Targeting on Gut Microbiota-Derived Metabolite Trimethylamine to Protect Adult Male Rat Offspring against Hypertension Programmed by Combined Maternal High-Fructose Intake and Dioxin Exposure

 ,
,  ,
, 
 , ,
, ,  and
and 
Abstract
:1. Introduction
2. Results
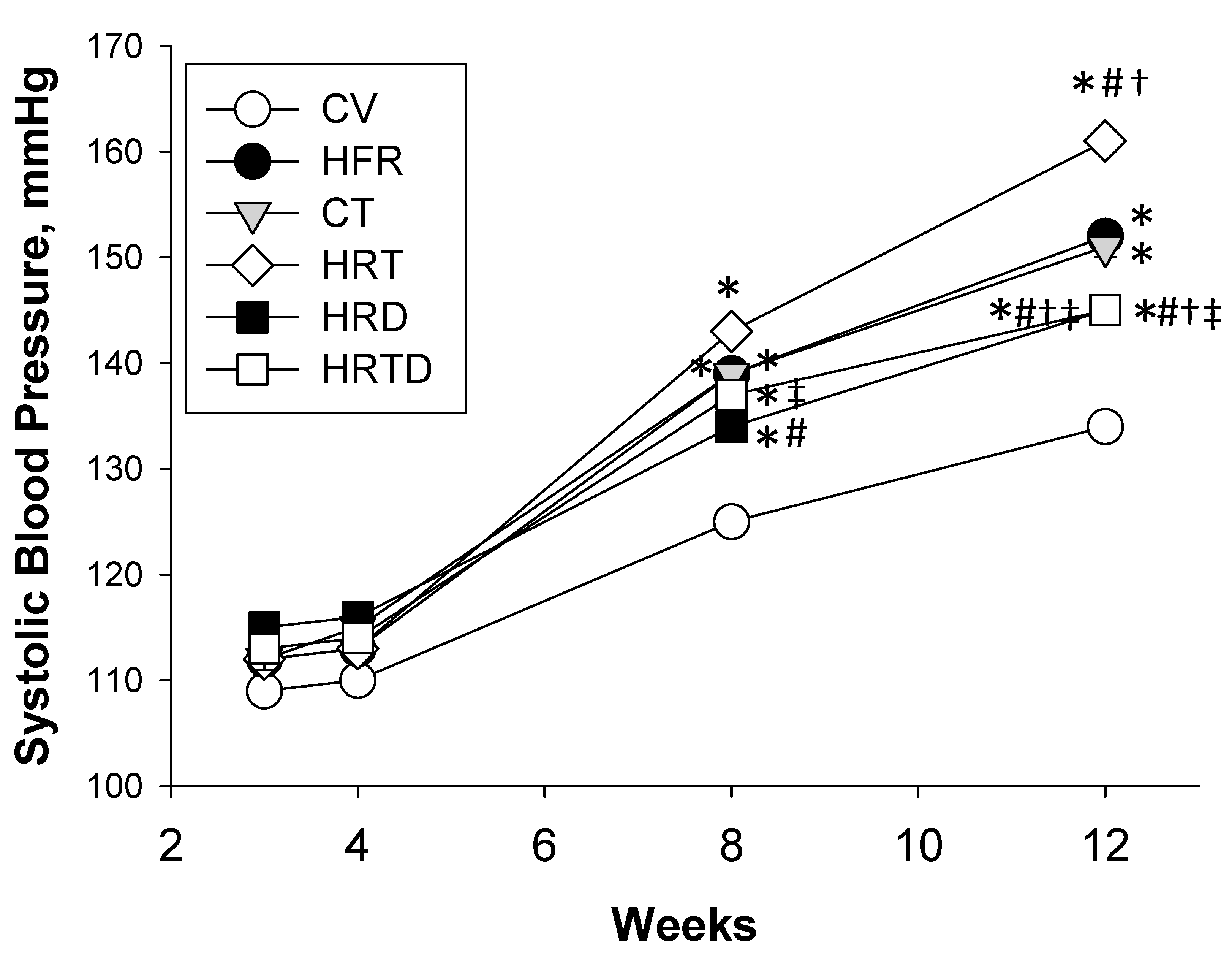
2.1. Blood Pressure and Renal Function
2.2. TMA, TMAO, and DMA
2.3. NO-Related Metabolites
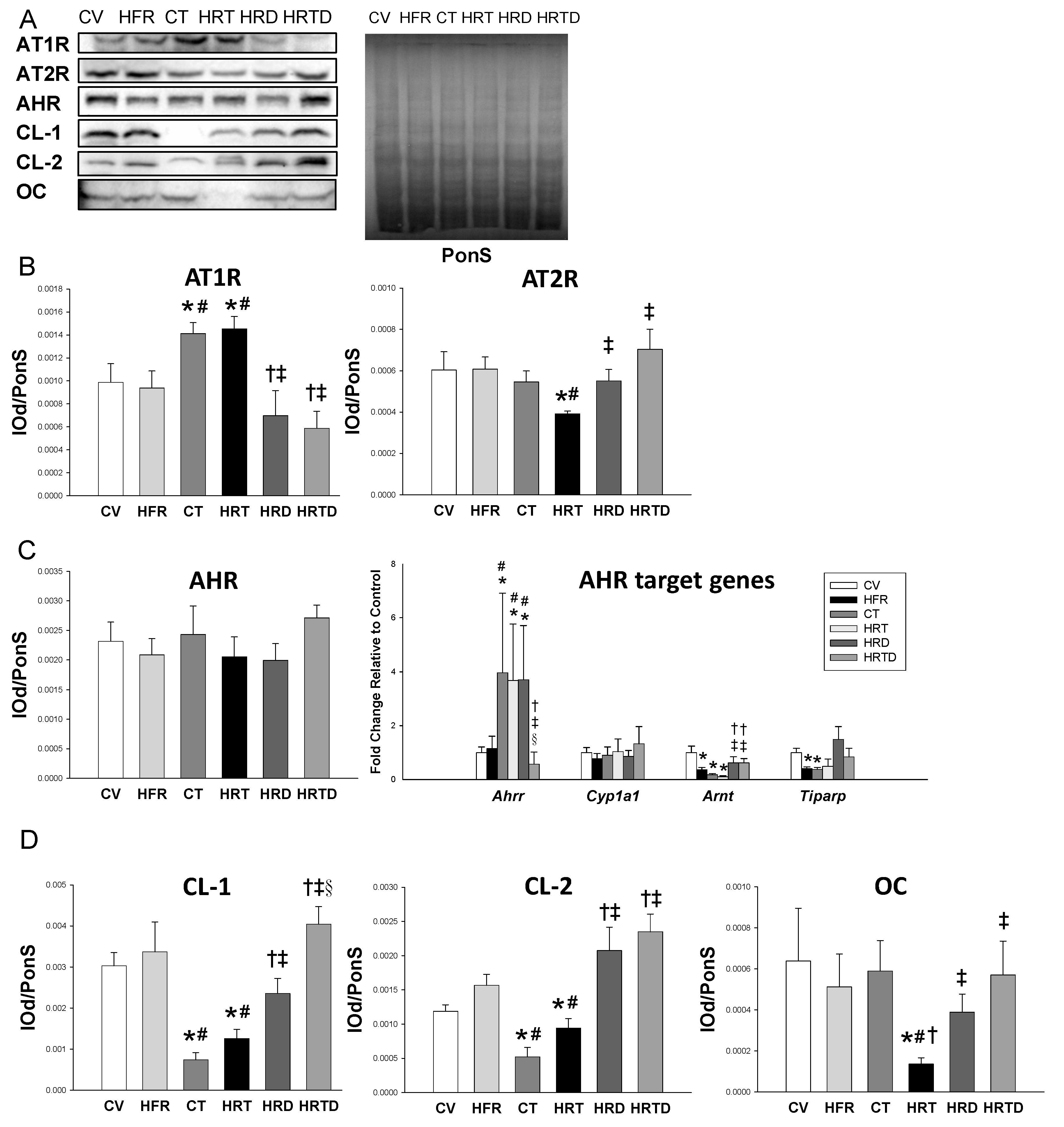
2.4. Renin-Angiotensin System
2.5. AHR Signaling Pathway
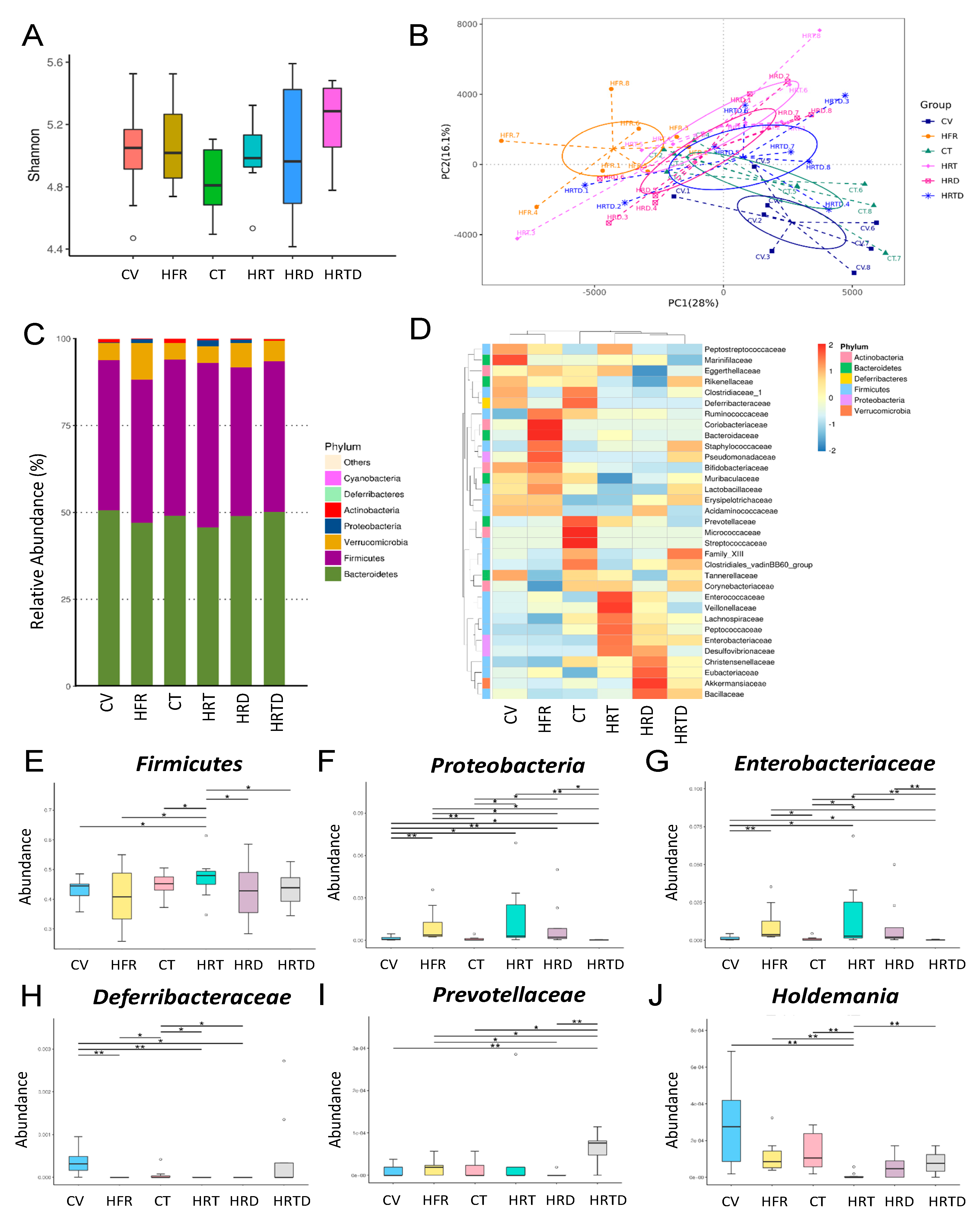
2.6. Gut Permeability and Microbiota Composition
3. Discussion
4. Materials and Methods
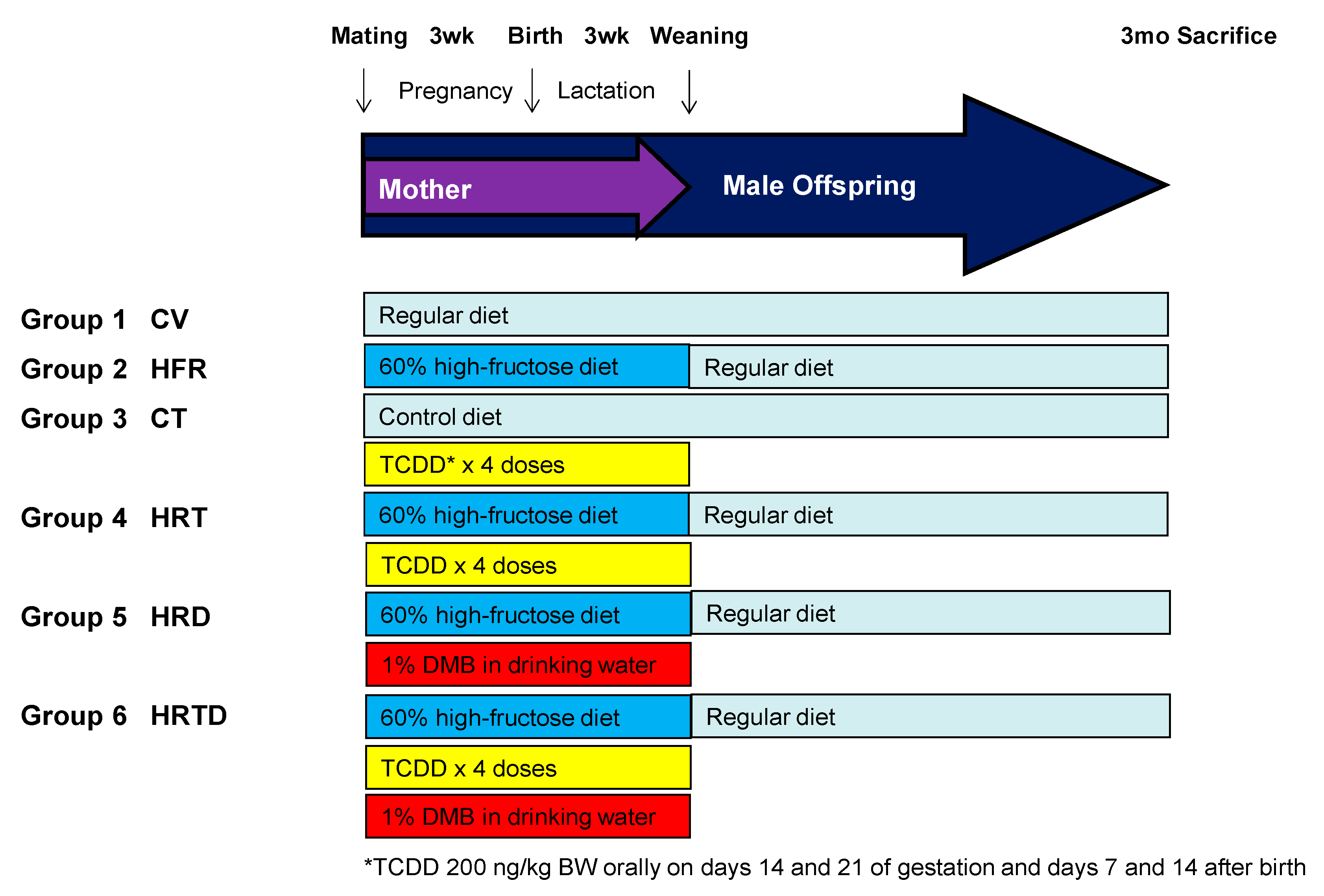
4.1. Animals and Experimental Design
4.2. Liquid Chromatography–Mass Spectrometry (LC–MS/MS) Analysis
4.3. High Performance Liquid Chromatography (HPLC)
4.4. Metagenomics Analysis of Gut Microbiota
4.5. Quantitative Real-Time PCR Analysis
4.6. Western Blot
4.7. Statistics
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADMA | Asymmetric dimethylarginine |
| AHR | Aryl hydrocarbon receptor |
| ANOSIM | Analysis of Similarities |
| AT1R | Angiotensin II type 1 receptor |
| AT2R | Angiotensin II type 2 receptor |
| CL-1 | Claudin-1 |
| CL-2 | Claudin-2 |
| DOHaD | Developmental origins of health and disease |
| DMA | Dimethylamine |
| DMB | 3,3-Dimethyl-1-butanol |
| FMO | Flavin-containing monooxygenase |
| OC | Occludin |
| PCA | Principal component analysis |
| PonS | Ponceau S red |
| RAS | Renin-angiotensin system |
| SD | Sprague–Dawley |
| SDMA | Symmetric dimethylarginine |
| TCDD | 2,3,7,8-tetrachlorodibenzo-p-dioxin |
| TMA | Trimethylamine |
| TMAO | Trimethylamine N-oxide |
References
- Nakagawa, T.; Tuttle, K.R.; Short, R.A.; Johnson, R.J. Hypothesis: Fructose-induced hyperuricemia as a causal mechanism for the epidemic of the metabolic syndrome. Nat. Clin. Pract. Nephrol. 2005, 1, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Tran, L.T.; Yuen, V.G.; McNeill, J.H. The fructose-fed rat: A review on the mechanisms of fructose-induced insulin resistance and hypertension. Mol. Cell Biochem. 2009, 332, 145–159. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.C.; Wu, K.L.H.; Leu, S.; Tain, Y.L. Translational insights on developmental origins of metabolic syndrome: Focus on fructose consumption. Biomed. J. 2018, 41, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Haugen, A.C.; Schug, T.T.; Collman, G.; Heindel, J.J. Evolution of DOHaD: The impact of environmental health sciences. J. Dev. Orig. Health Dis. 2015, 6, 55–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tain, Y.L.; Wu, K.L.; Lee, W.C.; Leu, S.; Chan, J.Y. Maternal fructose-intake-induced renal programming in adult male offspring. J. Nutr. Biochem. 2015, 26, 642–650. [Google Scholar] [CrossRef]
- Park, S.H.; Lim, J.E.; Park, H.; Jee, S.H. Body burden of persistent organic pollutants on hypertension: A meta-analysis. Environ. Sci. Pollut. Res. Int. 2016, 23, 14284–14293. [Google Scholar] [CrossRef]
- Aragon, A.C.; Goens, M.B.; Carbett, E.; Walker, M.K. Perinatal 2,3,7,8-tetrachlorodibenzo-p-dioxin exposure sensitizes offspring to angiotensin II-induced hypertension. Cardiovasc. Toxicol. 2008, 8, 145–154. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.N.; Lin, Y.J.; Lu, P.C.; Tain, Y.L. Maternal Resveratrol Therapy Protects Male Rat Offspring against Programmed Hypertension Induced by TCDD and Dexamethasone Exposures: Is It Relevant to Aryl Hydrocarbon Receptor? Int. J. Mol. Sci. 2018, 19, 2459. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.N.; Tain, Y.L. Regulation of Nitric Oxide Production in the Developmental Programming of Hypertension and Kidney Disease. Int. J. Mol. Sci. 2019, 20, 681. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.; Santisteban, M.M.; Rodriguez, V.; Li, E.; Ahmari, N.; Carvajal, J.M.; Zadeh, M.; Gong, M.; Qi, Y.; Zubcevic, J.; et al. Gut dysbiosis is linked to hypertension. Hypertension 2015, 65, 1331–1340. [Google Scholar] [CrossRef] [Green Version]
- Al Khodor, S.; Reichert, B.; Shatat, I.F. The Microbiome and Blood Pressure: Can Microbes Regulate Our Blood Pressure? Front. Pediatr. 2017, 5, 138. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.N.; Chang-Chien, G.P.; Lin, S.; Hou, C.Y.; Tain, Y.L. Targeting on Gut Microbial Metabolite Trimethylamine-N-Oxide and Short-Chain Fatty Acid to Prevent Maternal High-Fructose-Diet-Induced Developmental Programming of Hypertension in Adult Male Offspring. Mol. Nutr. Food Res. 2019, 63, 1900073. [Google Scholar] [CrossRef] [PubMed]
- Velasquez, M.T.; Ramezani, A.; Manal, A.; Raj, D.S. Trimethylamine N-Oxide: The Good, the Bad and the Unknown. Toxins 2016, 8, 326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, N. The role of endogenous aryl hydrocarbon receptor signaling in cardiovascular physiology. J. Cardiovasc. Dis. Res. 2011, 2, 91–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, C.N.; Lin, Y.J.; Hou, C.Y.; Tain, Y.L. Maternal Administration of Probiotic or Prebiotic Prevents Male Adult Rat Offspring against Developmental Programming of Hypertension Induced by High Fructose Consumption in Pregnancy and Lactation. Nutrients 2018, 10, 1229. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Zheng, X.; Feng, M.; Li, D.; Zhang, H. Gut Microbiota-Dependent Metabolite Trimethylamine N-Oxide Contributes to Cardiac Dysfunction in Western Diet-Induced Obese Mice. Front. Physiol. 2017, 8, 139. [Google Scholar] [CrossRef]
- Wagner, B.D.; Grunwald, G.K.; Zerbe, G.O.; Mikulich-Gilbertson, S.K.; Robertson, C.E.; Zemanick, E.T.; Harris, J.K. On the Use of Diversity Measures in Longitudinal Sequencing Studies of Microbial Communities. Front. Microbiol. 2018, 9, 1037. [Google Scholar] [CrossRef] [Green Version]
- Goodrich, J.K.; Di Rienzi, S.C.; Poole, A.C.; Koren, O.; Walters, W.A.; Caporaso, J.G.; Knight, R.; Ley, R.E. Conducting a microbiome study. Cell 2014, 158, 250–262. [Google Scholar] [CrossRef] [Green Version]
- Morris, E.K.; Caruso, T.; Buscot, F.; Fischer, M.; Hancock, C.; Maier, T.S.; Meiners, T.; Müller, C.; Obermaier, E.; Prati, D.; et al. Choosing and using diversity indices: Insights for ecological applications from the German Biodiversity Exploratories. Ecol. Evol. 2014, 4, 3514–3524. [Google Scholar] [CrossRef] [Green Version]
- Clarke, K.R.; Green, R.H. Statistical design and analysis for a ‘biological effects’ study. Mar. Ecol. Prog. Ser. 1988, 46, 213–226. [Google Scholar] [CrossRef]
- Jaworska, K.; Hering, D.; Mosieniak, G.; Bielak-Zmijewska, A.; Pilz, M.; Konwerski, M.; Gasecka, A.; Kapłon-Cieślicka, A.; Filipiak, K.; Sikora, E.; et al. TMA, A Forgotten Uremic Toxin, but Not TMAO, Is Involved in Cardiovascular Pathology. Toxins 2019, 11, 490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Te Riet, L.; van Esch, J.H.; Roks, A.J.; van den Meiracker, A.H.; Danser, A.H. Hypertension: Renin-angiotensin-aldosterone system alterations. Circ. Res. 2015, 116, 960–975. [Google Scholar] [CrossRef] [PubMed]
- Ufnal, M.; Jazwiec, R.; Dadlez, M.; Drapala, A.; Sikora, M.; Skrzypecki, J. Trimethylamine-N-oxide: A carnitine-derived metabolite that prolongs the hypertensive effect of angiotensin II in rats. Can. J. Cardiol. 2014, 30, 1700–1705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwapiszewska, G.; Johansen, A.K.Z.; Gomez-Arroyo, J.; Voelkel, N.F. Role of the Aryl Hydrocarbon Receptor/ARNT/Cytochrome P450 System in Pulmonary Vascular Diseases. Circ. Res. 2019, 125, 356–366. [Google Scholar] [CrossRef]
- Lindsey, S.; Papoutsakis, E.T. The aryl hydrocarbon receptor (AHR) transcription factor regulates megakaryocytic polyploidization. Br. J. Haematol. 2011, 152, 469–484. [Google Scholar] [CrossRef] [Green Version]
- Rath, S.; Heidrich, B.; Pieper, D.H.; Vital, M. Uncovering the trimethylamine-producing bacteria of the human gut microbiota. Microbiome 2017, 5, 54. [Google Scholar] [CrossRef] [Green Version]
- Hoyles, L.; Jimenez-Pranteda, M.L.; Chilloux, J.; Brial, F.; Myridakis, A.; Aranias, T.; Magnan, C.; Gibson, G.R.; Sanderson, J.D.; Nicholson, J.K.; et al. Metabolic retroconversion of trimethylamine N-oxide and the gut microbiota. Microbiome 2018, 6, 73. [Google Scholar] [CrossRef] [Green Version]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Li, H. The Role of Gut Microbiota in Atherosclerosis and Hypertension. Front. Pharmacol. 2018, 9, 1082. [Google Scholar] [CrossRef]
- Li, J.; Zhao, F.; Wang, Y.; Chen, J.; Tao, J.; Tian, G.; Wu, S.; Liu, W.; Cui, Q.; Geng, B.; et al. Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome 2017, 5, 14. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Roberts, A.B.; Buffa, J.A.; Levison, B.S.; Zhu, W.; Org, E.; Gu, X.; Huang, Y.; Zamanian-Daryoush, M.; Culley, M.K.; et al. Non-lethal Inhibition of Gut Microbial Trimethylamine Production for the Treatment of Atherosclerosis. Cell 2015, 163, 1585–1595. [Google Scholar] [CrossRef] [Green Version]
- Tain, Y.L.; Lee, W.C.; Wu, K.; Leu, S.; Chan, J.Y.H. Maternal high fructose intake increases the vulnerability to post-weaning high-fat diet induced programmed hypertension in male offspring. Nutrients 2018, 10, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faqi, A.S.; Dalsenter, P.R.; Merker, H.J.; Chahoud, I. Reproductive toxicity and tissue concentrations of low doses of 2,3,7,8-tetrachlorodibenzo-p-dioxin in male offspring rats exposed throughout pregnancy and lactation. Toxicol. Appl. Pharmacol. 1998, 150, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Reckelhoff, J.F. Gender differences in the regulation of blood pressure. Hypertension 2001, 37, 1199–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
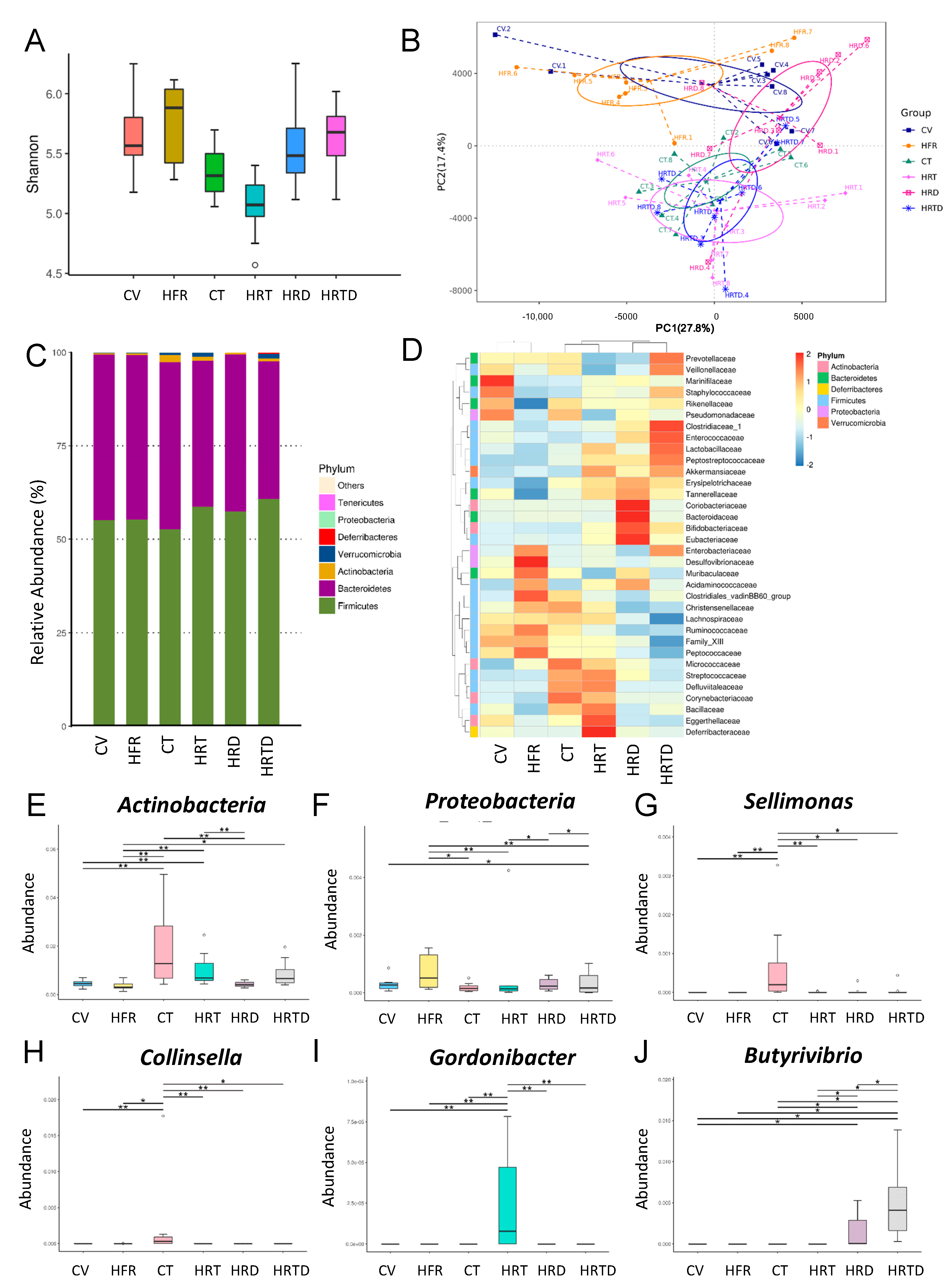{kind=link}
| Groups | CV | HFR | CT | HRT | HRD | HRTD |
|---|---|---|---|---|---|---|
| Mortality | 0% | 0% | 0% | 0% | 0% | 0% |
| Body weight (g) | 380 ± 9 | 366 ± 6 | 383 ± 17 | 390 ± 9 | 404 ± 13 # | 373 ± 7 |
| LKW (g) | 1.68 ± 0.05 | 1.59 ± 0.04 | 1.73 ± 0.08 | 1.79 ± 0.06 # | 1.69 ± 0.03 | 1.65 ± 0.04 |
| LKW/BW (g/g) | 0.44 ± 0.01 | 0.43 ± 0.01 | 0.45 ± 0.01 | 0.46 ± 0.1 | 0.42 ± 0.01 †,‡ | 0.44 ± 0.01 |
| SBP (mmHg) | 134 ± 0 | 152 ± 1 * | 151 ± 1 * | 161 ± 1*,#,† | 145 ± 1 *,#,†,‡ | 145 ± 1 *,#,†,‡ |
| Creatinine (μM) | 18.2 ± 1.9 | 23.3 ± 1.2 * | 30.5 ± 1.1 *,# | 21.2 ± 1.4 #,† | 19.4 ± 1.2 #,† | 22.7 ± 2.4 † |
| Groups | CV | HFR | CT | HRT | HRD | HRTD |
|---|---|---|---|---|---|---|
| Methylamines | ||||||
| TMA (ng/mL) | 174 ± 13 | 209 ± 6 * | 200 ± 12 | 210 ± 13 | 172 ± 15 | 212 ± 17 |
| TMAO (ng/mL) | 252 ± 19 | 234 ± 17 | 244 ± 13 | 219 ± 14 | 262 ± 10 ‡ | 247 ± 24 |
| DMA (ng/mL) | 96 ± 5 | 107 ± 4 | 92 ± 4 # | 88 ± 3 # | 87 ± 3 # | 114 ± 10 ‡,§ |
| NO-related metabolites | ||||||
| Citrulline (μM) | 62.7 ± 4.5 | 80.4 ± 8.4 | 65.3 ± 6 | 61.4 ± 6.7 | 44.8 ± 6.7 # | 50.1 ± 3.8 # |
| Arginine (μM) | 195 ± 14 | 214 ± 22 | 161 ± 16 | 172 ± 16 | 137 ± 13 *,# | 211 ± 19 § |
| ADMA (μM) | 1.5 ± 0.12 | 2.6 ± 0.25 * | 3.1 ± 0.2 * | 2 ± 0.25 † | 1.3 ± 0.21 #,†,‡ | 2.5 ± 0.18*,§ |
| SDMA (μM) | 0.6 ± 0.09 | 1.9 ± 0.26 * | 1.4 ± 0.29 * | 1.4 ± 0.28 * | 0.7 ± 0.17 #,‡ | 2.3 ± 0.45*,§ |
| Arginine-to-ADMA ratio | 137 ± 17 | 85 ± 7 * | 51 ± 5 *,# | 93 ± 14 † | 147 ± 43 | 88 ± 8 *,† |
| Gene | Forward (5′–3′) | Reverse (5′–3′) |
|---|---|---|
| Ahrr | cagcaacatggcttctttca | tgaagcactgcattccagac |
| Cyp1a1 | gcactctggacaaacacctg | atatccaccttctcgcctgg |
| Arnt | gtctccctcccagatgatga | gctggtagccaacagtagcc |
| Tiparp | gttgagggccaattaccaga | gctcctggcacataatccat |
| Rn18s | gccgcggtaattccagctcca | cccgcccgctcccaagatc |
| Antibody | Host | Source | Dilution |
|---|---|---|---|
| AT1R | Rabbit | Millipore | 1:500 |
| AT2R | Rabbit | Santa Cruz Biotechnology | 1:250 |
| AHR | Rabbit | Novus Biologicals | 1:1000 |
| CL-1 | Rabbit | Abcam | 1:500 |
| CL-2 | Rabbit | Thermo Fisher Scientific | 1:500 |
| OC | Rabbit | Thermo Fisher Scientific | 1:500 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, C.-N.; Chan, J.Y.H.; Yu, H.-R.; Lee, W.-C.; Wu, K.L.H.; Chang-Chien, G.-P.; Lin, S.; Hou, C.-Y.; Tain, Y.-L. Targeting on Gut Microbiota-Derived Metabolite Trimethylamine to Protect Adult Male Rat Offspring against Hypertension Programmed by Combined Maternal High-Fructose Intake and Dioxin Exposure. Int. J. Mol. Sci. 2020, 21, 5488. https://doi.org/10.3390/ijms21155488
Hsu C-N, Chan JYH, Yu H-R, Lee W-C, Wu KLH, Chang-Chien G-P, Lin S, Hou C-Y, Tain Y-L. Targeting on Gut Microbiota-Derived Metabolite Trimethylamine to Protect Adult Male Rat Offspring against Hypertension Programmed by Combined Maternal High-Fructose Intake and Dioxin Exposure. International Journal of Molecular Sciences. 2020; 21(15):5488. https://doi.org/10.3390/ijms21155488
Chicago/Turabian StyleHsu, Chien-Ning, Julie Y. H. Chan, Hong-Ren Yu, Wei-Chia Lee, Kay L. H. Wu, Guo-Ping Chang-Chien, Sufan Lin, Chih-Yao Hou, and You-Lin Tain. 2020. "Targeting on Gut Microbiota-Derived Metabolite Trimethylamine to Protect Adult Male Rat Offspring against Hypertension Programmed by Combined Maternal High-Fructose Intake and Dioxin Exposure" International Journal of Molecular Sciences 21, no. 15: 5488. https://doi.org/10.3390/ijms21155488
APA StyleHsu, C. -N., Chan, J. Y. H., Yu, H. -R., Lee, W. -C., Wu, K. L. H., Chang-Chien, G. -P., Lin, S., Hou, C. -Y., & Tain, Y. -L. (2020). Targeting on Gut Microbiota-Derived Metabolite Trimethylamine to Protect Adult Male Rat Offspring against Hypertension Programmed by Combined Maternal High-Fructose Intake and Dioxin Exposure. International Journal of Molecular Sciences, 21(15), 5488. https://doi.org/10.3390/ijms21155488