, M.; Breuss, J.; Xu, Q.; et al. Proteomic characterization of human early pro-angiogenic cells. J. Mol. Cell. Cardiol. 2011, 50, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Pula, G.; Mayr, U.; Evans, C.; Prokopi, M.; Vara, D.S.; Yin, X.; Astroulakis, Z.; Xiao, Q.; Hill, J.; Xu, Q.; et al. Proteomics identifies thymidine phosphorylase as a key regulator of the angiogenic potential of colony-forming units and endothelial progenitor cell cultures. Circ. Res. 2009, 104, 32–40. [Google Scholar] [CrossRef]
- Sullivan, K.M.; Bissonnette, R.; Yanagisawa, H.; Hussain, S.N.; Davis, E.C. Fibulin-5 functions as an endogenous angiogenesis inhibitor. Lab. Investig. 2007, 87, 818–827. [Google Scholar] [CrossRef] [PubMed]
- Chu, L.H.; Lee, E.; Bader, J.S.; Popel, A.S. Angiogenesis interactome and time course microarray data reveal the distinct activation patterns in endothelial cells. PLoS ONE 2014, 9, e110871. [Google Scholar] [CrossRef] [PubMed]
- Camare, C.; Pucelle, M.; Negre-Salvayre, A.; Salvayre, R. Angiogenesis in the atherosclerotic plaque. Redox Biol. 2017, 12, 18–34. [Google Scholar] [CrossRef]
- Mongiat, M.; Andreuzzi, E.; Tarticchio, G.; Paulitti, A. Extracellular Matrix, a Hard Player in Angiogenesis. Int. J. Mol. Sci. 2016, 17, 1822. [Google Scholar] [CrossRef] [Green Version]
- Hinz, B. The extracellular matrix and transforming growth factor-beta1: Tale of a strained relationship. Matrix Biol. 2015, 47, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Neve, A.; Cantatore, F.P.; Maruotti, N.; Corrado, A.; Ribatti, D. Extracellular matrix modulates angiogenesis in physiological and pathological conditions. BioMed Res. Int. 2014, 2014, 756078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eslava-Alcon, S.; Extremera-García, M.J.; González-Rovira, A.; Rosal-Vela, A.; Rojas-Torres, M.; Beltran-Camacho, L.; Sanchez-Gomar, I.; Jimenez-Palomares, M.; Alonso-Piñero, J.A.; Conejero, R.; et al. Molecular signatures of atherosclerotic plaques: An up-dated panel of protein related markers. J. Proteom. 2020, 221, 103757. [Google Scholar] [CrossRef] [PubMed]
- Walia, A.Y.J.F.; Huang, Y.-H.; Rosenblatt, M.I.; Chang, J.-H.; Azar, D.T. Endostatin’s emerging roles in angiogenesis, lymphangiogenesis, disease, and clinical applications. Biochim. Biophys. Acta 2015, 1850, 2422–2438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- John, H.R.K.; Standker, L.; Forssmann, W.G. Identification and characterization of novel endogenous proteolytic forms of the human angiogenesis inhibitors restin and endostatin. Biochim. Biophys. Acta 2005, 1747, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, N.; Anand-Apte, B.; Lee, M.; Sasaki, T.; Fukai, N.; Shapiro, R.; Que, I.; Lowik, C.; Timpl, R.; Olsen, B.R. Endostatin inhibits VEGF-induced endothelial cell migration and tumor growth independently of zinc binding. EMBO J. 1999, 18, 4414–4423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.J.; Lee, H.J.; Choi, S.H.; Jin, Y.B.; An, H.J.; Kang, J.H.; Yoon, S.S.; Lee, Y.S. Soluble HSPB1 regulates VEGF-mediated angiogenesis through their direct interaction. Angiogenesis 2012, 15, 229–242. [Google Scholar] [CrossRef] [PubMed]
- Law, E.W.; Cheung, A.K.; Kashuba, V.I.; Pavlova, T.V.; Zabarovsky, E.R.; Lung, H.L.; Cheng, Y.; Chua, D.; Lai-Wan Kwong, D.; Tsao, S.W.; et al. Anti-angiogenic and tumor-suppressive roles of candidate tumor-suppressor gene, Fibulin-2, in nasopharyngeal carcinoma. Oncogene 2012, 31, 728–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, L.; Palmsten, K.; MacDonald, B.; Kieran, M.W.; Potenta, S.; Vong, S.; Kalluri, R. Basement membrane derived fibulin-1 and fibulin-5 function as angiogenesis inhibitors and suppress tumor growth. Exp. Biol. Med. 2008, 233, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, T. Extracellular Interactions between Fibulins and Transforming Growth Factor (TGF)-beta in Physiological and Pathological Conditions. Int. J. Mol. Sci. 2018, 19, 2787. [Google Scholar] [CrossRef] [Green Version]
- Albig, A.R.; Schiemann, W.P. Fibulin-5 antagonizes vascular endothelial growth factor (VEGF) signaling and angiogenic sprouting by endothelial cells. DNA Cell Biol. 2004, 23, 367–379. [Google Scholar] [CrossRef]
- Singh, P.; Carraher, C.; Schwarzbauer, J.E. Assembly of fibronectin extracellular matrix. Annu. Rev. Cell Dev. Biol. 2010, 26, 397–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivaraman, K.; Shanthi, C. Matrikines for therapeutic and biomedical applications. Life Sci. 2018, 214, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Hocking, D.C.; Sottile, J.; Langenbach, K.J. Stimulation of integrin-mediated cell contractility by fibronectin polymerization. J. Biol. Chem. 2000, 275, 10673–10682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Li, Y.; Shen, Y.; Wang, A.; Wang, S.; Xie, T. The functions and applications of RGD in tumor therapy and tissue engineering. Int. J. Mol. Sci. 2013, 14, 13447–13462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, Q.; Qian, J.; Ge, L.; Shen, L.; Jia, J.; Jin, J.; Ge, J. Effect and mechanism of thrombospondin-1 on the angiogenesis potential in human endothelial progenitor cells: An in vitro study. PLoS ONE 2014, 9, e88213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawler, J.; Detmar, M. Tumor progression: The effects of thrombospondin-1 and -2. Int. J. Biochem. Cell Biol. 2004, 36, 1038–1045. [Google Scholar] [CrossRef]
- Munjal, C.; Opoka, A.M.; Osinska, H.; James, J.F.; Bressan, G.M.; Hinton, R.B. TGF-beta mediates early angiogenesis and latent fibrosis in an Emilin1-deficient mouse model of aortic valve disease. Dis. Models Mech. 2014, 7, 987–996. [Google Scholar] [CrossRef] [Green Version]
- Morgan-Rowe, L.; Nikitorowicz, J.; Shiwen, X.; Leask, A.; Tsui, J.; Abraham, D.; Stratton, R. Thrombospondin 1 in hypoxia-conditioned media blocks the growth of human microvascular endothelial cells and is increased in systemic sclerosis tissues. Fibrogenes. Tissue Repair 2011, 4, 13. [Google Scholar] [CrossRef] [Green Version]
- Robertson, I.B.; Horiguchi, M.; Zilberberg, L.; Dabovic, B.; Hadjiolova, K.; Rifkin, D.B. Latent TGF-beta-binding proteins. Matrix Biol. 2015, 47, 44–53. [Google Scholar] [CrossRef]
- Murphy-Ullrich, J.E.; Suto, M.J. Thrombospondin-1 regulation of latent TGF-beta activation: A therapeutic target for fibrotic disease. Matrix Biol. 2018, 68–69, 28–43. [Google Scholar] [CrossRef]
- Zacchigna, L.; Vecchione, C.; Notte, A.; Cordenonsi, M.; Dupont, S.; Maretto, S.; Cifelli, G.; Ferrari, A.; Maffei, A.; Fabbro, C.; et al. Emilin1 links TGF-beta maturation to blood pressure homeostasis. Cell 2006, 124, 929–942. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, G.; Cook, B.D.; Terushkin, V.; Pintucci, G.; Mignatti, P. Transforming growth factor-beta 1 (TGF-beta1) induces angiogenesis through vascular endothelial growth factor (VEGF)-mediated apoptosis. J. Cell. Physiol. 2009, 219, 449–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haibe, Y.; Kreidieh, M.; El Hajj, H.; Khalifeh, I.; Mukherji, D.; Temraz, S.; Shamseddine, A. Resistance Mechanisms to Anti-angiogenic Therapies in Cancer. Front. Oncol. 2020, 10, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerrero, P.A.; McCarty, J.H. TGF-β Activation and Signaling in Angiogenesis. In Physiologic and Pathologic Angiogenesis: Signaling Mechanisms and Targeted Therapy; Simionescu, D., Simionescu, A., Eds.; IntechOpen: London, UK, 2017. [Google Scholar]
- Lee, Y.H.; Albig, A.R.; Regner, M.; Schiemann, B.J.; Schiemann, W.P. Fibulin-5 initiates epithelial-mesenchymal transition (EMT) and enhances EMT induced by TGF-beta in mammary epithelial cells via a MMP-dependent mechanism. Carcinogenesis 2008, 29, 2243–2251. [Google Scholar] [CrossRef] [PubMed]
- Langer, H.F.; Chung, K.J.; Orlova, V.V.; Choi, E.Y.; Kaul, S.; Kruhlak, M.J.; Alatsatianos, M.; DeAngelis, R.A.; Roche, P.A.; Magotti, P.; et al. Complement-mediated inhibition of neovascularization reveals a point of convergence between innate immunity and angiogenesis. Blood 2010, 116, 4395–4403. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.G.; Jie, C.; Talbot, C. How PEDF prevents angiogenesis: A hypothesized pathway. Med. Hypotheses 2005, 64, 74–78. [Google Scholar] [CrossRef]
- Klose, R.; Adam, M.G.; Weis, E.M.; Moll, I.; Wustehube-Lausch, J.; Tetzlaff, F.; Oka, C.; Ehrmann, M.; Fischer, A. Inactivation of the serine protease HTRA1 inhibits tumor growth by deregulating angiogenesis. Oncogene 2018, 37, 4260–4272. [Google Scholar] [CrossRef]
- Liu, L.; Boffa, M.B.; Koschinsky, M.L. Apolipoprotein (a) inhibits in vitro tube formation in endothelial cells: Identification of roles for Kringle V and the plasminogen activation system. PLoS ONE 2013, 8, e52287. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharjee, P.S.; Huq, T.S.; Mandal, T.K.; Graves, R.A.; Muniruzzaman, S.; Clement, C.; McFerrin, H.E.; Hill, J.M. A novel peptide derived from human apolipoprotein E is an inhibitor of tumor growth and ocular angiogenesis. PLoS ONE 2011, 6, e15905. [Google Scholar] [CrossRef] [Green Version]
- Pola, C. Antitumor duality of ApoE. Nat. Med. 2012, 18, 1752. [Google Scholar] [CrossRef]
- Azoitei, N.; Diepold, K.; Brunner, C.; Rouhi, A.; Genze, F.; Becher, A.; Kestler, H.; van Lint, J.; Chiosis, G.; Koren, J.; et al. HSP90 supports tumor growth and angiogenesis through PRKD2 protein stabilization. Cancer Res. 2014, 74, 7125–7136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grochot-Przeczek, A.; Kotlinowski, J.; Kozakowska, M.; Starowicz, K.; Jagodzinska, J.; Stachurska, A.; Volger, O.L.; Bukowska-Strakova, K.; Florczyk, U.; Tertil, M.; et al. Heme oxygenase-1 is required for angiogenic function of bone marrow-derived progenitor cells: Role in therapeutic revascularization. Antioxid. Redox Signal. 2014, 20, 1677–1692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parma, L.; Baganha, F.; Quax, P.H.A.; de Vries, M.R. Plaque angiogenesis and intraplaque hemorrhage in atherosclerosis. Eur. J. Pharmacol. 2017, 816, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Slevin, M.; Krupinski, J.; Badimon, L. Controlling the angiogenic switch in developing atherosclerotic plaques: Possible targets for therapeutic intervention. J. Angiogenes. Res. 2009, 1, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, R.K.; Finn, A.V.; Kolodgie, F.D.; Gold, H.K.; Virmani, R. Antiangiogenic therapy for normalization of atherosclerotic plaque vasculature: A potential strategy for plaque stabilization. Nat. Clin. Pract. Cardiovasc. Med. 2007, 4, 491–502. [Google Scholar] [CrossRef]
- Patel, A. Does the Role of Angiogenesis Play a Role in Atherosclerosis and Plaque Instability? Anat. Physiol. 2014, 4, 147–158. [Google Scholar]
- Wang, L.; Chen, Q.; Ke, D.; Li, G. Ghrelin inhibits atherosclerotic plaque angiogenesis and promotes plaque stability in a rabbit atherosclerotic model. Peptides 2017, 90, 17–26. [Google Scholar] [CrossRef]
- Moulton, K.S.; Heller, E.; Konerding, M.A.; Flynn, E.; Palinski, W.; Folkman, J. Angiogenesis inhibitors endostatin or TNP-470 reduce intimal neovascularization and plaque growth in apolipoprotein E-deficient mice. Circulation 1999, 99, 1726–1732. [Google Scholar] [CrossRef]
- de Vries, M.R.; Parma, L.; Peters, H.A.B.; Schepers, A.; Hamming, J.F.; Jukema, J.W.; Goumans, M.; Guo, L.; Finn, A.V.; Virmani, R.; et al. Blockade of vascular endothelial growth factor receptor 2 inhibits intraplaque haemorrhage by normalization of plaque neovessels. J. Intern. Med. 2019, 285, 59–74. [Google Scholar] [CrossRef]
- Huang, Y.; Goel, S.; Duda, D.G.; Fukumura, D.; Jain, R.K. Vascular normalization as an emerging strategy to enhance cancer immunotherapy. Cancer Res. 2013, 73, 2943–2948. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Stylianopoulos, T.; Duda, D.G.; Fukumura, D.; Jain, R.K. Benefits of vascular normalization are dose and time dependent. Cancer Res. 2013, 73, 7144–7146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theelen, T.L.; Lappalainen, J.P.; Sluimer, J.C.; Gurzeler, E.; Cleutjens, J.P.; Gijbels, M.J.; Biessen, E.A.; Daemen, M.J.; Alitalo, K.; Yla-Herttuala, S. Angiopoietin-2 blocking antibodies reduce early atherosclerotic plaque development in mice. Atherosclerosis 2015, 241, 297–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Llamas, G.; Szalowska, E.; de Vries, M.P.; Weening, D.; Landman, K.; Hoek, A.; Wolffenbuttel, B.H.; Roelofsen, H.; Vonk, R.J. Characterization of the human visceral adipose tissue secretome. Mol. Cell. Proteom. 2007, 6, 589–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno-Luna, R.; Munoz-Hernandez, R.; Lin, R.Z.; Miranda, M.L.; Vallejo-Vaz, A.J.; Stiefel, P.; Praena-Fernandez, J.M.; Bernal-Bermejo, J.; Jimenez-Jimenez, L.M.; Villar, J.; et al. Maternal body-mass index and cord blood circulating endothelial colony-forming cells. J. Pediatr. 2014, 164, 566–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munoz-Hernandez, R.; Miranda, M.L.; Stiefel, P.; Lin, R.Z.; Praena-Fernandez, J.M.; Dominguez-Simeon, M.J.; Villar, J.; Moreno-Luna, R.; Melero-Martin, J.M. Decreased level of cord blood circulating endothelial colony-forming cells in preeclampsia. Hypertension 2014, 64, 165–171. [Google Scholar] [CrossRef] [Green Version]
- Vizcaino, J.A.; Csordas, A.; Del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T.; et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44, 11033. [Google Scholar] [CrossRef] [Green Version]





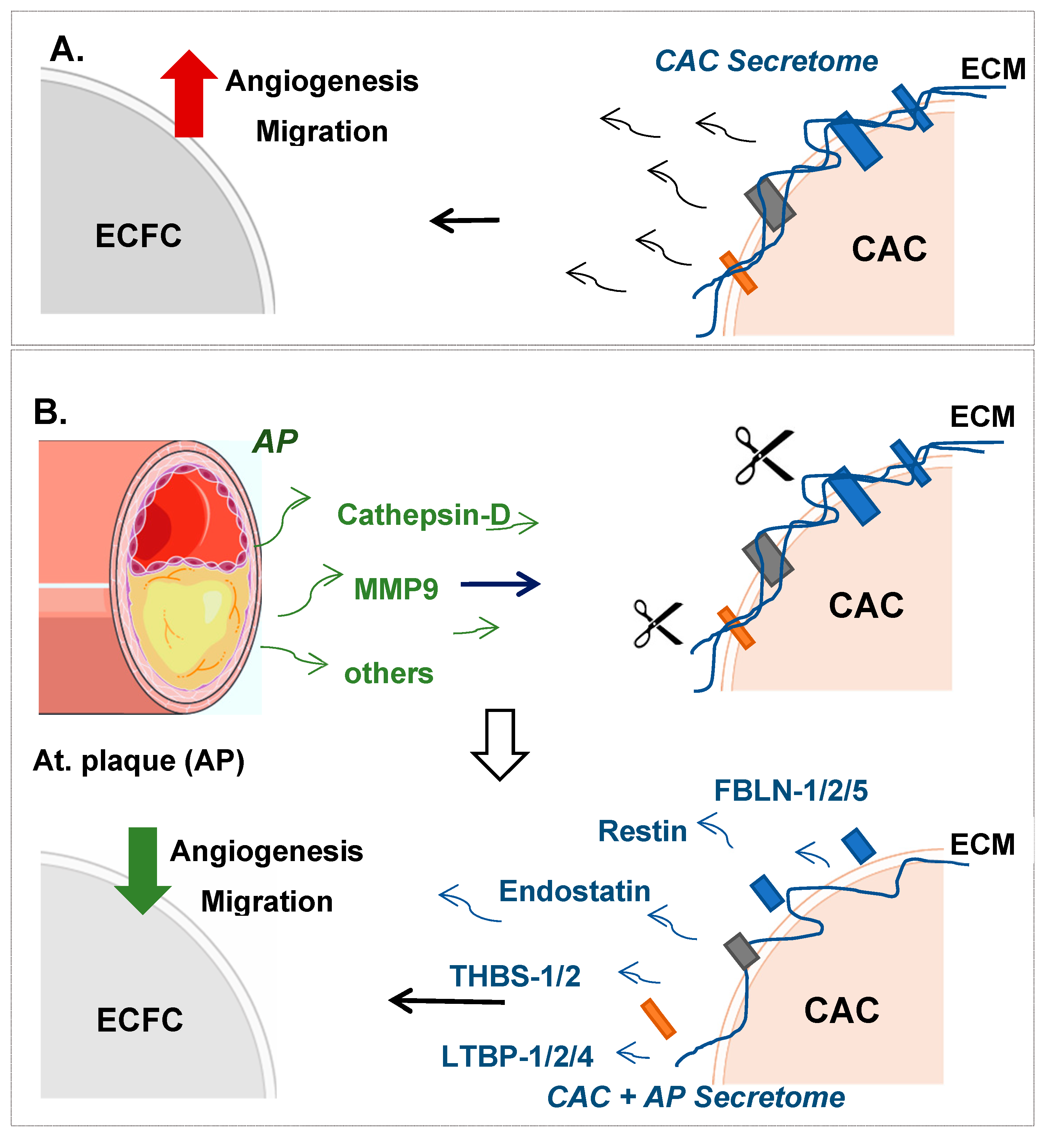
 ) ECFC angiogenesis and migration. (B) Incubation of CAC with atherosclerotic factors (Cathepsin-D, MMP9 and other proteases) promotes the rupture and degradation of ECM proteins. Further incubation of ECFC with the CAC released factors (fibulins, endostatin, restin, thrombospondins, LTBP1, etc.) inhibits the paracrine effect of CAC, since angiogenesis and migration of ECFC decreases (
) ECFC angiogenesis and migration. (B) Incubation of CAC with atherosclerotic factors (Cathepsin-D, MMP9 and other proteases) promotes the rupture and degradation of ECM proteins. Further incubation of ECFC with the CAC released factors (fibulins, endostatin, restin, thrombospondins, LTBP1, etc.) inhibits the paracrine effect of CAC, since angiogenesis and migration of ECFC decreases (  ) compared to ECFC stimulated with the secretome of control CAC (not affected by AP factors). Curved arrows represent the release of factors by CAC, the AP arteries or CAC after incubation with AP secretome. Straight arrows indicate the direction of the effect of the factors released by CAC control (A), AP or CAC+AP (B).
) ECFC angiogenesis and migration. (B) Incubation of CAC with atherosclerotic factors (Cathepsin-D, MMP9 and other proteases) promotes the rupture and degradation of ECM proteins. Further incubation of ECFC with the CAC released factors (fibulins, endostatin, restin, thrombospondins, LTBP1, etc.) inhibits the paracrine effect of CAC, since angiogenesis and migration of ECFC decreases ( ) compared to ECFC stimulated with the secretome of control CAC (not affected by AP factors). Curved arrows represent the release of factors by CAC, the AP arteries or CAC after incubation with AP secretome. Straight arrows indicate the direction of the effect of the factors released by CAC control (A), AP or CAC+AP (B).
) compared to ECFC stimulated with the secretome of control CAC (not affected by AP factors). Curved arrows represent the release of factors by CAC, the AP arteries or CAC after incubation with AP secretome. Straight arrows indicate the direction of the effect of the factors released by CAC control (A), AP or CAC+AP (B).
) ECFC angiogenesis and migration. (B) Incubation of CAC with atherosclerotic factors (Cathepsin-D, MMP9 and other proteases) promotes the rupture and degradation of ECM proteins. Further incubation of ECFC with the CAC released factors (fibulins, endostatin, restin, thrombospondins, LTBP1, etc.) inhibits the paracrine effect of CAC, since angiogenesis and migration of ECFC decreases ( ) compared to ECFC stimulated with the secretome of control CAC (not affected by AP factors). Curved arrows represent the release of factors by CAC, the AP arteries or CAC after incubation with AP secretome. Straight arrows indicate the direction of the effect of the factors released by CAC control (A), AP or CAC+AP (B).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
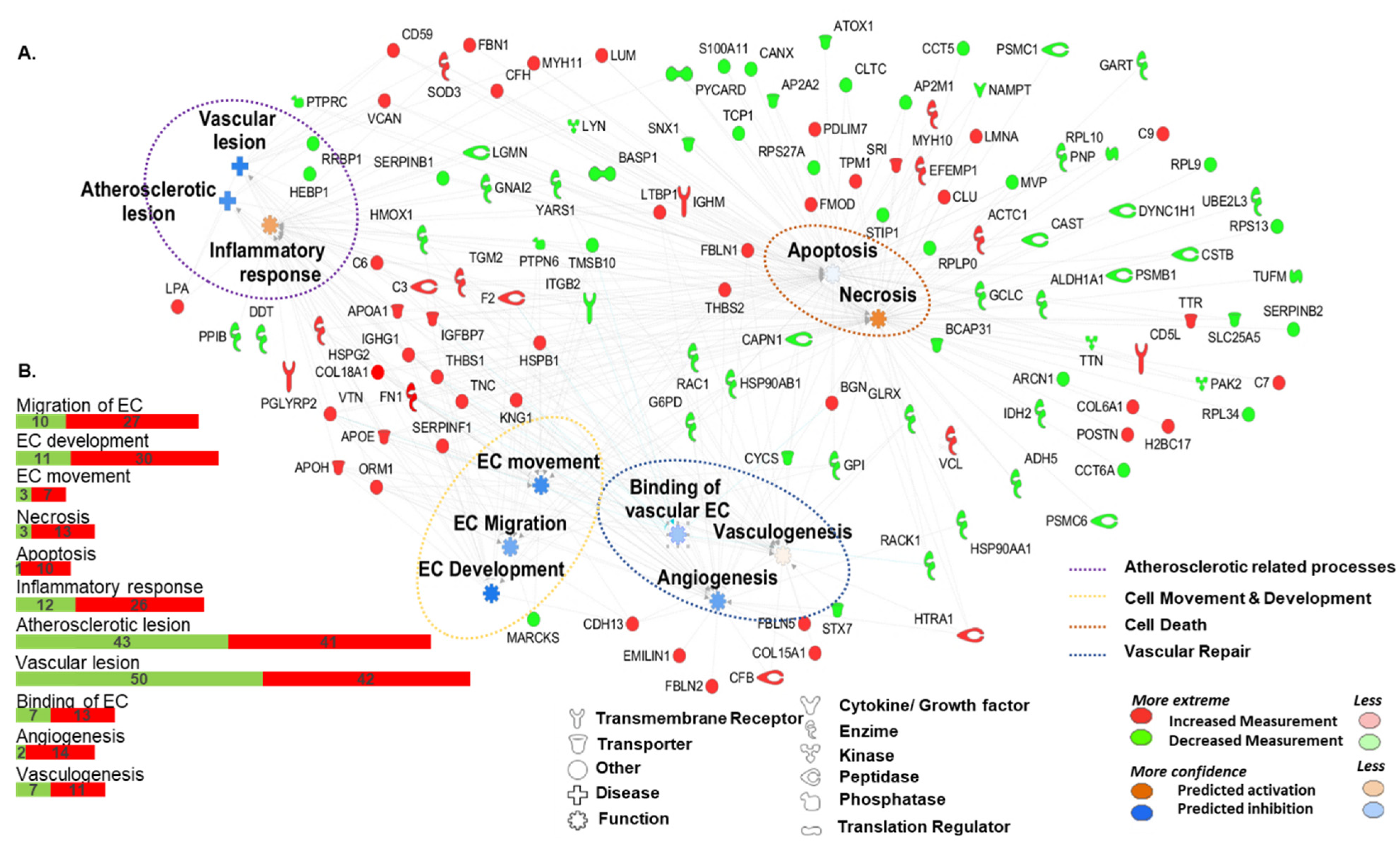
| Diseases or Functions | Molecules | p-Value | Gene Names | |
|---|---|---|---|---|
| CV System Development | EC Movement  | 20 | 1.01 × 10−8 | ↑APOE, ↑APOH, ↑CDH13, ↑COL18A1, ↑FN1, ↑HSPB1, ↑HSPG2, ↑KNG1, ↑ORM1, ↑SERPINF1, ↑THBS1, ↑THBS2, ↑VTN, ↓G6PD, ↓GPI, ↓HSP90AB1, ↓ITGB2, ↓MARCKS ↓PTPN6, ↓TMSB10 |
| EC Development | 16 | 4.54 × 10−6 | ↑APOA1, ↑APOE, ↑APOH, ↑C3, ↑CDH13, ↑COL18A1, ↑F2, ↑FN1, ↑HSPG2, ↑IGHG1, ↑KNG1, ↑SERPINF1, ↑THBS1, ↑VTN, ↓G6PD, ↓HMOX1 | |
| EC Migration | 18 | 7.64 × 10−8 | ↑APOE, ↑APOH, ↑CDH13, ↑COL18A1, ↑FN1, ↑HSPB1, ↑KNG1, ↑ORM1, ↑SERPINF1, ↑THBS1, ↑VTN, ↓G6PD, ↓GPI, ↓HSP90AB1, ↓ITGB2, ↓MARCKS ↓PTPN6, ↓TMSB10 | |
Vasculogenesis | 37 | 4.10 × 10−13 | ↑APOA1, ↑APOE, ↑APOH, ↑C3, ↑C6, ↑CDH13, ↑CFB, ↑COL15A1, ↑COL18A1, ↑F2, ↑FBLN1, ↑FBLN5, ↑FN1, ↑HSPG2, ↑HTRA1, ↑IGFBP7, IGHG1, ↑KNG1, ↑LTBP1, ↑MYH10, ↑ORM1, ↑SERPINF1, ↑TGM2, ↑THBS1, ↑THBS2, ↑TNC, ↑VTN, ↓CAPN1, ↓CYCS, ↓GCLC, ↓GLRX, ↓G6PD, ↓HMOX1, ↓ITGB2, ↓PTPN6, ↓STX7, ↓YARS | |
| Angiogenesis | 41 | 6.80 × 10−13 | ↑APOA1, ↑APOE, ↑APOH, ↑C3, ↑C6, ↑CDH13, ↑CFB, ↑COL15A1, ↑COL18A1, ↑EMILIN1, ↑F2, ↑FBLN1, ↑FBLN2, ↑FBLN5, ↑FN1, ↑HSPB1, ↑HSPG2, ↑HTRA1, ↑IGFBP7, ↑IGHG1, ↑KNG1, ↑LTBP1, ↑MYH10, ↑ORM1, ↑SERPINF1, ↑TGM2, ↑THBS1, ↑THBS2, ↑TNC, ↑VTN, ↓CAPN1, ↓CYCS, ↓GCLC, ↓GLRX, ↓G6PD, ↓HMOX1, ↓HSP90AA1, ↓ITGB2, ↓PTPN6, ↓STX7, ↓YARS | |
| Cell Death and survival | Necrosis | 92 | 2.54 × 10−23 | ↑APOE, ↑APOA1, ↑BGN, ↑C3, ↑C7, ↑C9, ↑CD59, ↑CD5L, ↑CFH, ↑CLU, ↑COL6A1, ↑COL18A1, ↑EFEMP1, ↑F2, ↑FBLN1, ↑FMOD, ↑FN1, ↑HIST1H2BO, ↑HSPB1, ↑HTRA1, ↑IGFBP7, ↑IGHG1, ↑IGHM, ↑KNG1, ↑LMNA, ↑LTBP1, ↑LUM, ↑MYH10, ↑MYH11, ↑PDLIM7, ↑POSTN, ↑SERPINF1, ↑SOD3, ↑SRI, ↑THBS1, ↑TNC, ↑THBS2, ↑TGM2, ↑TPM1, ↑TTR, ↑VCAN, ↑VTN, ↓ALDH1A1, ↓ADH5, ↓AP2A2, ↓AP2M1, ↓ARCN1, ↓BCAP31, ↓CAPN1, ↓CAST, ↓CCT5, ↓CCT6A, ↓CLTC, ↓CSTB, ↓CYCS, ↓DYNC1H1, ↓G6PD, ↓GCLC, ↓GLRX, ↓GNAI2, ↓GPI, ↓HMOX1, ↓HSP90AA1, ↓HSP90AB1, ↓IDH2, ↓ITGB2, ↓LGMN, ↓LYN, ↓MVP, ↓NAMPT, ↓PSMB1, ↓PSMC1, ↓PNP, ↓PSMC6, ↓PTPN6, ↓PTPRC, ↓PYCARD, ↓RACK 1, ↓RPL10, ↓RPL9, ↓RPL34, ↓RPS13, ↓SERPINB2, ↓STIP1, ↓SNX1, ↓S100A11, ↓TCP1, ↓TTN, ↓TMSB10, ↓TUFM, ↓UBE2L3, ↓YARS |
| Apoptosis | 84 | 2.10 × 10−18 | ↑APOA1, ↑APOE, ↑BGN, ↑C3, ↑C6, ↑CD59, ↑CD5L, ↑CLU, ↑COL18A1, ↑EFEMP1, ↑F2, ↑FBLN1, ↑FBN1, ↑FMOD, ↑FN1, ↑HTRA1, ↑HSPB1, ↑HSPG2, ↑IGFBP7, ↑IGHG1, ↑IGHM, ↑KNG1, ↑LMNA, ↑LTBP1, ↑LUM, ↑MYH10, ↑MYH11, ↑PDLIM7, ↑VCAN, ↑TTR, ↑SRI, ↑CFH, ↑TPM1, ↑SERPINF1, ↑SOD3, ↑TGM2, ↑THBS1, ↑THBS2, ↑TNC, ↑VCL, ↑VTN, ↓ADH5, ↓ALDH1A1, ↓AP2A2, ↓AP2M1, ↓ATOX1, ↓BASP1, ↓BCAP31, ↓CANX, ↓CAPN1, ↓CAST, ↓CLTC, ↓CSTB, ↓CYCS, ↓DYNC1H1, ↓GCLC, ↓GLRX, ↓GNAI2, ↓G6PD, ↓GPI, ↓HMOX1, ↓HSP90AA1, ↓HSP90AB1, ↓ITGB2, ↓LGMN, ↓LYN, ↓MVP, ↓NAMPT, ↓PSMB1, ↓PTPN6, ↓PTPRC, ↓PYCARD, ↓PNP, ↓RACK1, ↓RPL10, ↓RRBP1, ↓S100A11, ↓SERPINB1, ↓SNX1, ↓STIP1, ↓TCP1, ↓TMSB10, ↓TTN, ↓YARS | |
| CV Disease | Atherosclerotic Lesion | 11 | 1.13 × 10−6 | ↑APOA1, ↑APOE, ↑CD59, ↑COL18A1, ↑FN1, ↑HSPG2, ↑IGHG1, ↑LPA, ↑THBS1, ↑VCAN, ↓HMOX1 |
| Vascular Lesion | 16 | 7.59 × 10−9 | ↑APOA1, ↑APOE, ↑CD59, ↑COL18A1, ↑FBN1, ↑FN1, ↑HSPG2, ↑IGFBP7, ↑IGHG1, ↑LPA, ↑MYH11, ↑THBS1, ↑VCAN, ↓HMOX1, ↓PTPRC, ↓RRBP1 | |
| Inflammatory response | 38 | 9.27 × 10−14 | ↑APOA1, ↑APOE, ↑APOH, ↑C3, ↑C6, ↑CFH, ↑COL18A1, ↑F2, ↑FN1, ↑HSPB1, ↑HSPG2, ↑IGHG1, ↑IGHM, ↑KNG1, ↑LPA, ↑LTBP1, ↑LUM, ↑ORM1, ↑PGLYRP2, ↑SERPINF1, ↑SOD3, ↑TGM2, ↑THBS1, ↑THBS2, ↑TNC, ↑VTN, ↓GNAI2, ↓HEBP1, ↓HMOX1, ↓ITGB2, ↓LGMN, ↓LYN, ↓PPIB, ↓PTPN6, ↓PYCARD, ↓SERPINB1, ↓TMSB10, ↓YARS | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eslava-Alcon, S.; Extremera-García, M.J.; Sanchez-Gomar, I.; Beltrán-Camacho, L.; Rosal-Vela, A.; Muñoz, J.; Ibarz, N.; Alonso-Piñero, J.A.; Rojas-Torres, M.; Jiménez-Palomares, M.; et al. Atherosclerotic Pre-Conditioning Affects the Paracrine Role of Circulating Angiogenic Cells Ex-Vivo. Int. J. Mol. Sci. 2020, 21, 5256. https://doi.org/10.3390/ijms21155256
Eslava-Alcon S, Extremera-García MJ, Sanchez-Gomar I, Beltrán-Camacho L, Rosal-Vela A, Muñoz J, Ibarz N, Alonso-Piñero JA, Rojas-Torres M, Jiménez-Palomares M, et al. Atherosclerotic Pre-Conditioning Affects the Paracrine Role of Circulating Angiogenic Cells Ex-Vivo. International Journal of Molecular Sciences. 2020; 21(15):5256. https://doi.org/10.3390/ijms21155256
Chicago/Turabian StyleEslava-Alcon, Sara, Mª Jesús Extremera-García, Ismael Sanchez-Gomar, Lucía Beltrán-Camacho, Antonio Rosal-Vela, Javier Muñoz, Nuria Ibarz, Jose Angel Alonso-Piñero, Marta Rojas-Torres, Margarita Jiménez-Palomares, and et al. 2020. "Atherosclerotic Pre-Conditioning Affects the Paracrine Role of Circulating Angiogenic Cells Ex-Vivo" International Journal of Molecular Sciences 21, no. 15: 5256. https://doi.org/10.3390/ijms21155256
APA StyleEslava-Alcon, S., Extremera-García, M. J., Sanchez-Gomar, I., Beltrán-Camacho, L., Rosal-Vela, A., Muñoz, J., Ibarz, N., Alonso-Piñero, J. A., Rojas-Torres, M., Jiménez-Palomares, M., González-Rovira, A., Conejero, R., Doiz, E., Rodriguez-Piñero, M., Moreno-Luna, R., & Durán-Ruiz, M. C. (2020). Atherosclerotic Pre-Conditioning Affects the Paracrine Role of Circulating Angiogenic Cells Ex-Vivo. International Journal of Molecular Sciences, 21(15), 5256. https://doi.org/10.3390/ijms21155256