Control of Innate Immunity by Sialic Acids in the Nervous Tissue


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Brain Innate Immunity
3. Sialic Acids (Sias): Structure, Diversity, Biosynthesis Related Key Enzymes and Sialidases
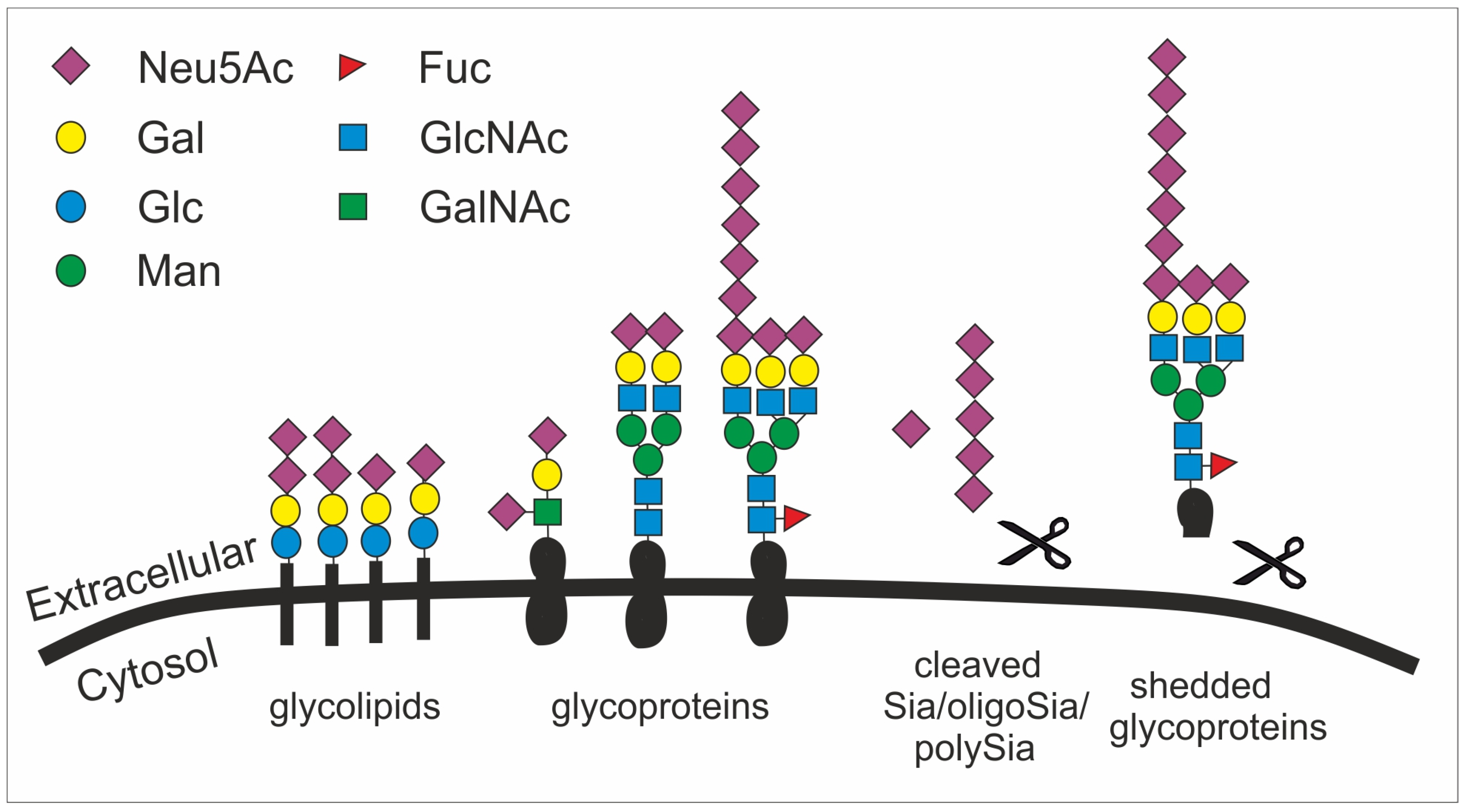
4. Sialic Acid (Sia)-Related Glycoconjugates, Expression Pattern and Function in the Brain
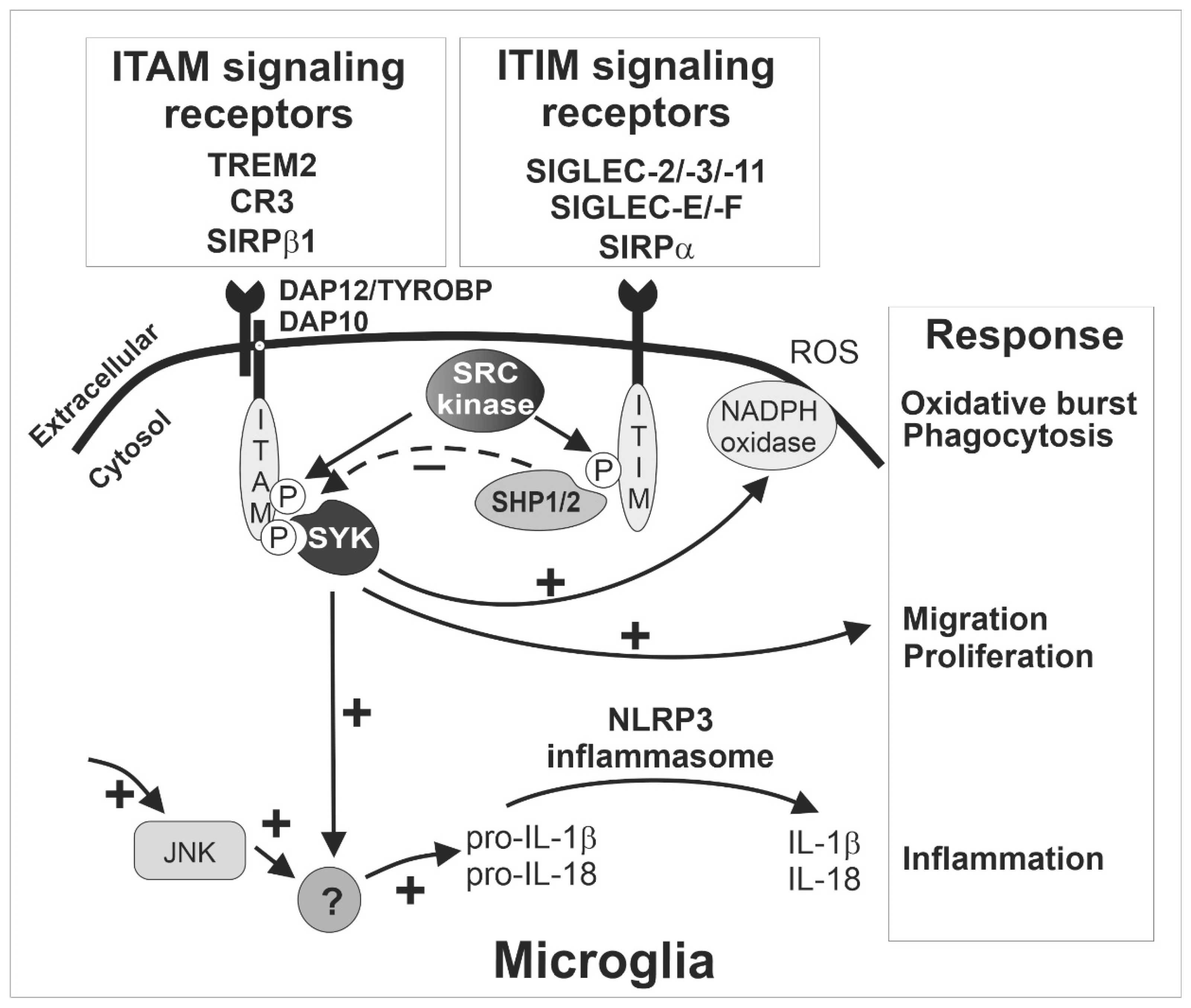
5. Sialic Acid (Sia)-Related Signaling: Complement, SIGLECS and Other Receptors
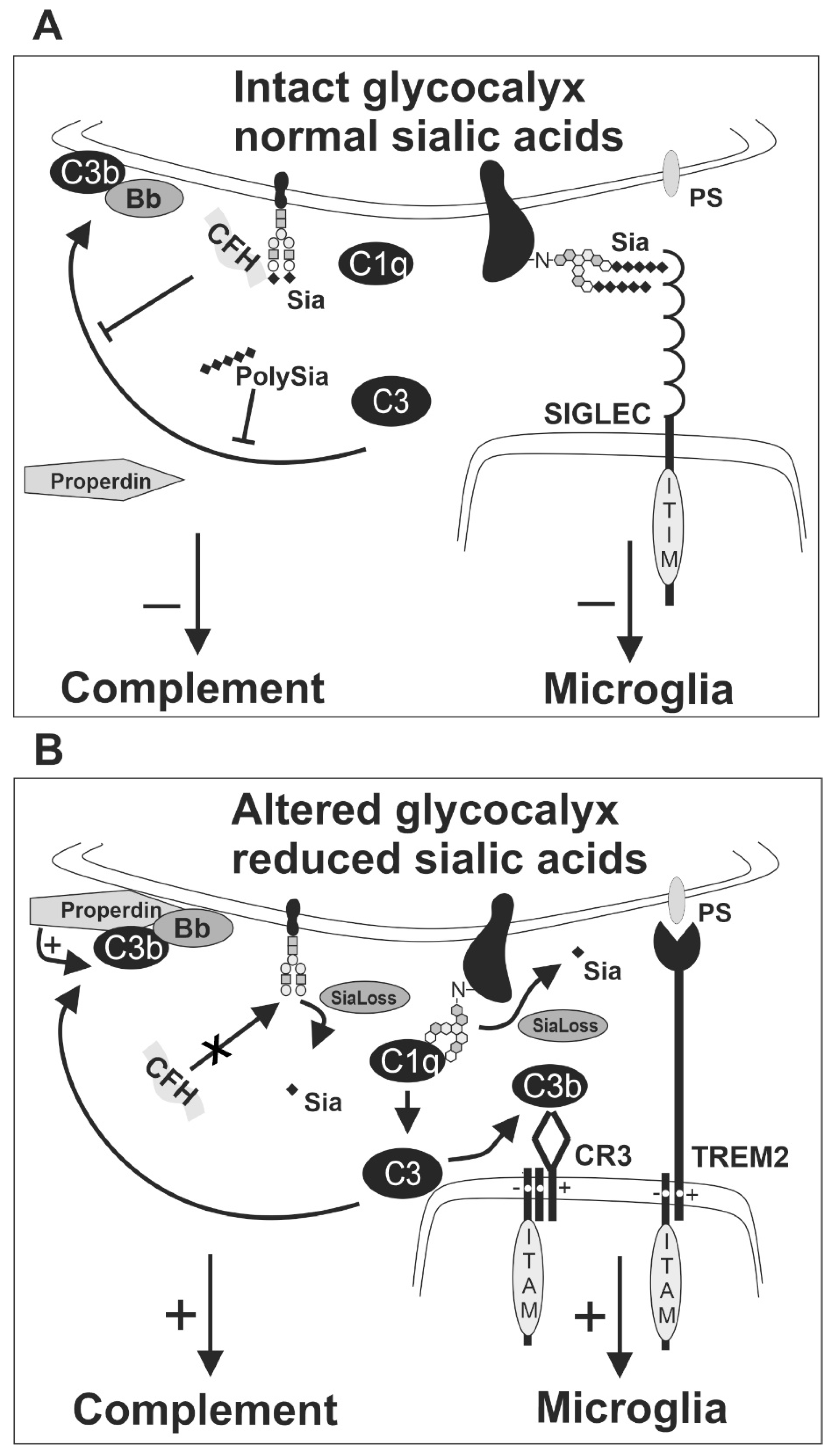
6. Effects of Desialylation on Brain Innate Immunity
7. Sia-Complement Axis and Sia-SIGLEC Axis in Brain Disorders
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| APP | Amyloid precursor protein |
| Bb | A subunit of complement factor B |
| BDNF | Brain-derived neurotrophic factor |
| C1 | Complement component 1 |
| C1q | Complement component 1q |
| C3 | Complement component 3 |
| C3a | Complement component 3a |
| C3b | Complement component 3b |
| CFB | Complement factor B |
| CFH | Complement factor H |
| CFI | Complement factor I |
| CMP | Cytidine 5’-monophospho |
| CNS | Central nervous system |
| CR3 | Complement receptor 3 |
| DAM | Disease-associated microglia |
| E. coli K1 | Escherichia coli K1 |
| Fuc | Fucose |
| Gal | Galactose |
| GalNAc | N-acetylgalactosamine |
| GBS | Group B Streptococcus |
| Glc | Glucose |
| GlcNAc | N-acetylglucosamine |
| GNE | Glucosamine (UDP-N-Acetyl)-2-epimerase/N-acetylmannosamine kinase |
| HSCT/DAP10 | Hematopoietic cell signal transducer |
| IL | Interleukin |
| ITAM | Immunoreceptor tyrosine-based activation motif |
| ITIM | Immuno-receptor tyrosine-based inhibitory motif |
| JNK | C-Jun NH2-terminal protein kinase |
| Kdn | Deaminoneuraminic acid |
| LPS | Lipopolysaccharides |
| MAG | Myelin-associated glycoprotein |
| Man | Mannose |
| ManNAc | N-Acetyl-D-mannosamine |
| ManNAc-6-P | N-Acetyl-mannosamine 6-phosphate |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NALP3/NLRP3 | NACHT, LRR and PYD domains-containing protein 3 |
| NCAM | Neural cell adhesion molecule |
| NEU1 | N-acetyl-alpha-neuramindase 1 |
| NEU2 | N-acetyl-alpha-neuraminidase 2 |
| NEU3 | N-acetyl-alpha-neuraminidase 3 |
| NEU4 | N-acetyl-alpha-neuraminidase 4 |
| Neu5Ac | N-acetylneuraminic acid |
| Neu5Gc | N-glycolylneuraminic acid |
| P | Phosphate |
| polySia avDP20 | Polysialic acid homopolymer with an average degree of polymerization of 20 |
| polySia | Polysialic acid |
| PS | Phosphatidylserine |
| RNA-seq | RNA sequencing |
| ROS | Reactive oxygen species |
| SHP-1 | Src homology region 2 domain-containing phosphatase-1 |
| SHP-2 | Src homology region 2 domain-containing phosphatase-2 |
| Sia | Sialic acid |
| SiaLoss | Desialylation |
| SIGLEC | Sialic acid-binding immunoglobulin-like lectin |
| SIRP | Signal regulartory protein |
| SYK | Spleen tyrosine kinase |
| TLR | Toll-like receptor |
| TREM2 | Triggering Receptor Expressed on Myeloid Cells 2 |
| TYROBP/DAP12 | TYRO protein tyrosine kinase binding protein/DNAX-activating protein of 12 kDa |
References
- Dennis, J.W. Genetic code asymmetry supports diversity through experimentation with posttranslational modifications. Curr. Opin. Chem. Biol. 2017, 41, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Severi, E.; Hood, D.W.; Thomas, G.H. Sialic acid utilization by bacterial pathogens. Microbiology 2007, 153, 2817–2822. [Google Scholar] [CrossRef] [Green Version]
- Varki, A. Since there are PAMPs and DAMPs, there must be SAMPs? Glycan “self-associated molecular patterns” dampen innate immunity, but pathogens can mimic them. Glycobiology 2011, 21, 1121–1124. [Google Scholar] [CrossRef] [PubMed]
- Ricklin, D.; Hajishengallis, G.; Yang, K.; Lambris, J.D. Complement: A key system for immune surveillance and homeostasis. Nat. Immunol. 2010, 11, 785–797. [Google Scholar] [CrossRef] [Green Version]
- Macauley, M.S.; Crocker, P.R.; Paulson, J.C. Siglec-mediated regulation of immune cell function in disease. Nat. Rev. Immunol. 2014, 14, 653–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Q.; Colodner, K.J.; Matousek, S.B.; Merry, K.; Hong, S.; Kenison, J.E.; Frost, J.L.; Le, K.X.; Li, S.; Dodart, J.C.; et al. Complement C3-Deficient Mice Fail to Display Age-Related Hippocampal Decline. J. Neurosci. 2015, 35, 13029–13042. [Google Scholar] [CrossRef] [PubMed]
- Kouser, L.; Abdul-Aziz, M.; Nayak, A.; Stover, C.M.; Sim, R.B.; Kishore, U. Properdin and factor h: Opposing players on the alternative complement pathway “see-saw”. Front. Immunol. 2013, 4, 93. [Google Scholar] [CrossRef] [Green Version]
- Blaum, B.S.; Hannan, J.P.; Herbert, A.P.; Kavanagh, D.; Uhrin, D.; Stehle, T. Structural basis for sialic acid-mediated self-recognition by complement factor H. Nat. Chem. Biol. 2015, 11, 77–82. [Google Scholar] [CrossRef]
- Crocker, P.R.; Paulson, J.C.; Varki, A. Siglecs and their roles in the immune system. Nat. Rev. Immunol. 2007, 7, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chow, R.; Deng, L.; Anderson, D.; Weidner, N.; Godwin, A.K.; Bewtra, C.; Zlotnik, A.; Bui, J.; Varki, A.; et al. Expression of Siglec-11 by human and chimpanzee ovarian stromal cells, with uniquely human ligands: Implications for human ovarian physiology and pathology. Glycobiology 2011, 21, 1038–1048. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.Y.; Tang, J.; Zheng, P.; Liu, Y. CD24 and Siglec-10 selectively repress tissue damage-induced immune responses. Science 2009, 323, 1722–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franceschi, C.; Olivieri, F.; Marchegiani, F.; Cardelli, M.; Cavallone, L.; Capri, M.; Salvioli, S.; Valensin, S.; De Benedictis, G.; Di Iorio, A.; et al. Genes involved in immune response/inflammation, IGF1/insulin pathway and response to oxidative stress play a major role in the genetics of human longevity: The lesson of centenarians. Mech. Ageing Dev. 2005, 126, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, F.; Pearce, O.M.; Wang, X.; Samraj, A.N.; Laubli, H.; Garcia, J.O.; Lin, H.; Fu, X.; Garcia-Bingman, A.; Secrest, P.; et al. Siglec receptors impact mammalian lifespan by modulating oxidative stress. eLife 2015, 4. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [Green Version]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Prinz, M. Microglia Heterogeneity in the Single-Cell Era. Cell Rep. 2020, 30, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Deczkowska, A.; Keren-Shaul, H.; Weiner, A.; Colonna, M.; Schwartz, M.; Amit, I. Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell 2018, 173, 1073–1081. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [Green Version]
- Linnartz, B.; Neumann, H. Microglial activatory (immunoreceptor tyrosine-based activation motif)- and inhibitory (immunoreceptor tyrosine-based inhibition motif)-signaling receptors for recognition of the neuronal glycocalyx. Glia 2013, 61, 37–46. [Google Scholar] [CrossRef]
- Quan, D.N.; Cooper, M.D.; Potter, J.L.; Roberts, M.H.; Cheng, H.; Jarvis, G.A. TREM-2 binds to lipooligosaccharides of Neisseria gonorrhoeae and is expressed on reproductive tract epithelial cells. Mucosal. Immunol. 2008, 1, 229–238. [Google Scholar] [CrossRef] [Green Version]
- Daws, M.R.; Sullam, P.M.; Niemi, E.C.; Chen, T.T.; Tchao, N.K.; Seaman, W.E. Pattern recognition by TREM-2: Binding of anionic ligands. J. Immunol. 2003, 171, 594–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, F.L.; Wang, Y.; Tom, I.; Gonzalez, L.C.; Sheng, M. TREM2 Binds to Apolipoproteins, Including APOE and CLU/APOJ, and Thereby Facilitates Uptake of Amyloid-Beta by Microglia. Neuron 2016, 91, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Lessard, C.B.; Malnik, S.L.; Zhou, Y.; Ladd, T.B.; Cruz, P.E.; Ran, Y.; Mahan, T.E.; Chakrabaty, P.; Holtzman, D.M.; Ulrich, J.D.; et al. High-affinity interactions and signal transduction between Abeta oligomers and TREM2. EMBO Mol. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Konishi, H.; Kiyama, H. Microglial TREM2/DAP12 Signaling: A Double-Edged Sword in Neural Diseases. Front. Cell Neurosci. 2018, 12, 206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 107–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rayaprolu, S.; Mullen, B.; Baker, M.; Lynch, T.; Finger, E.; Seeley, W.W.; Hatanpaa, K.J.; Lomen-Hoerth, C.; Kertesz, A.; Bigio, E.H.; et al. TREM2 in neurodegeneration: Evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson’s disease. Mol. Neurodegener. 2013, 8, 19. [Google Scholar] [CrossRef] [Green Version]
- Borroni, B.; Ferrari, F.; Galimberti, D.; Nacmias, B.; Barone, C.; Bagnoli, S.; Fenoglio, C.; Piaceri, I.; Archetti, S.; Bonvicini, C.; et al. Heterozygous TREM2 mutations in frontotemporal dementia. Neurobiol. Aging 2014, 35, 934.e7–934.e10. [Google Scholar] [CrossRef]
- Zhang, B.; Gaiteri, C.; Bodea, L.G.; Wang, Z.; McElwee, J.; Podtelezhnikov, A.A.; Zhang, C.; Xie, T.; Tran, L.; Dobrin, R.; et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 2013, 153, 707–720. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.; Klaus, C.; Pricop-Jeckstadt, M.; Miller, J.A.; Struebing, F.L. A Microglial Signature Directing Human Aging and Neurodegeneration-Related Gene Networks. Front. Neurosci. 2019, 13, 2. [Google Scholar] [CrossRef]
- Paloneva, J.; Kestila, M.; Wu, J.; Salminen, A.; Bohling, T.; Ruotsalainen, V.; Hakola, P.; Bakker, A.B.; Phillips, J.H.; Pekkarinen, P.; et al. Loss-of-function mutations in TYROBP (DAP12) result in a presenile dementia with bone cysts. Nat. Genet. 2000, 25, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Paloneva, J.; Manninen, T.; Christman, G.; Hovanes, K.; Mandelin, J.; Adolfsson, R.; Bianchin, M.; Bird, T.; Miranda, R.; Salmaggi, A.; et al. Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am. J. Hum. Genet. 2002, 71, 656–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linnartz-Gerlach, B.; Bodea, L.G.; Klaus, C.; Ginolhac, A.; Halder, R.; Sinkkonen, L.; Walter, J.; Colonna, M.; Neumann, H. TREM2 triggers microglial density and age-related neuronal loss. Glia 2019, 67, 539–550. [Google Scholar] [CrossRef] [Green Version]
- Haure-Mirande, J.V.; Audrain, M.; Fanutza, T.; Kim, S.H.; Klein, W.L.; Glabe, C.; Readhead, B.; Dudley, J.T.; Blitzer, R.D.; Wang, M.; et al. Deficiency of TYROBP, an adapter protein for TREM2 and CR3 receptors, is neuroprotective in a mouse model of early Alzheimer’s pathology. Acta Neuropathol. 2017, 134, 769–788. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, M.E.; Lowery, R.L.; Majewska, A.K. Microglial interactions with synapses are modulated by visual experience. PLoS Biol. 2010, 8, e1000527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.D.; Mehalow, A.K.; Huberman, A.D.; Stafford, B.; et al. The classical complement cascade mediates CNS synapse elimination. Cell 2007, 131, 1164–1178. [Google Scholar] [CrossRef] [Green Version]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef] [Green Version]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef] [Green Version]
- Filipello, F.; Morini, R.; Corradini, I.; Zerbi, V.; Canzi, A.; Michalski, B.; Erreni, M.; Markicevic, M.; Starvaggi-Cucuzza, C.; Otero, K.; et al. The Microglial Innate Immune Receptor TREM2 Is Required for Synapse Elimination and Normal Brain Connectivity. Immunity 2018, 48, 979–991.e8. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef] [Green Version]
- Linnartz, B.; Kopatz, J.; Tenner, A.J.; Neumann, H. Sialic acid on the neuronal glycocalyx prevents complement C1 binding and complement receptor-3-mediated removal by microglia. J. Neurosci. 2012, 32, 946–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linnartz-Gerlach, B.; Schuy, C.; Shahraz, A.; Tenner, A.J.; Neumann, H. Sialylation of neurites inhibits complement-mediated macrophage removal in a human macrophage-neuron Co-Culture System. Glia 2016, 64, 35–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varki, A. Are humans prone to autoimmunity? Implications from evolutionary changes in hominin sialic acid biology. J. Autoimmun. 2017, 83, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Inoue, S.; Kitajima, K.; Inoue, Y. Identification of 2-keto-3-deoxy-D-glycero—Galactonononic acid (KDN, deaminoneuraminic acid) residues in mammalian tissues and human lung carcinoma cells. Chemical evidence of the occurrence of KDN glycoconjugates in mammals. J. Biol. Chem. 1996, 271, 24341–24344. [Google Scholar] [CrossRef] [Green Version]
- Hara, S.; Takemori, Y.; Yamaguchi, M.; Nakamura, M.; Ohkura, Y. Fluorometric high-performance liquid chromatography of N-acetyl- and N-glycolylneuraminic acids and its application to their microdetermination in human and animal sera, glycoproteins, and glycolipids. Anal. Biochem. 1987, 164, 138–145. [Google Scholar] [CrossRef]
- Bardor, M.; Nguyen, D.H.; Diaz, S.; Varki, A. Mechanism of uptake and incorporation of the non-human sialic acid N-glycolylneuraminic acid into human cells. J. Biol. Chem. 2005, 280, 4228–4237. [Google Scholar] [CrossRef] [Green Version]
- Tangvoranuntakul, P.; Gagneux, P.; Diaz, S.; Bardor, M.; Varki, N.; Varki, A.; Muchmore, E. Human uptake and incorporation of an immunogenic nonhuman dietary sialic acid. Proc. Natl. Acad. Sci. USA 2003, 100, 12045–12050. [Google Scholar] [CrossRef] [Green Version]
- Angata, T.; Varki, A. Chemical diversity in the sialic acids and related alpha-keto acids: An evolutionary perspective. Chem. Rev. 2002, 102, 439–469. [Google Scholar] [CrossRef]
- Collins, B.E.; Blixt, O.; DeSieno, A.R.; Bovin, N.; Marth, J.D.; Paulson, J.C. Masking of CD22 by cis ligands does not prevent redistribution of CD22 to sites of cell contact. Proc. Natl. Acad. Sci. USA 2004, 101, 6104–6109. [Google Scholar] [CrossRef] [Green Version]
- Keppler, O.T.; Hinderlich, S.; Langner, J.; Schwartz-Albiez, R.; Reutter, W.; Pawlita, M. UDP-GlcNAc 2-epimerase: A regulator of cell surface sialylation. Science 1999, 284, 1372–1376. [Google Scholar] [CrossRef]
- Li, Y.; Chen, X. Sialic acid metabolism and sialyltransferases: Natural functions and applications. Appl. Microbiol. Biotechnol. 2012, 94, 887–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munster-Kuhnel, A.K.; Tiralongo, J.; Krapp, S.; Weinhold, B.; Ritz-Sedlacek, V.; Jacob, U.; Gerardy-Schahn, R. Structure and function of vertebrate CMP-sialic acid synthetases. Glycobiology 2004, 14, 43R–51R. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Varki, A. Advances in the biology and chemistry of sialic acids. ACS Chem. Biol. 2010, 5, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Schnaar, R.L.; Gerardy-Schahn, R.; Hildebrandt, H. Sialic acids in the brain: Gangliosides and polysialic acid in nervous system development, stability, disease, and regeneration. Physiol. Rev. 2014, 94, 461–518. [Google Scholar] [CrossRef] [Green Version]
- Mockl, L.; Horst, A.K.; Kolbe, K.; Lindhorst, T.K.; Brauchle, C. Microdomain Formation Controls Spatiotemporal Dynamics of Cell-Surface Glycoproteins. ChemBioChem 2015, 16, 2023–2028. [Google Scholar] [CrossRef]
- Kelm, S.; Schauer, R.; Manuguerra, J.C.; Gross, H.J.; Crocker, P.R. Modifications of cell surface sialic acids modulate cell adhesion mediated by sialoadhesin and CD22. Glycoconj. J. 1994, 11, 576–585. [Google Scholar] [CrossRef]
- Varki, A.; Gagneux, P. Multifarious roles of sialic acids in immunity. Ann. N. Y. Acad. Sci. 2012, 1253, 16–36. [Google Scholar] [CrossRef] [Green Version]
- Lubbers, J.; Rodriguez, E.; van Kooyk, Y. Modulation of Immune Tolerance via Siglec-Sialic Acid Interactions. Front. Immunol. 2018, 9, 2807. [Google Scholar] [CrossRef] [Green Version]
- Bassaganas, S.; Perez-Garay, M.; Peracaula, R. Cell surface sialic acid modulates extracellular matrix adhesion and migration in pancreatic adenocarcinoma cells. Pancreas 2014, 43, 109–117. [Google Scholar] [CrossRef]
- Kiermaier, E.; Moussion, C.; Veldkamp, C.T.; Gerardy-Schahn, R.; de Vries, I.; Williams, L.G.; Chaffee, G.R.; Phillips, A.J.; Freiberger, F.; Imre, R.; et al. Polysialylation controls dendritic cell trafficking by regulating chemokine recognition. Science 2016, 351, 186–190. [Google Scholar] [CrossRef] [Green Version]
- Sato, C.; Kitajima, K. Sialic Acids in Neurology. Adv. Carbohydr. Chem. Biochem. 2019, 76, 1–64. [Google Scholar] [CrossRef] [PubMed]
- Neu, U.; Bauer, J.; Stehle, T. Viruses and sialic acids: Rules of engagement. Curr. Opin. Struct. Biol. 2011, 21, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Robbins, J.B.; McCracken, G.H., Jr.; Gotschlich, E.C.; Orskov, F.; Orskov, I.; Hanson, L.A. Escherichia coli K1 capsular polysaccharide associated with neonatal meningitis. N. Engl. J. Med. 1974, 290, 1216–1220. [Google Scholar] [CrossRef] [PubMed]
- Troy, F.A., II. The chemistry and biosynthesis of selected bacterial capsular polymers. Annu. Rev. Microbiol. 1979, 33, 519–560. [Google Scholar] [CrossRef] [PubMed]
- Pluschke, G.; Mayden, J.; Achtman, M.; Levine, R.P. Role of the capsule and the O antigen in resistance of O18:K1 Escherichia coli to complement-mediated killing. Infect. Immun. 1983, 42, 907–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahler, C.M.; Martin, L.E.; Shih, G.C.; Rahman, M.M.; Carlson, R.W.; Stephens, D.S. The (alpha2—>8)-linked polysialic acid capsule and lipooligosaccharide structure both contribute to the ability of serogroup B Neisseria meningitidis to resist the bactericidal activity of normal human serum. Infect. Immun. 1998, 66, 5939–5947. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, F.; Landig, C.S.; Siddiqui, S.; Secundino, I.; Olson, J.; Varki, N.; Nizet, V.; Varki, A. Paired Siglec receptors generate opposite inflammatory responses to a human-specific pathogen. EMBO J. 2017, 36, 751–760. [Google Scholar] [CrossRef] [Green Version]
- Wei, M.; Wang, P.G. Desialylation in physiological and pathological processes: New target for diagnostic and therapeutic development. Prog. Mol. Biol. Transl. Sci. 2019, 162, 25–57. [Google Scholar] [CrossRef]
- Miyagi, T.; Yamaguchi, K. Mammalian sialidases: Physiological and pathological roles in cellular functions. Glycobiology 2012, 22, 880–896. [Google Scholar] [CrossRef] [Green Version]
- Pshezhetsky, A.V.; Ashmarina, M. Keeping it trim: Roles of neuraminidases in CNS function. Glycoconj. J. 2018, 35, 375–386. [Google Scholar] [CrossRef] [Green Version]
- Varki, A. Sialic acids in human health and disease. Trends Mol. Med. 2008, 14, 351–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnaar, R.L. Gangliosides of the Vertebrate Nervous System. J. Mol. Biol. 2016, 428, 3325–3336. [Google Scholar] [CrossRef] [Green Version]
- Norton, W.T.; Poduslo, S.E. Myelination in rat brain: Changes in myelin composition during brain maturation. J. Neurochem. 1973, 21, 759–773. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.K.; Macala, L.J.; Taki, T.; Weinfield, H.M.; Yu, F.S. Developmental changes in ganglioside composition and synthesis in embryonic rat brain. J. Neurochem. 1988, 50, 1825–1829. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Wu, Y.P.; Sandhoff, R.; Werth, N.; Mizukami, H.; Ellis, J.M.; Dupree, J.L.; Geyer, R.; Sandhoff, K.; Proia, R.L. Interruption of ganglioside synthesis produces central nervous system degeneration and altered axon-glial interactions. Proc. Natl. Acad. Sci. USA 2005, 102, 2725–2730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magistretti, P.J.; Geisler, F.H.; Schneider, J.S.; Li, P.A.; Fiumelli, H.; Sipione, S. Gangliosides: Treatment Avenues in Neurodegenerative Disease. Front. Neurol. 2019, 10, 859. [Google Scholar] [CrossRef] [Green Version]
- Tettamanti, G.; Bonali, F.; Marchesini, S.; Zambotti, V. A new procedure for the extraction, purification and fractionation of brain gangliosides. Biochim. Biophys. Acta 1973, 296, 160–170. [Google Scholar] [CrossRef]
- Schnaar, R.L. Glycolipid-mediated cell-cell recognition in inflammation and nerve regeneration. Arch. Biochem. Biophys. 2004, 426, 163–172. [Google Scholar] [CrossRef]
- Kracun, I.; Rosner, H.; Drnovsek, V.; Heffer-Lauc, M.; Cosovic, C.; Lauc, G. Human brain gangliosides in development, aging and disease. Int. J. Dev. Biol. 1991, 35, 289–295. [Google Scholar]
- Vajn, K.; Viljetic, B.; Degmecic, I.V.; Schnaar, R.L.; Heffer, M. Differential distribution of major brain gangliosides in the adult mouse central nervous system. PLoS ONE 2013, 8, e75720. [Google Scholar] [CrossRef] [Green Version]
- Saville, J.T.; Thai, H.N.; Lehmann, R.J.; Derrick-Roberts, A.L.; Fuller, M. Subregional brain distribution of simple and complex glycosphingolipids in the mucopolysaccharidosis type I (Hurler syndrome) mouse: Impact of diet. J. Neurochem. 2017, 141, 287–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maguire, T.M.; Breen, K.C. A decrease in neural sialyltransferase activity in Alzheimer’s disease. Dementia 1995, 6, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Cremer, H.; Lange, R.; Christoph, A.; Plomann, M.; Vopper, G.; Roes, J.; Brown, R.; Baldwin, S.; Kraemer, P.; Scheff, S.; et al. Inactivation of the N-CAM gene in mice results in size reduction of the olfactory bulb and deficits in spatial learning. Nature 1994, 367, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Galuska, S.P.; Rollenhagen, M.; Kaup, M.; Eggers, K.; Oltmann-Norden, I.; Schiff, M.; Hartmann, M.; Weinhold, B.; Hildebrandt, H.; Geyer, R.; et al. Synaptic cell adhesion molecule SynCAM 1 is a target for polysialylation in postnatal mouse brain. Proc. Natl. Acad. Sci. USA 2010, 107, 10250–10255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curreli, S.; Arany, Z.; Gerardy-Schahn, R.; Mann, D.; Stamatos, N.M. Polysialylated neuropilin-2 is expressed on the surface of human dendritic cells and modulates dendritic cell-T lymphocyte interactions. J. Biol. Chem. 2007, 282, 30346–30356. [Google Scholar] [CrossRef] [Green Version]
- Sumida, M.; Hane, M.; Yabe, U.; Shimoda, Y.; Pearce, O.M.; Kiso, M.; Miyagi, T.; Sawada, M.; Varki, A.; Kitajima, K.; et al. Rapid Trimming of Cell Surface Polysialic Acid (PolySia) by Exovesicular Sialidase Triggers Release of Preexisting Surface Neurotrophin. J. Biol. Chem. 2015, 290, 13202–13214. [Google Scholar] [CrossRef] [Green Version]
- Werneburg, S.; Buettner, F.F.; Erben, L.; Mathews, M.; Neumann, H.; Muhlenhoff, M.; Hildebrandt, H. Polysialylation and lipopolysaccharide-induced shedding of E-selectin ligand-1 and neuropilin-2 by microglia and THP-1 macrophages. Glia 2016, 64, 1314–1330. [Google Scholar] [CrossRef]
- Angata, K.; Long, J.M.; Bukalo, O.; Lee, W.; Dityatev, A.; Wynshaw-Boris, A.; Schachner, M.; Fukuda, M.; Marth, J.D. Sialyltransferase ST8Sia-II assembles a subset of polysialic acid that directs hippocampal axonal targeting and promotes fear behavior. J. Biol. Chem. 2004, 279, 32603–32613. [Google Scholar] [CrossRef] [Green Version]
- Karlstetter, M.; Kopatz, J.; Aslanidis, A.; Shahraz, A.; Caramoy, A.; Linnartz-Gerlach, B.; Lin, Y.; Luckoff, A.; Fauser, S.; Duker, K.; et al. Polysialic acid blocks mononuclear phagocyte reactivity, inhibits complement activation, and protects from vascular damage in the retina. EMBO Mol. Med. 2017, 9, 154–166. [Google Scholar] [CrossRef]
- Klaus, C.; Hansen, J.N.; Ginolhac, A.; Gerard, D.; Gnanapragassam, V.S.; Horstkorte, R.; Rossdam, C.; Buettner, F.F.R.; Sauter, T.; Sinkkonen, L.; et al. Reduced sialylation triggers homeostatic synapse and neuronal loss in middle-aged mice. Neurobiol. Aging 2020, 88, 91–107. [Google Scholar] [CrossRef]
- Colley, K.J.; Kitajima, K.; Sato, C. Polysialic acid: Biosynthesis, novel functions and applications. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 498–532. [Google Scholar] [CrossRef] [PubMed]
- Sato, C.; Kitajima, K. Disialic, oligosialic and polysialic acids: Distribution, functions and related disease. J. Biochem. 2013, 154, 115–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merle, N.S.; Church, S.E.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part I—Molecular Mechanisms of Activation and Regulation. Front. Immunol. 2015, 6, 262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harboe, M.; Mollnes, T.E. The alternative complement pathway revisited. J. Cell Mol. Med. 2008, 12, 1074–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.Y.; Cortes, C.; Ferreira, V.P. Properdin: A multifaceted molecule involved in inflammation and diseases. Mol. Immunol. 2018, 102, 58–72. [Google Scholar] [CrossRef]
- Hyvarinen, S.; Meri, S.; Jokiranta, T.S. Disturbed sialic acid recognition on endothelial cells and platelets in complement attack causes atypical hemolytic uremic syndrome. Blood 2016, 127, 2701–2710. [Google Scholar] [CrossRef] [Green Version]
- Xue, X.; Wu, J.; Ricklin, D.; Forneris, F.; Di Crescenzio, P.; Schmidt, C.Q.; Granneman, J.; Sharp, T.H.; Lambris, J.D.; Gros, P. Regulator-dependent mechanisms of C3b processing by factor I allow differentiation of immune responses. Nat. Struct. Mol. Biol. 2017, 24, 643–651. [Google Scholar] [CrossRef]
- Abeln, M.; Albers, I.; Peters-Bernard, U.; Flachsig-Schulz, K.; Kats, E.; Kispert, A.; Tomlinson, S.; Gerardy-Schahn, R.; Munster-Kuhnel, A.; Weinhold, B. Sialic acid is a critical fetal defense against maternal complement attack. J. Clin. Investig. 2019, 129, 422–436. [Google Scholar] [CrossRef] [Green Version]
- Fujita, T.; Satomura, A.; Hidaka, M.; Ohsawa, I.; Endo, M.; Ohi, H. Inhibitory effect of free sialic acid on complement activation and its significance in hypocomplementemic glomerulonephritis. J. Clin. Lab. Anal. 1999, 13, 173–179. [Google Scholar] [CrossRef]
- Schmidt, C.Q.; Hipgrave Ederveen, A.L.; Harder, M.J.; Wuhrer, M.; Stehle, T.; Blaum, B.S. Biophysical analysis of sialic acid recognition by the complement regulator Factor H. Glycobiology 2018, 28, 765–773. [Google Scholar] [CrossRef]
- Parente, R.; Clark, S.J.; Inforzato, A.; Day, A.J. Complement factor H in host defense and immune evasion. Cell Mol. Life Sci. 2017, 74, 1605–1624. [Google Scholar] [CrossRef] [PubMed]
- Duan, S.; Paulson, J.C. Siglecs as Immune Cell Checkpoints in Disease. Annu. Rev. Immunol. 2020, 38, 365–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crocker, P.R. Siglecs in innate immunity. Curr. Opin. Pharmacol. 2005, 5, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Linnartz, B.; Wang, Y.; Neumann, H. Microglial immunoreceptor tyrosine-based activation and inhibition motif signaling in neuroinflammation. Int. J. Alzheimers Dis. 2010, 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitzig, F.; Martinez-Barriocanal, A.; Lopez-Botet, M.; Sayos, J. Cloning of two new splice variants of Siglec-10 and mapping of the interaction between Siglec-10 and SHP-1. Biochem. Biophys. Res. Commun. 2002, 296, 355–362. [Google Scholar] [CrossRef]
- Walter, R.B.; Hausermann, P.; Raden, B.W.; Teckchandani, A.M.; Kamikura, D.M.; Bernstein, I.D.; Cooper, J.A. Phosphorylated ITIMs enable ubiquitylation of an inhibitory cell surface receptor. Traffic 2008, 9, 267–279. [Google Scholar] [CrossRef]
- Avril, T.; Freeman, S.D.; Attrill, H.; Clarke, R.G.; Crocker, P.R. Siglec-5 (CD170) can mediate inhibitory signaling in the absence of immunoreceptor tyrosine-based inhibitory motif phosphorylation. J. Biol. Chem. 2005, 280, 19843–19851. [Google Scholar] [CrossRef] [Green Version]
- Avril, T.; Floyd, H.; Lopez, F.; Vivier, E.; Crocker, P.R. The membrane-proximal immunoreceptor tyrosine-based inhibitory motif is critical for the inhibitory signaling mediated by Siglecs-7 and -9, CD33-related Siglecs expressed on human monocytes and NK cells. J. Immunol. 2004, 173, 6841–6849. [Google Scholar] [CrossRef] [Green Version]
- Angata, T.; Kerr, S.C.; Greaves, D.R.; Varki, N.M.; Crocker, P.R.; Varki, A. Cloning and characterization of human Siglec-11. A recently evolved signaling molecule that can interact with SHP-1 and SHP-2 and is expressed by tissue macrophages, including brain microglia. J. Biol. Chem. 2002, 277, 24466–24474. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Maoui, M.; Wu, L.; Banville, D.; Shen, S. mSiglec-E, a novel mouse CD33-related siglec (sialic acid-binding immunoglobulin-like lectin) that recruits Src homology 2 (SH2)-domain-containing protein tyrosine phosphatases SHP-1 and SHP-2. Biochem. J. 2001, 353, 483–492. [Google Scholar] [CrossRef]
- Chen, G.Y.; Brown, N.K.; Zheng, P.; Liu, Y. Siglec-G/10 in self-nonself discrimination of innate and adaptive immunity. Glycobiology 2014, 24, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Mazharian, A.; Mori, J.; Wang, Y.J.; Heising, S.; Neel, B.G.; Watson, S.P.; Senis, Y.A. Megakaryocyte-specific deletion of the protein-tyrosine phosphatases Shp1 and Shp2 causes abnormal megakaryocyte development, platelet production, and function. Blood 2013, 121, 4205–4220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wishcamper, C.A.; Coffin, J.D.; Lurie, D.I. Lack of the protein tyrosine phosphatase SHP-1 results in decreased numbers of glia within the motheaten (me/me) mouse brain. J. Comp. Neurol. 2001, 441, 118–133. [Google Scholar] [CrossRef]
- Ehrman, L.A.; Nardini, D.; Ehrman, S.; Rizvi, T.A.; Gulick, J.; Krenz, M.; Dasgupta, B.; Robbins, J.; Ratner, N.; Nakafuku, M.; et al. The protein tyrosine phosphatase Shp2 is required for the generation of oligodendrocyte progenitor cells and myelination in the mouse telencephalon. J. Neurosci. 2014, 34, 3767–3778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Brooks, D.M.; Lurie, D.I. Lipopolysaccharide-activated SHP-1-deficient motheaten microglia release increased nitric oxide, TNF-alpha, and IL-1beta. Glia 2006, 53, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Lurie, D.I. Loss of SHP-1 phosphatase alters cytokine expression in the mouse hindbrain following cochlear ablation. Cytokine 2004, 28, 1–9. [Google Scholar] [CrossRef]
- Lyons, B.L.; Smith, R.S.; Hurd, R.E.; Hawes, N.L.; Burzenski, L.M.; Nusinowitz, S.; Hasham, M.G.; Chang, B.; Shultz, L.D. Deficiency of SHP-1 protein-tyrosine phosphatase in “viable motheaten” mice results in retinal degeneration. Invest. Ophthalmol. Vis. Sci. 2006, 47, 1201–1209. [Google Scholar] [CrossRef]
- Zhu, Y.; Shen, J.; Sun, T.; Jiang, H.; Xu, K.; Samuthrat, T.; Xie, Y.; Weng, Y.; Li, Y.; Xie, Q.; et al. Loss of Shp2 within radial glia is associated with cerebral cortical dysplasia, glial defects of cerebellum and impaired sensorymotor development in newborn mice. Mol. Med. Rep. 2018, 17, 3170–3177. [Google Scholar] [CrossRef]
- Rapoport, E.M.; Sapot’ko, Y.B.; Pazynina, G.V.; Bojenko, V.K.; Bovin, N.V. Sialoside-binding macrophage lectins in phagocytosis of apoptotic bodies. Biochemistry 2005, 70, 330–338. [Google Scholar] [CrossRef]
- Wang, Y.; Neumann, H. Alleviation of neurotoxicity by microglial human Siglec-11. J. Neurosci. 2010, 30, 3482–3488. [Google Scholar] [CrossRef] [Green Version]
- Claude, J.; Linnartz-Gerlach, B.; Kudin, A.P.; Kunz, W.S.; Neumann, H. Microglial CD33-related Siglec-E inhibits neurotoxicity by preventing the phagocytosis-associated oxidative burst. J. Neurosci. 2013, 33, 18270–18276. [Google Scholar] [CrossRef] [PubMed]
- Griciuc, A.; Serrano-Pozo, A.; Parrado, A.R.; Lesinski, A.N.; Asselin, C.N.; Mullin, K.; Hooli, B.; Choi, S.H.; Hyman, B.T.; Tanzi, R.E. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 2013, 78, 631–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pluvinage, J.V.; Haney, M.S.; Smith, B.A.H.; Sun, J.; Iram, T.; Bonanno, L.; Li, L.; Lee, D.P.; Morgens, D.W.; Yang, A.C.; et al. CD22 blockade restores homeostatic microglial phagocytosis in ageing brains. Nature 2019, 568, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Hara, H.; Tsuchiya, K.; Kawamura, I.; Fang, R.; Hernandez-Cuellar, E.; Shen, Y.; Mizuguchi, J.; Schweighoffer, E.; Tybulewicz, V.; Mitsuyama, M. Phosphorylation of the adaptor ASC acts as a molecular switch that controls the formation of speck-like aggregates and inflammasome activity. Nat. Immunol. 2013, 14, 1247–1255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, K.; Ruland, J. Kinases conquer the inflammasomes. Nat. Immunol. 2013, 14, 1207–1208. [Google Scholar] [CrossRef] [PubMed]
- Prochnicki, T.; Latz, E. Inflammasomes on the Crossroads of Innate Immune Recognition and Metabolic Control. Cell Metab. 2017, 26, 71–93. [Google Scholar] [CrossRef] [Green Version]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef]
- Tsai, C.M.; Riestra, A.M.; Ali, S.R.; Fong, J.J.; Liu, J.Z.; Hughes, G.; Varki, A.; Nizet, V. Siglec-14 Enhances NLRP3-Inflammasome Activation in Macrophages. J. Innate. Immun. 2019, 333–343. [Google Scholar] [CrossRef]
- Fong, J.J.; Tsai, C.M.; Saha, S.; Nizet, V.; Varki, A.; Bui, J.D. Siglec-7 engagement by GBS beta-protein suppresses pyroptotic cell death of natural killer cells. Proc. Natl. Acad. Sci. USA 2018, 115, 10410–10415. [Google Scholar] [CrossRef] [Green Version]
- Demina, E.P.; Pierre, W.C.; Nguyen, A.L.A.; Londono, I.; Reiz, B.; Zou, C.; Chakraberty, R.; Cairo, C.W.; Pshezhetsky, A.V.; Lodygensky, G.A. Persistent reduction in sialylation of cerebral glycoproteins following postnatal inflammatory exposure. J. Neuroinflam. 2018, 15, 336. [Google Scholar] [CrossRef] [Green Version]
- Goswami, K.; Koner, B.C. Level of sialic acid residues in platelet proteins in diabetes, aging, and Hodgkin’s lymphoma: A potential role of free radicals in desialylation. Biochem. Biophys. Res. Commun. 2002, 297, 502–505. [Google Scholar] [CrossRef]
- Granados-Duran, P.; Lopez-Avalos, M.D.; Hughes, T.R.; Johnson, K.; Morgan, B.P.; Tamburini, P.P.; Fernandez-Llebrez, P.; Grondona, J.M. Complement system activation contributes to the ependymal damage induced by microbial neuraminidase. J. Neuroinflam. 2016, 13, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allendorf, D.H.; Puigdellivol, M.; Brown, G.C. Activated microglia desialylate their surface, stimulating complement receptor 3-mediated phagocytosis of neurons. Glia 2020, 68, 989–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradshaw, E.M.; Chibnik, L.B.; Keenan, B.T.; Ottoboni, L.; Raj, T.; Tang, A.; Rosenkrantz, L.L.; Imboywa, S.; Lee, M.; Von Korff, A.; et al. CD33 Alzheimer’s disease locus: Altered monocyte function and amyloid biology. Nat. Neurosci. 2013, 16, 848–850. [Google Scholar] [CrossRef] [PubMed]
- Wielgat, P.; Holownia, A.; Braszko, J.J. Lipopolysaccharide changes sialylation pattern in the mouse central nervous system. J. Physiol. Pharmacol. 2012, 63, 555–561. [Google Scholar] [PubMed]
- Amith, S.R.; Jayanth, P.; Franchuk, S.; Finlay, T.; Seyrantepe, V.; Beyaert, R.; Pshezhetsky, A.V.; Szewczuk, M.R. Neu1 desialylation of sialyl alpha-2,3-linked beta-galactosyl residues of TOLL-like receptor 4 is essential for receptor activation and cellular signaling. Cell. Signal. 2010, 22, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Kappagantula, S.; Andrews, M.R.; Cheah, M.; Abad-Rodriguez, J.; Dotti, C.G.; Fawcett, J.W. Neu3 sialidase-mediated ganglioside conversion is necessary for axon regeneration and is blocked in CNS axons. J. Neurosci. 2014, 34, 2477–2492. [Google Scholar] [CrossRef] [Green Version]
- Mountney, A.; Zahner, M.R.; Lorenzini, I.; Oudega, M.; Schramm, L.P.; Schnaar, R.L. Sialidase enhances recovery from spinal cord contusion injury. Proc. Natl. Acad. Sci. USA 2010, 107, 11561–11566. [Google Scholar] [CrossRef] [Green Version]
- Nomura, K.; Vilalta, A.; Allendorf, D.H.; Hornik, T.C.; Brown, G.C. Activated Microglia Desialylate and Phagocytose Cells via Neuraminidase, Galectin-3, and Mer Tyrosine Kinase. J. Immunol. 2017, 198, 4792–4801. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.; De Aragao, C.B.P.; Velasco-Martin, J.P.; Priestman, D.A.; Wu, H.Y.; Takahashi, K.; Yamaguchi, K.; Sturiale, L.; Garozzo, D.; Platt, F.M.; et al. Neuraminidases 3 and 4 regulate neuronal function by catabolizing brain gangliosides. FASEB J. 2017, 31, 3467–3483. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.Y.; Morreau, H.; Rottier, R.; Davis, D.; Bonten, E.; Gillemans, N.; Wenger, D.; Grosveld, F.G.; Doherty, P.; Suzuki, K.; et al. Mouse model for the lysosomal disorder galactosialidosis and correction of the phenotype with overexpressing erythroid precursor cells. Genes Dev. 1995, 9, 2623–2634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annunziata, I.; Patterson, A.; Helton, D.; Hu, H.; Moshiach, S.; Gomero, E.; Nixon, R.; d’Azzo, A. Lysosomal NEU1 deficiency affects amyloid precursor protein levels and amyloid-beta secretion via deregulated lysosomal exocytosis. Nat. Commun. 2013, 4, 2734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, C.; Hulkova, H.; Dridi, L.; Dormoy-Raclet, V.; Grigoryeva, L.; Choi, Y.; Langford-Smith, A.; Wilkinson, F.L.; Ohmi, K.; DiCristo, G.; et al. Neuroinflammation, mitochondrial defects and neurodegeneration in mucopolysaccharidosis III type C mouse model. Brain 2015, 138, 336–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohmi, K.; Kudo, L.C.; Ryazantsev, S.; Zhao, H.Z.; Karsten, S.L.; Neufeld, E.F. Sanfilippo syndrome type B, a lysosomal storage disease, is also a tauopathy. Proc. Natl. Acad. Sci. USA 2009, 106, 8332–8337. [Google Scholar] [CrossRef] [Green Version]
- Schmid, J.A.; Mach, L.; Paschke, E.; Glossl, J. Accumulation of sialic acid in endocytic compartments interferes with the formation of mature lysosomes. Impaired proteolytic processing of cathepsin B in fibroblasts of patients with lysosomal sialic acid storage disease. J. Biol. Chem. 1999, 274, 19063–19071. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, K.; Khatua, B.; Mandal, C. Sialic Acid-Siglec-E Interactions during Pseudomonas aeruginosa Infection of Macrophages Interferes with Phagosome Maturation by Altering Intracellular Calcium Concentrations. Front. Immunol. 2020, 11, 332. [Google Scholar] [CrossRef]
- Jackson, J.; Jambrina, E.; Li, J.; Marston, H.; Menzies, F.; Phillips, K.; Gilmour, G. Targeting the Synapse in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 735. [Google Scholar] [CrossRef] [Green Version]
- Schafer, D.P.; Stevens, B. Synapse elimination during development and disease: Immune molecules take centre stage. Biochem. Soc. Trans. 2010, 38, 476–481. [Google Scholar] [CrossRef] [Green Version]
- Rajendran, L.; Paolicelli, R.C. Microglia-Mediated Synapse Loss in Alzheimer’s Disease. J. Neurosci. 2018, 38, 2911–2919. [Google Scholar] [CrossRef] [Green Version]
- Fricker, M.; Tolkovsky, A.M.; Borutaite, V.; Coleman, M.; Brown, G.C. Neuronal Cell Death. Physiol. Rev. 2018, 98, 813–880. [Google Scholar] [CrossRef]
- Schwarzkopf, M.; Knobeloch, K.P.; Rohde, E.; Hinderlich, S.; Wiechens, N.; Lucka, L.; Horak, I.; Reutter, W.; Horstkorte, R. Sialylation is essential for early development in mice. Proc. Natl. Acad. Sci. USA 2002, 99, 5267–5270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddiqui, S.S.; Matar, R.; Merheb, M.; Hodeify, R.; Vazhappilly, C.G.; Marton, J.; Shamsuddin, S.A.; Al Zouabi, H. Siglecs in Brain Function and Neurological Disorders. Cells 2019, 8, 1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wielgat, P.; Braszko, J.J. The participation of sialic acids in microglia-neuron interactions. Cell. Immunol. 2012, 273, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Bornhofft, K.F.; Goldammer, T.; Rebl, A.; Galuska, S.P. Siglecs: A journey through the evolution of sialic acid-binding immunoglobulin-type lectins. Dev. Comp. Immunol. 2018, 86, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Malik, M.; Simpson, J.F.; Parikh, I.; Wilfred, B.R.; Fardo, D.W.; Nelson, P.T.; Estus, S. CD33 Alzheimer’s risk-altering polymorphism, CD33 expression, and exon 2 splicing. J. Neurosci. 2013, 33, 13320–13325. [Google Scholar] [CrossRef] [Green Version]
- Bertram, L.; Lange, C.; Mullin, K.; Parkinson, M.; Hsiao, M.; Hogan, M.F.; Schjeide, B.M.; Hooli, B.; Divito, J.; Ionita, I.; et al. Genome-wide association analysis reveals putative Alzheimer’s disease susceptibility loci in addition to APOE. Am. J. Hum. Genet. 2008, 83, 623–632. [Google Scholar] [CrossRef] [Green Version]
- Hollingworth, P.; Harold, D.; Sims, R.; Gerrish, A.; Lambert, J.C.; Carrasquillo, M.M.; Abraham, R.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet. 2011, 43, 429–435. [Google Scholar] [CrossRef] [Green Version]
- Naj, A.C.; Jun, G.; Beecham, G.W.; Wang, L.S.; Vardarajan, B.N.; Buros, J.; Gallins, P.J.; Buxbaum, J.D.; Jarvik, G.P.; Crane, P.K.; et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat. Genet. 2011, 43, 436–441. [Google Scholar] [CrossRef] [Green Version]
- Walker, D.G.; Whetzel, A.M.; Serrano, G.; Sue, L.I.; Beach, T.G.; Lue, L.F. Association of CD33 polymorphism rs3865444 with Alzheimer’s disease pathology and CD33 expression in human cerebral cortex. Neurobiol. Aging 2015, 36, 571–582. [Google Scholar] [CrossRef] [Green Version]
- Estus, S.; Shaw, B.C.; Devanney, N.; Katsumata, Y.; Press, E.E.; Fardo, D.W. Evaluation of CD33 as a genetic risk factor for Alzheimer’s disease. Acta Neuropathol. 2019, 138, 187–199. [Google Scholar] [CrossRef]
- Raj, T.; Ryan, K.J.; Replogle, J.M.; Chibnik, L.B.; Rosenkrantz, L.; Tang, A.; Rothamel, K.; Stranger, B.E.; Bennett, D.A.; Evans, D.A.; et al. CD33: Increased inclusion of exon 2 implicates the Ig V-set domain in Alzheimer’s disease susceptibility. Hum. Mol. Genet. 2014, 23, 2729–2736. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, S.S.; Springer, S.A.; Verhagen, A.; Sundaramurthy, V.; Alisson-Silva, F.; Jiang, W.; Ghosh, P.; Varki, A. The Alzheimer’s disease-protective CD33 splice variant mediates adaptive loss of function via diversion to an intracellular pool. J. Biol. Chem. 2017, 292, 15312–15320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, B.A.; Srinivasan, K.; Ayalon, G.; Meilandt, W.J.; Lin, H.; Huntley, M.A.; Cao, Y.; Lee, S.H.; Haddick, P.C.G.; Ngu, H.; et al. Diverse Brain Myeloid Expression Profiles Reveal Distinct Microglial Activation States and Aspects of Alzheimer’s Disease Not Evident in Mouse Models. Cell Rep. 2018, 22, 832–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funikov, S.Y.; Rezvykh, A.P.; Mazin, P.V.; Morozov, A.V.; Maltsev, A.V.; Chicheva, M.M.; Vikhareva, E.A.; Evgen’ev, M.B.; Ustyugov, A.A. FUS(1-359) transgenic mice as a model of ALS: Pathophysiological and molecular aspects of the proteinopathy. Neurogenetics 2018, 19, 189–204. [Google Scholar] [CrossRef]
- Cougnoux, A.; Drummond, R.A.; Collar, A.L.; Iben, J.R.; Salman, A.; Westgarth, H.; Wassif, C.A.; Cawley, N.X.; Farhat, N.Y.; Ozato, K.; et al. Microglia activation in Niemann-Pick disease, type C1 is amendable to therapeutic intervention. Hum. Mol. Genet. 2018, 27, 2076–2089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahraz, A.; Kopatz, J.; Mathy, R.; Kappler, J.; Winter, D.; Kapoor, S.; Schutza, V.; Scheper, T.; Gieselmann, V.; Neumann, H. Anti-inflammatory activity of low molecular weight polysialic acid on human macrophages. Sci. Rep. 2015, 5, 16800. [Google Scholar] [CrossRef] [Green Version]
- Brinkman-Van der Linden, E.C.; Angata, T.; Reynolds, S.A.; Powell, L.D.; Hedrick, S.M.; Varki, A. CD33/Siglec-3 binding specificity, expression pattern, and consequences of gene deletion in mice. Mol. Cell Biol. 2003, 23, 4199–4206. [Google Scholar] [CrossRef] [Green Version]
- Bhattacherjee, A.; Rodrigues, E.; Jung, J.; Luzentales-Simpson, M.; Enterina, J.R.; Galleguillos, D.; St Laurent, C.D.; Nakhaei-Nejad, M.; Fuchsberger, F.F.; Streith, L.; et al. Repression of phagocytosis by human CD33 is not conserved with mouse CD33. Commun. Biol. 2019, 2, 450. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, A.C.; Weis, J.H. Comparative functional evolution of human and mouse CR1 and CR2. J. Immunol. 2008, 181, 2953–2959. [Google Scholar] [CrossRef]
- Pouw, R.B.; Vredevoogd, D.W.; Kuijpers, T.W.; Wouters, D. Of mice and men: The factor H protein family and complement regulation. Mol. Immunol. 2015, 67, 12–20. [Google Scholar] [CrossRef]
- Cserhalmi, M.; Papp, A.; Brandus, B.; Uzonyi, B.; Jozsi, M. Regulation of regulators: Role of the complement factor H-related proteins. Semin. Immunol. 2019, 45, 101341. [Google Scholar] [CrossRef] [PubMed]
- Altman, M.O.; Gagneux, P. Absence of Neu5Gc and Presence of Anti-Neu5Gc Antibodies in Humans—An Evolutionary Perspective. Front. Immunol. 2019, 10, 789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perota, A.; Galli, C. N-Glycolylneuraminic Acid (Neu5Gc) Null Large Animals by Targeting the CMP-Neu5Gc Hydroxylase (CMAH). Front. Immunol. 2019, 10, 2396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, L.R.; Pearce, O.M.; Tessier, M.B.; Assar, S.; Smutova, V.; Pajunen, M.; Sumida, M.; Sato, C.; Kitajima, K.; Finne, J.; et al. Metabolism of vertebrate amino sugars with N-glycolyl groups: Resistance of alpha2-8-linked N-glycolylneuraminic acid to enzymatic cleavage. J. Biol. Chem. 2012, 287, 28917–28931. [Google Scholar] [CrossRef] [Green Version]
- Banda, K.; Gregg, C.J.; Chow, R.; Varki, N.M.; Varki, A. Metabolism of vertebrate amino sugars with N-glycolyl groups: Mechanisms underlying gastrointestinal incorporation of the non-human sialic acid xeno-autoantigen N-glycolylneuraminic acid. J. Biol. Chem. 2012, 287, 28852–28864. [Google Scholar] [CrossRef] [Green Version]
- Casadesus, A.V.; Fernandez-Marrero, Y.; Clavell, M.; Gomez, J.A.; Hernandez, T.; Moreno, E.; Lopez-Requena, A. A shift from N-glycolyl- to N-acetyl-sialic acid in the GM3 ganglioside impairs tumor development in mouse lymphocytic leukemia cells. Glycoconj. J. 2013, 30, 687–699. [Google Scholar] [CrossRef]
- Dhar, C.; Sasmal, A.; Varki, A. From “Serum Sickness” to “Xenosialitis”: Past, Present, and Future Significance of the Non-human Sialic Acid Neu5Gc. Front. Immunol. 2019, 10, 807. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liao, H.; Klaus, C.; Neumann, H. Control of Innate Immunity by Sialic Acids in the Nervous Tissue. Int. J. Mol. Sci. 2020, 21, 5494. https://doi.org/10.3390/ijms21155494
Liao H, Klaus C, Neumann H. Control of Innate Immunity by Sialic Acids in the Nervous Tissue. International Journal of Molecular Sciences. 2020; 21(15):5494. https://doi.org/10.3390/ijms21155494
Chicago/Turabian StyleLiao, Huan, Christine Klaus, and Harald Neumann. 2020. "Control of Innate Immunity by Sialic Acids in the Nervous Tissue" International Journal of Molecular Sciences 21, no. 15: 5494. https://doi.org/10.3390/ijms21155494
APA StyleLiao, H., Klaus, C., & Neumann, H. (2020). Control of Innate Immunity by Sialic Acids in the Nervous Tissue. International Journal of Molecular Sciences, 21(15), 5494. https://doi.org/10.3390/ijms21155494