The Functional Roles and Applications of Immunoglobulins in Neurodegenerative Disease



Abstract
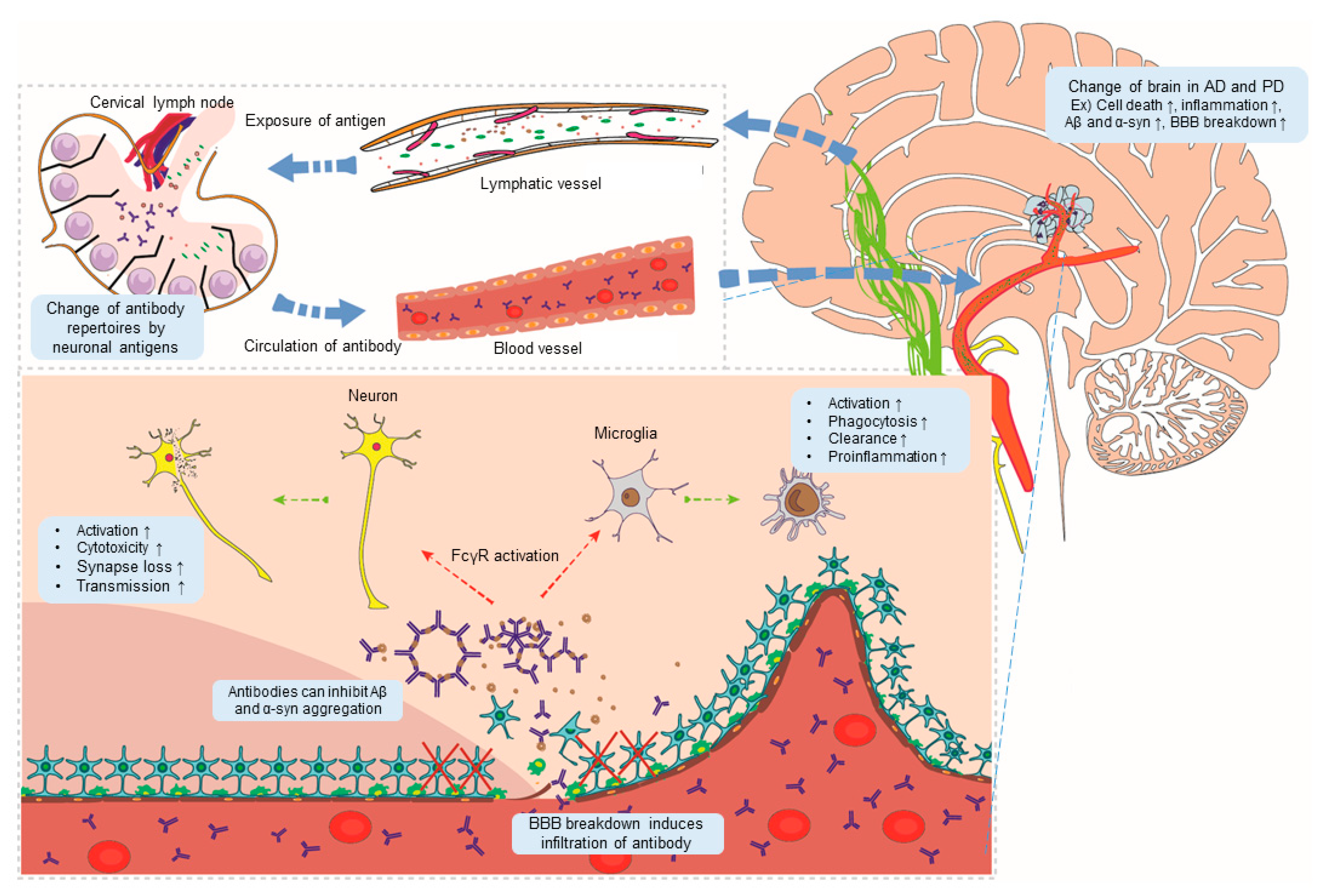
:1. Introduction
2. Role of Igs in AD
2.1. Alteration of Adaptive Immune Responses in AD
2.2. Evidence for an Association between Ig Responses and AD
2.3. BBB Breakdown and Ig Infiltration of the CNS
2.4. Protective Role of Natural Antibodies in AD
2.5. Pathogenic Natural Antibodies in AD
2.6. Diagnostic Application of Antibodies for AD
2.7. Therapeutic Application of Antibodies in AD
3. Role of Igs in PD
3.1. Changes to the Adaptive Immune System in PD
3.2. Evidence for an Association Between Ig Response and PD
3.3. BBB Breakdown in PD
3.4. Protective Role of Natural Antibodies in PD
3.5. Pathogenic Role of Natural Antibodies in PD
3.6. Diagnostic Application of Antibodies in PD
3.7. Therapeutic Application of Antibodies in PD
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Ig | Immunoglobulin |
| SS | Sjögren’s syndrome |
| SLE | Systemic lupus erythematosus |
| RA | Rheumatoid arthritis |
| AD | Alzheimer’s disease |
| PD | Parkinson’s disease |
| CNS | Central nervous system |
| CSF | Cerebrospinal fluid |
| NK | Natural killer |
| BBB | Blood–brain barrier |
| α7 nAChR | Nicotinic acetylcholine receptor α7 |
| Syk | Spleen tyrosine kinase |
| FcγR | Fc gamma receptor |
| SGs | Stress granules |
| Aβ | Beta-amyloid |
| SN | Substantia nigra |
| α-syn | α-Synuclein |
| TH | Tyrosine hydroxylase |
| MAG | Myelin-associated glycoprotein |
References
- Casali, P.; Schettino, E.W. Structure and function of natural antibodies. Curr. Top. Microbiol. Immunol. 1996, 210, 167–179. [Google Scholar] [PubMed]
- Elkon, K.; Casali, P. Nature and functions of autoantibodies. Nat. Clin. Pract. Rheumatol. 2008, 4, 491–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Zhurbenko, N.; Quach, T.D.; Hopkins, T.J.; Rothstein, T.L.; Hernandez, A.M. Human B-1 Cells and B-1 Cell Antibodies Change With Advancing Age. Front. Immunol. 2019, 10, 483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sim, K.Y.; Park, S.H.; Choi, K.Y.; Park, J.E.; Lee, J.S.; Kim, B.C.; Gwak, J.; Song, W.K.; Lee, K.H.; Park, S.G. High-throughput epitope profiling of antibodies in the plasma of Alzheimer’s disease patients using random peptide microarrays. Sci. Rep. 2019, 9, 4587. [Google Scholar] [CrossRef]
- Neiman, M.; Hellstrom, C.; Just, D.; Mattsson, C.; Fagerberg, L.; Schuppe-Koistinen, I.; Gummesson, A.; Bergstrom, G.; Kallioniemi, O.; Achour, A.; et al. Individual and stable autoantibody repertoires in healthy individuals. Autoimmunity 2019, 52, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madi, A.; Hecht, I.; Bransburg-Zabary, S.; Merbl, Y.; Pick, A.; Zucker-Toledano, M.; Quintana, F.J.; Tauber, A.I.; Cohen, I.R.; Ben-Jacob, E. Organization of the autoantibody repertoire in healthy newborns and adults revealed by system level informatics of antigen microarray data. Proc. Natl. Acad. Sci. USA 2009, 106, 14484–14489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, W.T.; Chang, C.; Gershwin, M.E.; Lian, Z.X. Development of autoantibodies precedes clinical manifestations of autoimmune diseases: A comprehensive review. J. Autoimmun. 2017, 83, 95–112. [Google Scholar] [CrossRef]
- Ludwig, R.J.; Vanhoorelbeke, K.; Leypoldt, F.; Kaya, Z.; Bieber, K.; McLachlan, S.M.; Komorowski, L.; Luo, J.; Cabral-Marques, O.; Hammers, C.M.; et al. Mechanisms of Autoantibody-Induced Pathology. Front. Immunol. 2017, 8, 603. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, C.; Kokkonen, H.; Johansson, M.; Hallmans, G.; Wadell, G.; Rantapaa-Dahlqvist, S. Autoantibodies predate the onset of systemic lupus erythematosus in northern Sweden. Arthritis Res. Ther. 2011, 13, R30. [Google Scholar] [CrossRef] [Green Version]
- Shmerling, R.H. Autoantibodies in systemic lupus erythematosus—There before you know it. N. Engl. J. Med. 2003, 349, 1499–1500. [Google Scholar] [CrossRef]
- Lichtbroun, A.S. Positive Findings on an Early Autoantibody Panel in Sjogren’s Syndrome Often Predate Positive Findings on Classic Tests: Comment on the Article by Theander et al. Arthritis Rheumatol. 2016, 68, 2054–2055. [Google Scholar] [CrossRef] [PubMed]
- Gronwall, C.; Silverman, G.J. Natural IgM: Beneficial autoantibodies for the control of inflammatory and autoimmune disease. J. Clin. Immunol. 2014, 34, S12–S21. [Google Scholar] [CrossRef] [Green Version]
- Grossmayer, G.E.; Munoz, L.E.; Weber, C.K.; Franz, S.; Voll, R.E.; Kern, P.M.; Kalden, J.R.; Schett, G.; Herrmann, M.; Gaipl, U.S. IgG autoantibodies bound to surfaces of necrotic cells and complement C4 comprise the phagocytosis promoting activity for necrotic cells of systemic lupus erythaematosus sera. Ann. Rheum. Dis. 2008, 67, 1626–1632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarmiento, L.F.; Munoz, L.E.; Chirinos, P.; Bianco, N.E.; Zabaleta-Lanz, M.E. Opsonization by anti-dsDNA antibodies of apoptotic cells in systemic lupus erythematosus. Autoimmunity 2007, 40, 337–339. [Google Scholar] [CrossRef] [PubMed]
- Manfredi, A.A.; Rovere, P.; Galati, G.; Heltai, S.; Bozzolo, E.; Soldini, L.; Davoust, J.; Balestrieri, G.; Tincani, A.; Sabbadini, M.G. Apoptotic cell clearance in systemic lupus erythematosus. I. Opsonization by antiphospholipid antibodies. Arthritis Rheum. 1998, 41, 205–214. [Google Scholar] [CrossRef]
- Maderna, P.; Godson, C. Phagocytosis of apoptotic cells and the resolution of inflammation. Biochim. Biophys. Acta 2003, 1639, 141–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louveau, A.; Harris, T.H.; Kipnis, J. Revisiting the Mechanisms of CNS Immune Privilege. Trends Immunol. 2015, 36, 569–577. [Google Scholar] [CrossRef] [Green Version]
- Korin, B.; Ben-Shaanan, T.L.; Schiller, M.; Dubovik, T.; Azulay-Debby, H.; Boshnak, N.T.; Koren, T.; Rolls, A. High-dimensional, single-cell characterization of the brain’s immune compartment. Nat. Neurosci. 2017, 20, 1300–1309. [Google Scholar] [CrossRef]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Corrigendum: Structural and functional features of central nervous system lymphatic vessels. Nature 2016, 533, 278. [Google Scholar] [CrossRef] [Green Version]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef]
- Benkler, M.; Agmon-Levin, N.; Hassin-Baer, S.; Cohen, O.S.; Ortega-Hernandez, O.D.; Levy, A.; Moscavitch, S.D.; Szyper-Kravitz, M.; Damianovich, M.; Blank, M.; et al. Immunology, autoimmunity, and autoantibodies in Parkinson’s disease. Clin. Rev. Allergy Immunol. 2012, 42, 164–171. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, M.R. Add Alzheimer’s disease to the list of autoimmune diseases. Med. Hypotheses 2005, 64, 458–463. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Li, G.; Xu, J.; Gao, S.; Chen, X. The Challenge of the Pathogenesis of Parkinson’s Disease: Is Autoimmunity the Culprit? Front. Immunol. 2018, 9, 2047. [Google Scholar] [CrossRef] [PubMed]
- DeMarshall, C.A.; Han, M.; Nagele, E.P.; Sarkar, A.; Acharya, N.K.; Godsey, G.; Goldwaser, E.L.; Kosciuk, M.; Thayasivam, U.; Belinka, B.; et al. Potential utility of autoantibodies as blood-based biomarkers for early detection and diagnosis of Parkinson’s disease. Immunol. Lett. 2015, 168, 80–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeMarshall, C.A.; Nagele, E.P.; Sarkar, A.; Acharya, N.K.; Godsey, G.; Goldwaser, E.L.; Kosciuk, M.; Thayasivam, U.; Han, M.; Belinka, B.; et al. Detection of Alzheimer’s disease at mild cognitive impairment and disease progression using autoantibodies as blood-based biomarkers. Alzheimers Dement. (Amsterdam) 2016, 3, 51–62. [Google Scholar] [CrossRef] [Green Version]
- Alzheimer’s Association. 2020 Alzheimer’s disease facts and figures. Alzheimers Dement. 2020, 16, 391. [Google Scholar] [CrossRef]
- Kametani, F.; Hasegawa, M. Reconsideration of Amyloid Hypothesis and Tau Hypothesis in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 25. [Google Scholar] [CrossRef] [Green Version]
- Gate, D.; Saligrama, N.; Leventhal, O.; Yang, A.C.; Unger, M.S.; Middeldorp, J.; Chen, K.; Lehallier, B.; Channappa, D.; De Los Santos, M.B.; et al. Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer’s disease. Nature 2020, 577, 399–404. [Google Scholar] [CrossRef]
- Stowe, A.M.; Ireland, S.J.; Ortega, S.B.; Chen, D.; Huebinger, R.M.; Tarumi, T.; Harris, T.S.; Cullum, C.M.; Rosenberg, R.; Monson, N.L.; et al. Adaptive lymphocyte profiles correlate to brain Abeta burden in patients with mild cognitive impairment. J. Neuroinflamm. 2017, 14, 149. [Google Scholar] [CrossRef] [Green Version]
- Bulati, M.; Buffa, S.; Martorana, A.; Gervasi, F.; Camarda, C.; Azzarello, D.M.; Monastero, R.; Caruso, C.; Colonna-Romano, G. Double negative (IgG+IgD-CD27-) B cells are increased in a cohort of moderate-severe Alzheimer’s disease patients and show a pro-inflammatory trafficking receptor phenotype. J. Alzheimers Dis. 2015, 44, 1241–1251. [Google Scholar] [CrossRef]
- Sollvander, S.; Ekholm-Pettersson, F.; Brundin, R.M.; Westman, G.; Kilander, L.; Paulie, S.; Lannfelt, L.; Sehlin, D. Increased Number of Plasma B Cells Producing Autoantibodies Against Abeta42 Protofibrils in Alzheimer’s Disease. J. Alzheimers Dis. 2015, 48, 63–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabatino, J.J., Jr.; Probstel, A.K.; Zamvil, S.S. B cells in autoimmune and neurodegenerative central nervous system diseases. Nat. Rev. Neurosci. 2019, 20, 728–745. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, M.R. Evidence linking neuronal cell death to autoimmunity in Alzheimer’s disease. Brain Res. 2003, 982, 19–30. [Google Scholar] [CrossRef]
- Zotova, E.; Bharambe, V.; Cheaveau, M.; Morgan, W.; Holmes, C.; Harris, S.; Neal, J.W.; Love, S.; Nicoll, J.A.; Boche, D. Inflammatory components in human Alzheimer’s disease and after active amyloid-beta42 immunization. Brain 2013, 136, 2677–2696. [Google Scholar] [CrossRef] [Green Version]
- Bouras, C.; Riederer, B.M.; Kovari, E.; Hof, P.R.; Giannakopoulos, P. Humoral immunity in brain aging and Alzheimer’s disease. Brain Res. Brain Res. Rev. 2005, 48, 477–487. [Google Scholar] [CrossRef]
- Maftei, M.; Thurm, F.; Schnack, C.; Tumani, H.; Otto, M.; Elbert, T.; Kolassa, I.T.; Przybylski, M.; Manea, M.; von Arnim, C.A. Increased levels of antigen-bound beta-amyloid autoantibodies in serum and cerebrospinal fluid of Alzheimer’s disease patients. PLoS ONE 2013, 8, e68996. [Google Scholar] [CrossRef]
- Bartos, A.; Fialova, L.; Svarcova, J.; Ripova, D. Patients with Alzheimer disease have elevated intrathecal synthesis of antibodies against tau protein and heavy neurofilament. J. Neuroimmunol. 2012, 252, 100–105. [Google Scholar] [CrossRef]
- Koval, L.; Lykhmus, O.; Kalashnyk, O.; Bachinskaya, N.; Kravtsova, G.; Soldatkina, M.; Zouridakis, M.; Stergiou, C.; Tzartos, S.; Tsetlin, V.; et al. The presence and origin of autoantibodies against α4 and α7 nicotinic acetylcholine receptors in the human blood: Possible relevance to Alzheimer’s pathology. J. Alzheimers Dis. 2011, 25, 747–761. [Google Scholar] [CrossRef]
- Báez-Pagán, C.A.; Delgado-Vélez, M.; Lasalde-Dominicci, J.A. Activation of the Macrophage α7 Nicotinic Acetylcholine Receptor and Control of Inflammation. J. Neuroimmune Pharmacol. 2015, 10, 468–476. [Google Scholar] [CrossRef] [Green Version]
- Parada, E.; Egea, J.; Romero, A.; del Barrio, L.; García, A.G.; López, M.G. Poststress treatment with PNU282987 can rescue SH-SY5Y cells undergoing apoptosis via α7 nicotinic receptors linked to a Jak2/Akt/HO-1 signaling pathway. Free Radic. Biol. Med. 2010, 49, 1815–1821. [Google Scholar] [CrossRef]
- Parri, H.R.; Dineley, K.T. Nicotinic Acetylcholine Receptor Interaction with Ab-Amyloid: Molecular, Cellular, and Physiological Consequences. Curr. Alzheimer Res. 2010, 7, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Lykhmus, O.; Mishra, N.; Koval, L.; Kalashnyk, O.; Gergalova, G.; Uspenska, K.; Komisarenko, S.; Soreq, H.; Skok, M. Molecular Mechanisms Regulating LPS-Induced Inflammation in the Brain. Front. Mol. Neurosci. 2016, 9, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, B.; Tsolaki, M.; Batruch, I.; Anastasiou, A.; Frontistis, A.; Prassas, I.; Diamandis, E.P. Putative autoantibodies in the cerebrospinal fluid of Alzheimer’s disease patients. F1000Research 2019, 8, 1900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Li, L. Autoantibodies in Alzheimer’s disease: Potential biomarkers, pathogenic roles, and therapeutic implications. J. Biomed. Res. 2016, 30, 361–372. [Google Scholar]
- Papuc, E.; Krupski, W.; Kurys-Denis, E.; Rejdak, K. Antibodies against small heat-shock proteins in Alzheimer’s disease as a part of natural human immune repertoire or activation of humoral response? J. Neural. Transm. 2016, 123, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louveau, A.; Da Mesquita, S.; Kipnis, J. Lymphatics in Neurological Disorders: A Neuro-Lympho-Vascular Component of Multiple Sclerosis and Alzheimer’s Disease? Neuron 2016, 91, 957–973. [Google Scholar] [CrossRef] [Green Version]
- Ahn, J.H.; Cho, H.; Kim, J.H.; Kim, S.H.; Ham, J.S.; Park, I.; Suh, S.H.; Hong, S.P.; Song, J.H.; Hong, Y.K.; et al. Meningeal lymphatic vessels at the skull base drain cerebrospinal fluid. Nature 2019, 572, 62–66. [Google Scholar] [CrossRef]
- Sampson, J.H.; Gunn, M.D.; Fecci, P.E.; Ashley, D.M. Brain immunology and immunotherapy in brain tumours. Nat. Rev. Cancer 2020, 20, 12–25. [Google Scholar] [CrossRef]
- Lang, K.; Pruss, H. Frequencies of neuronal autoantibodies in healthy controls: Estimation of disease specificity. Neurol. Neuroimmunol. Neuroinflamm. 2017, 4, e386. [Google Scholar] [CrossRef] [Green Version]
- Kheirkhah, R.; D, C.; Sieber, F.; Oh, E.; Nagele, R.G. The origin and nature of the complex autoantibody profile in cerebrospinal fluid. Brain Behav. Immun. Health 2020, 2, 100032. [Google Scholar] [CrossRef]
- Britschgi, M.; Olin, C.E.; Johns, H.T.; Takeda-Uchimura, Y.; LeMieux, M.C.; Rufibach, K.; Rajadas, J.; Zhang, H.; Tomooka, B.; Robinson, W.H.; et al. Neuroprotective natural antibodies to assemblies of amyloidogenic peptides decrease with normal aging and advancing Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 12145–12150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glass, L.J.; Sinclair, D.; Boerrigter, D.; Naude, K.; Fung, S.J.; Brown, D.; Catts, V.S.; Tooney, P.; O’Donnell, M.; Lenroot, R.; et al. Brain antibodies in the cortex and blood of people with schizophrenia and controls. Transl. Psychiatry 2017, 7, e1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montagne, A.; Zhao, Z.; Zlokovic, B.V. Alzheimer’s disease: A matter of blood-brain barrier dysfunction? J. Exp. Med. 2017, 214, 3151–3169. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.R.; Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer’s disease. Biochim. Biophys. Acta 2016, 1862, 887–900. [Google Scholar] [CrossRef]
- Fullerton, S.M.; Shirman, G.A.; Strittmatter, W.J.; Matthew, W.D. Impairment of the blood-nerve and blood-brain barriers in apolipoprotein e knockout mice. Exp. Neurol. 2001, 169, 13–22. [Google Scholar] [CrossRef]
- Ryu, J.K.; McLarnon, J.G. A leaky blood-brain barrier, fibrinogen infiltration and microglial reactivity in inflamed Alzheimer’s disease brain. J. Cell. Mol. Med. 2009, 13, 2911–2925. [Google Scholar] [CrossRef] [Green Version]
- Morch, M.T.; Sorensen, S.F.; Khorooshi, R.; Asgari, N.; Owens, T. Selective localization of IgG from cerebrospinal fluid to brain parenchyma. J. Neuroinflamm. 2018, 15, 110. [Google Scholar] [CrossRef] [Green Version]
- Villasenor, R.; Ozmen, L.; Messaddeq, N.; Gruninger, F.; Loetscher, H.; Keller, A.; Betsholtz, C.; Freskgard, P.O.; Collin, L. Trafficking of Endogenous Immunoglobulins by Endothelial Cells at the Blood-Brain Barrier. Sci. Rep. 2016, 6, 25658. [Google Scholar] [CrossRef]
- McBrayer, M.; Nixon, R.A. Lysosome and calcium dysregulation in Alzheimer’s disease: Partners in crime. Biochem. Soc. Trans. 2013, 41, 1495–1502. [Google Scholar] [CrossRef] [Green Version]
- Marsh, S.E.; Abud, E.M.; Lakatos, A.; Karimzadeh, A.; Yeung, S.T.; Davtyan, H.; Fote, G.M.; Lau, L.; Weinger, J.G.; Lane, T.E.; et al. The adaptive immune system restrains Alzheimer’s disease pathogenesis by modulating microglial function. Proc. Natl. Acad. Sci. USA 2016, 113, E1316–E1325. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Xu, J.; Gao, J.; Chen, P.; Yin, M.; Zhao, W. Decreased immunoglobulin G in brain regions of elder female APOE4-TR mice accompany with Abeta accumulation. Immun. Ageing 2019, 16, 2. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Geahlen, R.L. Stress Granules Modulate SYK to Cause Microglial Cell Dysfunction in Alzheimer’s Disease. EBioMedicine 2015, 2, 1785–1798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valls-Comamala, V.; Guivernau, B.; Bonet, J.; Puig, M.; Peralvarez-Marin, A.; Palomer, E.; Fernandez-Busquets, X.; Altafaj, X.; Tajes, M.; Puig-Pijoan, A.; et al. The antigen-binding fragment of human gamma immunoglobulin prevents amyloid beta-peptide folding into beta-sheet to form oligomers. Oncotarget 2017, 8, 41154–41165. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Le Pichon, C.E.; Adolfsson, O.; Gafner, V.; Pihlgren, M.; Lin, H.; Solanoy, H.; Brendza, R.; Ngu, H.; Foreman, O.; et al. Antibody-Mediated Targeting of Tau In Vivo Does Not Require Effector Function and Microglial Engagement. Cell Rep. 2016, 16, 1690–1700. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Koudstaal, W.; Fletcher, L.; Costa, M.; van Winsen, M.; Siregar, B.; Inganas, H.; Kim, J.; Keogh, E.; Macedo, J.; et al. Naturally occurring antibodies isolated from PD patients inhibit synuclein seeding in vitro and recognize Lewy pathology. Acta Neuropathol. 2019, 137, 825–836. [Google Scholar] [CrossRef] [Green Version]
- Bae, E.J.; Lee, H.J.; Rockenstein, E.; Ho, D.H.; Park, E.B.; Yang, N.Y.; Desplats, P.; Masliah, E.; Lee, S.J. Antibody-aided clearance of extracellular alpha-synuclein prevents cell-to-cell aggregate transmission. J. Neurosci. 2012, 32, 13454–13469. [Google Scholar] [CrossRef]
- Fuller, J.P.; Stavenhagen, J.B.; Teeling, J.L. New roles for Fc receptors in neurodegeneration-the impact on Immunotherapy for Alzheimer’s Disease. Front. Neurosci. 2014, 8, 235. [Google Scholar] [CrossRef] [Green Version]
- Andoh, T.; Kuraishi, Y. Direct action of immunoglobulin G on primary sensory neurons through Fc gamma receptor I. FASEB J. 2004, 18, 182–184. [Google Scholar] [CrossRef]
- Teeling, J.L.; Carare, R.O.; Glennie, M.J.; Perry, V.H. Intracerebral immune complex formation induces inflammation in the brain that depends on Fc receptor interaction. Acta Neuropathol. 2012, 124, 479–490. [Google Scholar] [CrossRef] [Green Version]
- Dinkins, M.B.; Dasgupta, S.; Wang, G.; Zhu, G.; He, Q.; Kong, J.N.; Bieberich, E. The 5XFAD Mouse Model of Alzheimer’s Disease Exhibits an Age-Dependent Increase in Anti-Ceramide IgG and Exogenous Administration of Ceramide Further Increases Anti-Ceramide Titers and Amyloid Plaque Burden. J. Alzheimers Dis. 2015, 46, 55–61. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.Y.; Tan, M.S.; Yu, J.T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar] [PubMed]
- Wang, X.J.; Yan, Z.Q.; Lu, G.Q.; Stuart, S.; Chen, S.D. Parkinson disease IgG and C5a-induced synergistic dopaminergic neurotoxicity: Role of microglia. Neurochem. Int. 2007, 50, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Theodore, S.; Standaert, D.G. Fcgamma receptors are required for NF-kappaB signaling, microglial activation and dopaminergic neurodegeneration in an AAV-synuclein mouse model of Parkinson’s disease. Mol. Neurodegener. 2010, 5, 42. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.R.; Cha, S.-H.; Kang, S.-J.; Kim, J.-B.; Jou, I.; Park, S.M. Prion-like Propagation of α-Synuclein Is Regulated by the FcγRIIB-SHP-1/2 Signaling Pathway in Neurons. Cell Rep. 2018, 22, 136–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruhns, P.; Jonsson, F. Mouse and human FcR effector functions. Immunol. Rev. 2015, 268, 25–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsinelos, T.; Tuck, B.J.; Mukadam, A.S.; McEwan, W.A. The Role of Antibodies and Their Receptors in Protection Against Ordered Protein Assembly in Neurodegeneration. Front. Immunol. 2019, 10, 1139. [Google Scholar] [CrossRef]
- Stamou, M.; Lein, P.J. Commentary: Fc Gamma Receptors are Expressed in the Developing Rat Brain and Activate Downstream Signaling Molecules upon Cross-Linking with Immune Complex. J. Neurol. Neuromed. 2019, 4, 26–29. [Google Scholar] [CrossRef] [Green Version]
- Murinello, S.; Mullins, R.F.; Lotery, A.J.; Perry, V.H.; Teeling, J.L. Fcgamma receptor upregulation is associated with immune complex inflammation in the mouse retina and early age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2014, 55, 247–258. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, H.A.; Mosier, D.R.; Zou, L.L.; Siklos, L.; Alexianu, M.E.; Engelhardt, J.I.; Beers, D.R.; Le, W.D.; Appel, S.H. Immunoglobulin Fc gamma receptor promotes immunoglobulin uptake, immunoglobulin-mediated calcium increase, and neurotransmitter release in motor neurons. J. Neurosci. Res. 2002, 69, 110–116. [Google Scholar] [CrossRef]
- Kam, T.I.; Song, S.; Gwon, Y.; Park, H.; Yan, J.J.; Im, I.; Choi, J.W.; Choi, T.Y.; Kim, J.; Song, D.K.; et al. FcgammaRIIb mediates amyloid-beta neurotoxicity and memory impairment in Alzheimer’s disease. J. Clin. Investig. 2013, 123, 2791–2802. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Vizarra, P.; Lopez-Franco, O.; Mallavia, B.; Higuera-Matas, A.; Lopez-Parra, V.; Ortiz-Munoz, G.; Ambrosio, E.; Egido, J.; Almeida, O.F.; Gomez-Guerrero, C. Immunoglobulin G Fc receptor deficiency prevents Alzheimer-like pathology and cognitive impairment in mice. Brain 2012, 135, 2826–2837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quan, Y.; Moller, T.; Weinstein, J.R. Regulation of Fcgamma receptors and immunoglobulin G-mediated phagocytosis in mouse microglia. Neurosci. Lett. 2009, 464, 29–33. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Hou, X.; Feng, Q.; Li, Y.; Wang, X.; Qin, L.; Yang, P. Lupus serum IgG induces microglia activation through Fc fragment dependent way and modulated by B-cell activating factor. J. Transl. Med. 2019, 17, 426. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, J.I.; Le, W.D.; Siklos, L.; Obal, I.; Boda, K.; Appel, S.H. Stereotaxic injection of IgG from patients with Alzheimer disease initiates injury of cholinergic neurons of the basal forebrain. Arch. Neurol. 2000, 57, 681–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, A.; Adua, E.; Ugrina, I.; Laws, S.; Wang, W. Unravelling Immunoglobulin G Fc N-Glycosylation: A Dynamic Marker Potentiating Predictive, Preventive and Personalised Medicine. Int. J. Mol. Sci. 2018, 19, 390. [Google Scholar] [CrossRef] [Green Version]
- Russell, A.C.; Šimurina, M.; Garcia, M.T.; Novokmet, M.; Wang, Y.; Rudan, I.; Campbell, H.; Lauc, G.; Thomas, M.G.; Wang, W. The N-glycosylation of immunoglobulin G as a novel biomarker of Parkinson’s disease. Glycobiology 2017, 27, 501–510. [Google Scholar] [CrossRef]
- Bacskai, B.J.; Kajdasz, S.T.; McLellan, M.E.; Games, D.; Seubert, P.; Schenk, D.; Hyman, B.T. Non-Fc-mediated mechanisms are involved in clearance of amyloid-beta in vivo by immunotherapy. J. Neurosci. 2002, 22, 7873–7878. [Google Scholar] [CrossRef] [Green Version]
- Tamura, Y.; Hamajima, K.; Matsui, K.; Yanoma, S.; Narita, M.; Tajima, N.; Xin, K.Q.; Klinman, D.; Okuda, K. The F(ab)′2 fragment of an Abeta-specific monoclonal antibody reduces Abeta deposits in the brain. Neurobiol. Dis. 2005, 20, 541–549. [Google Scholar] [CrossRef]
- Li, X.W.; Li, X.X.; Liu, Q.S.; Cheng, Y. Blood and Cerebrospinal Fluid Autoantibody to Abeta Levels in Patients with Alzheimer’s Disease: A Meta-Analysis Study. J. Mol. Neurosci. 2020, 70, 1208–1215. [Google Scholar] [CrossRef]
- Hromadkova, L.; Ovsepian, S.V. Tau-Reactive Endogenous Antibodies: Origin, Functionality, and Implications for the Pathophysiology of Alzheimer’s Disease. J. Immunol. Res. 2019, 2019, 7406810. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.Y.; Li, W.W.; Yang, H.M.; Manucat-Tan, N.B.; Wang, J.; Wang, Y.R.; Sun, B.L.; Hu, Z.C.; Zhang, L.L.; Tan, L.; et al. Naturally Occurring Antibodies to Tau Exists in Human Blood and Are Not Changed in Alzheimer’s Disease. Neurotox. Res. 2020, 37, 1029–1035. [Google Scholar] [CrossRef] [PubMed]
- DeMarshall, C.; Oh, E.; Kheirkhah, R.; Sieber, F.; Zetterberg, H.; Blennow, K.; Nagele, R.G. Detection of early-stage Alzheimer’s pathology using blood-based autoantibody biomarkers in elderly hip fracture repair patients. PLoS ONE 2019, 14, e0225178. [Google Scholar] [CrossRef] [PubMed]
- Counts, S.E.; Ikonomovic, M.D.; Mercado, N.; Vega, I.E.; Mufson, E.J. Biomarkers for the Early Detection and Progression of Alzheimer’s Disease. Neurotherapeutics 2017, 14, 35–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davydova, T.V.; Voskresenskaya, N.I.; Fomina, V.G.; Vetrile, L.A.; Doronina, O.A. Induction of autoantibodies to glutamate in patients with Alzheimer’s disease. Bull. Exp. Biol. Med. 2007, 143, 182–183. [Google Scholar] [CrossRef]
- Gruden, M.A.; Davidova, T.B.; Malisauskas, M.; Sewell, R.D.; Voskresenskaya, N.I.; Wilhelm, K.; Elistratova, E.I.; Sherstnev, V.V.; Morozova-Roche, L.A. Differential neuroimmune markers to the onset of Alzheimer’s disease neurodegeneration and dementia: Autoantibodies to Abeta((25-35)) oligomers, S100b and neurotransmitters. J. Neuroimmunol. 2007, 186, 181–192. [Google Scholar] [CrossRef]
- Hempel, P.; Heinig, B.; Jerosch, C.; Decius, I.; Karczewski, P.; Kassner, U.; Kunze, R.; Steinhagen-Thiessen, E.; Bimmler, M. Immunoadsorption of Agonistic Autoantibodies Against alpha1-Adrenergic Receptors in Patients With Mild to Moderate Dementia. Ther. Apher. Dial. 2016, 20, 523–529. [Google Scholar] [CrossRef]
- Honig, L.S.; Vellas, B.; Woodward, M.; Boada, M.; Bullock, R.; Borrie, M.; Hager, K.; Andreasen, N.; Scarpini, E.; Liu-Seifert, H.; et al. Trial of Solanezumab for Mild Dementia Due to Alzheimer’s Disease. N. Engl. J. Med. 2018, 378, 321–330. [Google Scholar] [CrossRef]
- Vandenberghe, R.; Rinne, J.O.; Boada, M.; Katayama, S.; Scheltens, P.; Vellas, B.; Tuchman, M.; Gass, A.; Fiebach, J.B.; Hill, D.; et al. Bapineuzumab for mild to moderate Alzheimer’s disease in two global, randomized, phase 3 trials. Alzheimers Res. Ther. 2016, 8, 18. [Google Scholar] [CrossRef] [Green Version]
- Ostrowitzki, S.; Lasser, R.A.; Dorflinger, E.; Scheltens, P.; Barkhof, F.; Nikolcheva, T.; Ashford, E.; Retout, S.; Hofmann, C.; Delmar, P.; et al. A phase III randomized trial of gantenerumab in prodromal Alzheimer’s disease. Alzheimers Res. Ther. 2017, 9, 95. [Google Scholar] [CrossRef]
- Klein, G.; Delmar, P.; Voyle, N.; Rehal, S.; Hofmann, C.; Abi-Saab, D.; Andjelkovic, M.; Ristic, S.; Wang, G.; Bateman, R.; et al. Gantenerumab reduces amyloid-beta plaques in patients with prodromal to moderate Alzheimer’s disease: A PET substudy interim analysis. Alzheimers Res. Ther. 2019, 11, 101. [Google Scholar] [CrossRef] [Green Version]
- Vellas, B.; Black, R.; Thal, L.J.; Fox, N.C.; Daniels, M.; McLennan, G.; Tompkins, C.; Leibman, C.; Pomfret, M.; Grundman, M.; et al. Long-term follow-up of patients immunized with AN1792: Reduced functional decline in antibody responders. Curr. Alzheimer Res. 2009, 6, 144–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicoll, J.A.R.; Buckland, G.R.; Harrison, C.H.; Page, A.; Harris, S.; Love, S.; Neal, J.W.; Holmes, C.; Boche, D. Persistent neuropathological effects 14 years following amyloid-beta immunization in Alzheimer’s disease. Brain 2019, 142, 2113–2126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chantran, Y.; Capron, J.; Alamowitch, S.; Aucouturier, P. Anti-Abeta Antibodies and Cerebral Amyloid Angiopathy Complications. Front. Immunol. 2019, 10, 1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.K.; Chao, S.P.; Hu, C.J. Clinical trials of new drugs for Alzheimer disease. J. Biomed. Sci. 2020, 27, 18. [Google Scholar] [CrossRef] [PubMed]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef]
- Garretti, F.; Agalliu, D.; Lindestam Arlehamn, C.S.; Sette, A.; Sulzer, D. Autoimmunity in Parkinson’s Disease: The Role of α-Synuclein-Specific T Cells. Front. Immunol. 2019, 10, 303. [Google Scholar] [CrossRef] [Green Version]
- Schröder, J.B.; Pawlowski, M.; Meyer Zu Hörste, G.; Gross, C.C.; Wiendl, H.; Meuth, S.G.; Ruck, T.; Warnecke, T. Immune Cell Activation in the Cerebrospinal Fluid of Patients With Parkinson’s Disease. Front. Neurol. 2018, 9, 1081. [Google Scholar] [CrossRef] [Green Version]
- Kustrimovic, N.; Comi, C.; Magistrelli, L.; Rasini, E.; Legnaro, M.; Bombelli, R.; Aleksic, I.; Blandini, F.; Minafra, B.; Riboldazzi, G.; et al. Parkinson’s disease patients have a complex phenotypic and functional Th1 bias: Cross-sectional studies of CD4+ Th1/Th2/T17 and Treg in drug-naïve and drug-treated patients. J. Neuroinflamm. 2018, 15, 205. [Google Scholar] [CrossRef]
- Pisani, V.; Stefani, A.; Pierantozzi, M.; Natoli, S.; Stanzione, P.; Franciotta, D.; Pisani, A. Increased blood-cerebrospinal fluid transfer of albumin in advanced Parkinson’s disease. J. Neuroinflamm. 2012, 9, 188. [Google Scholar] [CrossRef] [Green Version]
- Pienaar, I.S.; Lee, C.H.; Elson, J.L.; McGuinness, L.; Gentleman, S.M.; Kalaria, R.N.; Dexter, D.T. Deep-brain stimulation associates with improved microvascular integrity in the subthalamic nucleus in Parkinson’s disease. Neurobiol. Dis. 2015, 74, 392–405. [Google Scholar] [CrossRef]
- Orr, C.F.; Rowe, D.B.; Mizuno, Y.; Mori, H.; Halliday, G.M. A possible role for humoral immunity in the pathogenesis of Parkinson’s disease. Brain 2005, 128, 2665–2674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folke, J.; Rydbirk, R.; Løkkegaard, A.; Salvesen, L.; Hejl, A.-M.; Starhof, C.; Bech, S.; Winge, K.; Christensen, S.; Pedersen, L.Ø.; et al. Distinct Autoimmune Anti-α-Synuclein Antibody Patterns in Multiple System Atrophy and Parkinson’s Disease. Front. Immunol. 2019, 10, 2253. [Google Scholar] [CrossRef] [PubMed]
- Scott, K.M.; Kouli, A.; Yeoh, S.L.; Clatworthy, M.R.; Williams-Gray, C.H. A Systematic Review and Meta-Analysis of Alpha Synuclein Auto-Antibodies in Parkinson’s Disease. Front. Neurol. 2018, 9, 815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brudek, T.; Winge, K.; Folke, J.; Christensen, S.; Fog, K.; Pakkenberg, B.; Pedersen, L.O. Autoimmune antibody decline in Parkinson’s disease and Multiple System Atrophy; A step towards immunotherapeutic strategies. Mol. Neurodegener. 2017, 12, 44. [Google Scholar] [CrossRef]
- Kortekaas, R.; Leenders, K.L.; van Oostrom, J.C.; Vaalburg, W.; Bart, J.; Willemsen, A.T.; Hendrikse, N.H. Blood-brain barrier dysfunction in parkinsonian midbrain in vivo. Ann. Neurol. 2005, 57, 176–179. [Google Scholar] [CrossRef] [PubMed]
- Ham, J.H.; Yi, H.; Sunwoo, M.K.; Hong, J.Y.; Sohn, Y.H.; Lee, P.H. Cerebral microbleeds in patients with Parkinson’s disease. J. Neurol. 2014, 261, 1628–1635. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.T.; Woulfe, J.M. Striatal blood-brain barrier permeability in Parkinson’s disease. J. Cereb. Blood Flow Metab. 2015, 35, 747–750. [Google Scholar] [CrossRef] [Green Version]
- Desai Bradaric, B.; Patel, A.; Schneider, J.A.; Carvey, P.M.; Hendey, B. Evidence for angiogenesis in Parkinson’s disease, incidental Lewy body disease, and progressive supranuclear palsy. J. Neural. Transm. 2012, 119, 59–71. [Google Scholar] [CrossRef] [Green Version]
- Masliah, E.; Rockenstein, E.; Mante, M.; Crews, L.; Spencer, B.; Adame, A.; Patrick, C.; Trejo, M.; Ubhi, K.; Rohn, T.T.; et al. Passive immunization reduces behavioral and neuropathological deficits in an alpha-synuclein transgenic model of Lewy body disease. PLoS ONE 2011, 6, e19338. [Google Scholar] [CrossRef] [Green Version]
- Spencer, B.; Valera, E.; Rockenstein, E.; Overk, C.; Mante, M.; Adame, A.; Zago, W.; Seubert, P.; Barbour, R.; Schenk, D.; et al. Anti-alpha-synuclein immunotherapy reduces alpha-synuclein propagation in the axon and degeneration in a combined viral vector and transgenic model of synucleinopathy. Acta Neuropathol. Commun. 2017, 5, 7. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Le, W.D.; Appel, S.H. Role of Fcgamma receptors in nigral cell injury induced by Parkinson disease immunoglobulin injection into mouse substantia nigra. Exp. Neurol. 2002, 176, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Papuć, E.; Rejdak, K. Anti-MAG autoantibodies are increased in Parkinson’s disease but not in atypical parkinsonism. J. Neural. Transm. 2017, 124, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Papuć, E.; Kurzepa, J.; Kurys-Denis, E.; Grabarska, A.; Krupski, W.; Rejdak, K. Humoral response against glial derived antigens in Parkinson’s disease. Neurosci. Lett. 2014, 566, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Magy, L.; Kabore, R.; Mathis, S.; Lebeau, P.; Ghorab, K.; Caudie, C.; Vallat, J.M. Heterogeneity of Polyneuropathy Associated with Anti-MAG Antibodies. J. Immunol. Res. 2015, 2015, 450391. [Google Scholar] [CrossRef] [PubMed]
- Garces-Sanchez, M.; Dyck, P.J.; Kyle, R.A.; Zeldenrust, S.; Wu, Y.; Ladha, S.S.; Klein, C.J. Antibodies to myelin-associated glycoprotein (anti-Mag) in IgM amyloidosis may influence expression of neuropathy in rare patients. Muscle Nerve 2008, 37, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Grambalova, Z.; Kaiserova, M.; Vastik, M.; Mensikova, K.; Otruba, P.; Zapletalova, J.; Dufek, J.; Kanovsky, P. Peripheral neuropathy in Parkinson’s disease. Neuro. Endocrinol. Lett. 2015, 36, 363–367. [Google Scholar] [PubMed]
- Feng, N.; Simanski, S.; Islam, K.; Hynan, L.S.; Kodadek, T.; German, D.C. Antibody biomarker for de novo Parkinson disease: Attempted validation. NPJ Parkinsons Dis. 2018, 4, 28. [Google Scholar] [CrossRef]
- Akhtar, R.S.; Licata, J.P.; Luk, K.C.; Shaw, L.M.; Trojanowski, J.Q.; Lee, V.M. Measurements of auto-antibodies to α-synuclein in the serum and cerebral spinal fluids of patients with Parkinson’s disease. J. Neurochem. 2018, 145, 489–503. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.R.; Xie, X.X.; Ji, M.; Yu, X.L.; Zhu, J.; Zhang, L.X.; Liu, X.G.; Wei, C.; Li, G.; Liu, R.T. Naturally occurring autoantibodies against α-synuclein rescues memory and motor deficits and attenuates α-synuclein pathology in mouse model of Parkinson’s disease. Neurobiol. Dis. 2019, 124, 202–217. [Google Scholar] [CrossRef]
- George, S.; Brundin, P. Immunotherapy in Parkinson’s Disease: Micromanaging Alpha-Synuclein Aggregation. J. Parkinsons Dis. 2015, 5, 413–424. [Google Scholar] [CrossRef] [Green Version]
- Schenk, D.B.; Koller, M.; Ness, D.K.; Griffith, S.G.; Grundman, M.; Zago, W.; Soto, J.; Atiee, G.; Ostrowitzki, S.; Kinney, G.G. First-in-human assessment of PRX002, an anti-alpha-synuclein monoclonal antibody, in healthy volunteers. Mov. Disord. 2017, 32, 211–218. [Google Scholar] [CrossRef]
- McFarthing, K.; Simuni, T. Clinical Trial Highlights: Targetting Alpha-Synuclein. J. Parkinsons. Dis. 2019, 9, 5–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, J.; Kim, H.J.; Jeon, B. Immunotherapy Targeting Neurodegenerative Proteinopathies: Alpha-Synucleinopathies and Tauopathies. J. Mov. Disord. 2020, 13, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, D.; Kordower, J.H. Immunotherapy in Parkinson’s disease: Current status and future directions. Neurobiol. Dis. 2019, 132, 104587. [Google Scholar] [CrossRef] [PubMed]
- Liliang, P.C.; Liang, C.L.; Lu, K.; Yang, S.N.; Hsieh, M.T.; Tai, Y.C.; Wang, K.W. Population-based study suggests an increased risk of Alzheimer’s disease in Sjogren’s syndrome. Clin. Rheumatol. 2018, 37, 935–941. [Google Scholar] [CrossRef] [Green Version]
- Wotton, C.J.; Goldacre, M.J. Associations between specific autoimmune diseases and subsequent dementia: Retrospective record-linkage cohort study, UK. J. Epidemiol. Commun. Health 2017, 71, 576–583. [Google Scholar] [CrossRef]
- Chang, C.C.; Lin, T.M.; Chang, Y.S.; Chen, W.S.; Sheu, J.J.; Chen, Y.H.; Chen, J.H. Autoimmune rheumatic diseases and the risk of Parkinson disease: A nationwide population-based cohort study in Taiwan. Ann. Med. 2018, 50, 83–90. [Google Scholar] [CrossRef]
- Liu, F.C.; Huang, W.Y.; Lin, T.Y.; Shen, C.H.; Chou, Y.C.; Lin, C.L.; Lin, K.T.; Kao, C.H. Inverse Association of Parkinson Disease With Systemic Lupus Erythematosus: A Nationwide Population-based Study. Medicine 2015, 94, e2097. [Google Scholar] [CrossRef]


{kind=link}
| Protective Role | ||||
| Disease | Function | Antibody Target | Effector Cell | Reference |
| AD | Enhance the phagocytosis of Aβ | Non-specific | Microglia | [60,61,62] |
| Inhibit Aβ aggregation | Fab-mediated function | Cortical Neuron Endothelial cell | [63] | |
| Neutralize Aβ-induced toxicity | Oligomeric Aβ1-42 | Primary Neuron | [51] | |
| Reduce tau toxicity | Tau | Microglia | [64] | |
| PD | Neutralize the intracellular α-syn aggregation | α-syn | Neuron | [65] |
| Remove the extracellular α-syn proteins | Microglia | [66] | ||
| Prevent α-syn transfer between neuron and astrocyte | ||||
| Pathogenic Role | ||||
| Disease | Function | Antibody Target | Effector Cell | Reference |
| AD | Induce the neuroinflammation as Immune complexes | FcγR-mediated function | FcγR-expressing cells in the CNS | [67,68,69] |
| Increase Aβ plaque formation | Ceramide | Neuron Endothelial cell | [70] | |
| Release the proinflammatory cytokines | FcγR-mediated function | Microglia | [71] | |
| Release the proinflammatory cytokines | Tau | [64] | ||
| PD | Induce neuronal damage via microglial activation | FcγR-mediated function | Microglia | [72,73] |
| Promote the α-syn transfer | Neuron | [74] | ||
| Drug | Antibody Type (Target) | FDA Status | NCT (Patients) |
|---|---|---|---|
| Solanezumab | Passive (Mid-domain of the Aβ peptide) | Phase 3 | NCT02008357 (Older individuals who may be at risk for memory loss) NCT02760602 (Prodromal AD) |
| Bapineuzumab | Passive (N-terminal region of Aβ) | Discontinued | NA |
| Gantenerumab | Passive (Conformational epitope on Aβ fibrils) | Phase 3 | NCT03443973 and NCT03444870 (Early AD) |
| Crenezumab | Passive (Multiple forms of aggregated Aβ) | Phase 2 | NCT02670083 (Prodromal to mild AD) NCT01998841 (Autosomal-dominant AD) NCT01397578 (Mild to moderate AD) |
| Aducanumab | Passive (Conformational epitope on Aβ) | Phase 3 | NCT02477800 and NCT02484547 (Early AD) |
| BAN2401 | Passive (Large, soluble Aβ protofibrils) | Phase 3 | NCT01767311 and NCT03887455 (Early AD) |
| AN-1792 | Active (Synthetic full-length Aβ peptide with QS-21 adjuvant) | Discontinued | NA |
| Gosuranemab | Passive (N-terminal fragments of tau) | Phase 2 | NCT03352557 (Early AD) |
| AADvac1 | Active (Synthetic tau peptide withaluminum hydroxide adjuvant) | Phase 2 | NCT01117818 (Early AD) |
| Prasinezumab | Passive (Against aggregated α-syn) | Phase 2 | NCT02157714 and NCT03100149 (PD) |
| BIIB054 | Passive (α-syn residues 1-10) | Phase 2 | NCT03318523 and NCT02459886 (PD) |
| ABBV-0805 | Passive (Oligomeric/protofibrillar α-syn) | Phase1 | NCT04127695 (PD) |
| Activa Affitope (PD01A) | Active (Synthetic α-syn peptide with adjuvant) | Phase1 | NCT02618941 (PD) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sim, K.-Y.; Im, K.C.; Park, S.-G. The Functional Roles and Applications of Immunoglobulins in Neurodegenerative Disease. Int. J. Mol. Sci. 2020, 21, 5295. https://doi.org/10.3390/ijms21155295
Sim K-Y, Im KC, Park S-G. The Functional Roles and Applications of Immunoglobulins in Neurodegenerative Disease. International Journal of Molecular Sciences. 2020; 21(15):5295. https://doi.org/10.3390/ijms21155295
Chicago/Turabian StyleSim, Kyu-Young, Kyeong Chan Im, and Sung-Gyoo Park. 2020. "The Functional Roles and Applications of Immunoglobulins in Neurodegenerative Disease" International Journal of Molecular Sciences 21, no. 15: 5295. https://doi.org/10.3390/ijms21155295
APA StyleSim, K. -Y., Im, K. C., & Park, S. -G. (2020). The Functional Roles and Applications of Immunoglobulins in Neurodegenerative Disease. International Journal of Molecular Sciences, 21(15), 5295. https://doi.org/10.3390/ijms21155295