Physicochemical Evidence that Francisella FupA and FupB Proteins Are Porins

 , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
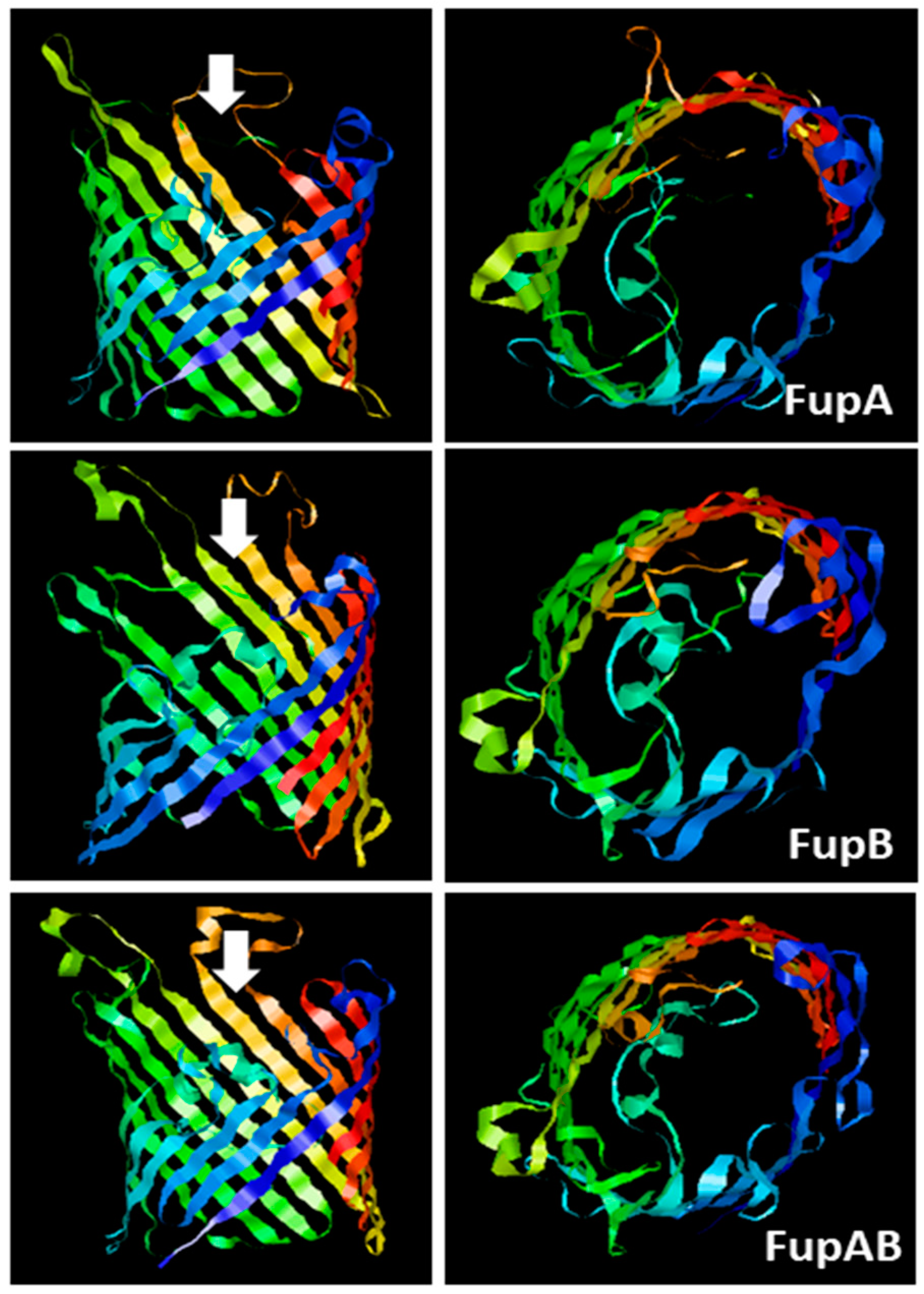
2.1. Structural Modelling of Fup Proteins Predicted Their Folding as Porins
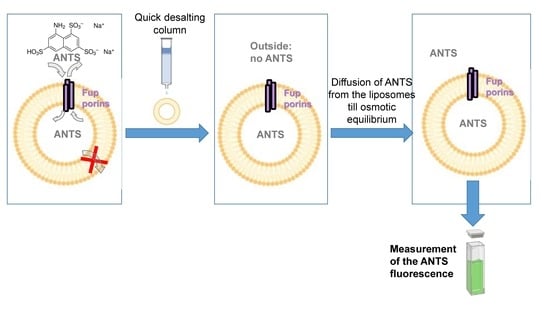
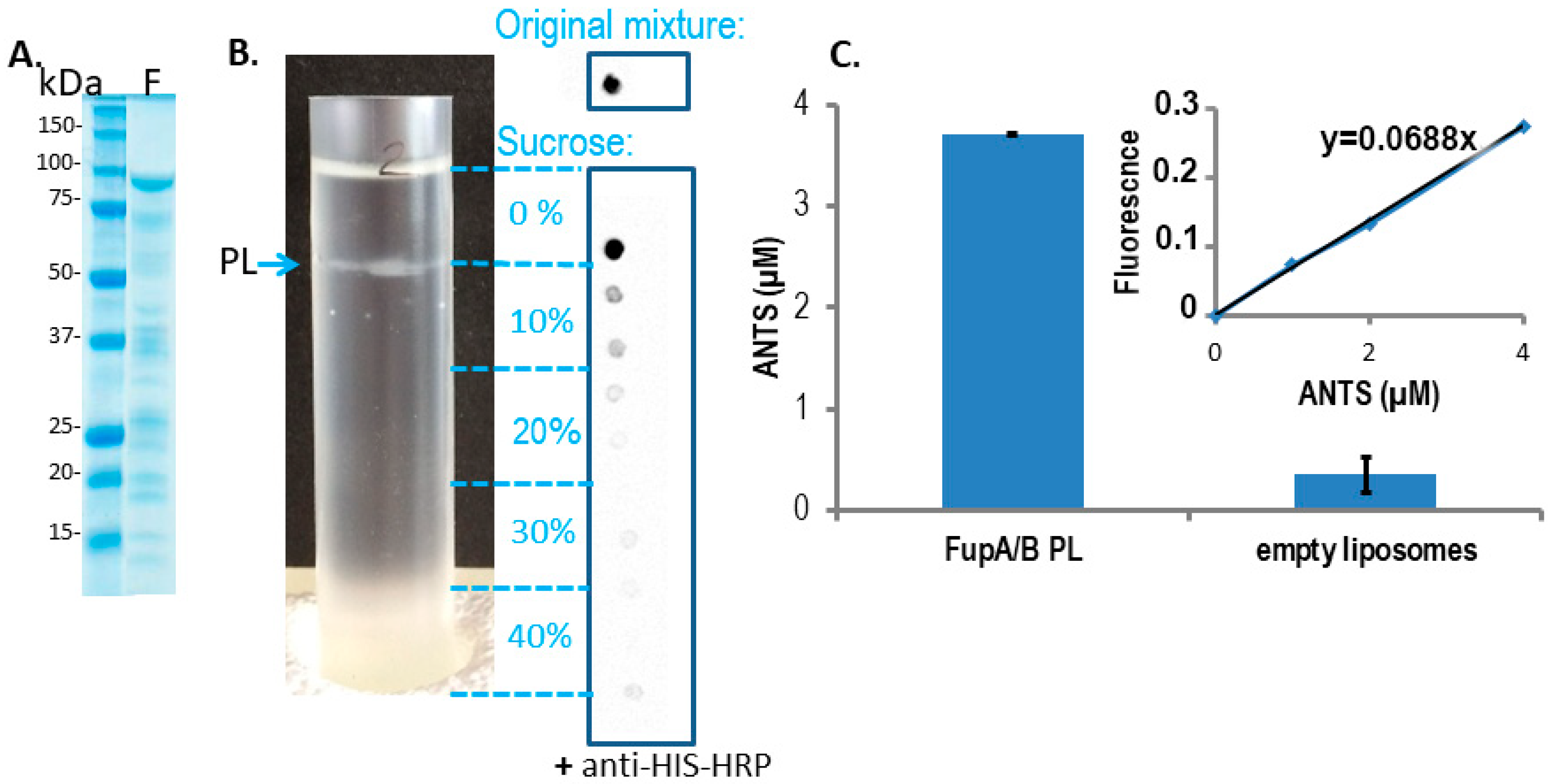
2.2. Fluorescence Assays on Proteoliposomes Containing FupA/B Suggested Porin Activity
- The intensity of the FupA/B light signal emitted by the PL dot was quantified as being 73% of that emitted by the dot corresponding to the starting material (FupA/B-MBP-His tagged protein at 100 μg/mL) (Figure 2B). Given that the sucrose gradient fractions were of 1 mL, we estimated that the whole PL fraction contained 73 μg of FupA/B protein. Knowing that the molar mass of MBP-FupA/B is 100 kDa (Reference [9] and Figure 2A), we estimated that there were 73 × 10−11 moles of FupA/B, i.e., 4.38 × 1014 molecules, of FupA/B inserted in the total number of PLs.
- We calculated the number of lipid molecules per PL by dividing the whole surface occupied by lipids in one PL by the mean surface of one phospholipid. Dynamic Light Scattering (DLS) measured the mean radius of PLs as being 63.5 ± 3.6 nm (mean ± SD; n = 3). Given that PLs contain 2 layers of lipids, we calculated the whole surface occupied by lipids in one PL as 2 × (4π R2), thus as 1.01 × 105 nm2. Knowing that the mean surface of a phospholipid is estimated to be 0.65 nm2 [18], we estimated the number of lipid molecules per PL as being 1.01 × 105/0.65 = 1.55 × 105.
- To calculate the number of formed PLs, we divided the total number of lipid molecules found in all the PLs by the number of lipid molecules per PL. The liposomes were prepared with 2 mg, thus 2.85 µM, i.e., 1.71 × 1018 molecules of lipids. Assuming that there was no loss of lipids between the liposome formation step and that of PLs, the number of formed PLs was thus estimated to be 1.71 × 1018/1.55 × 105 = 1.03 × 1013.
- Finally, the number of FupA/B monomers per PL was calculated as the total number of FupA/B proteins in the whole number of PLs, divided by the estimated number of PLs, thus 4.38 × 1014/1.03 × 1013 = 39.7.
- Knowing that the PLs’ radius estimated by DLS was 63.5 ± 3.6 nm, we calculated the volume of any given PL as 4/3 × π × R3 = 1.072 × 10−21 m3 = 1.072 × 10−18 L.
- Knowing that 2 mg of lipids had been used for the experiment, with the production of 1.013 × 1013 PLs, the total volume occupied by the formed PLs in the 1 mL working fraction was calculated as 1.013 × 1013 × 1.072 × 10−18 = 1.04 × 10−5 L = 11.04 μL. The total volume of PLs thus represented 1.10% of the 1 mL working fraction.
- Given that PLs were incubated in a 20 mM ANTS solution and that the total volume of PLs represented 1.10% of the total volume (1 mL), we reasoned that the maximum concentration of ANTS reached inside the PLs would have been 20 × 103 × 1.10% = 220 μM if their membrane had been permeable to the dye. In contrast, we obtained a concentration of 3.70 ± 0.01 μM ANTS within the PLs (Figure 2C), which represented only 1.7% of the ANTS molecules that would have diffused passively through a permeable membrane.
- Given the ANTS concentration calculated within the PLs (3.70 ± 0.01 µM) and the calculated volume of one PL (1.072 × 10−18 L), we calculated that each PL would contain 4.02 × 10−24 moles of ANTS, i.e., 2.42 molecules of ANTS, while it would have contained 142 molecules if the PL membrane had been permeable to ANTS.
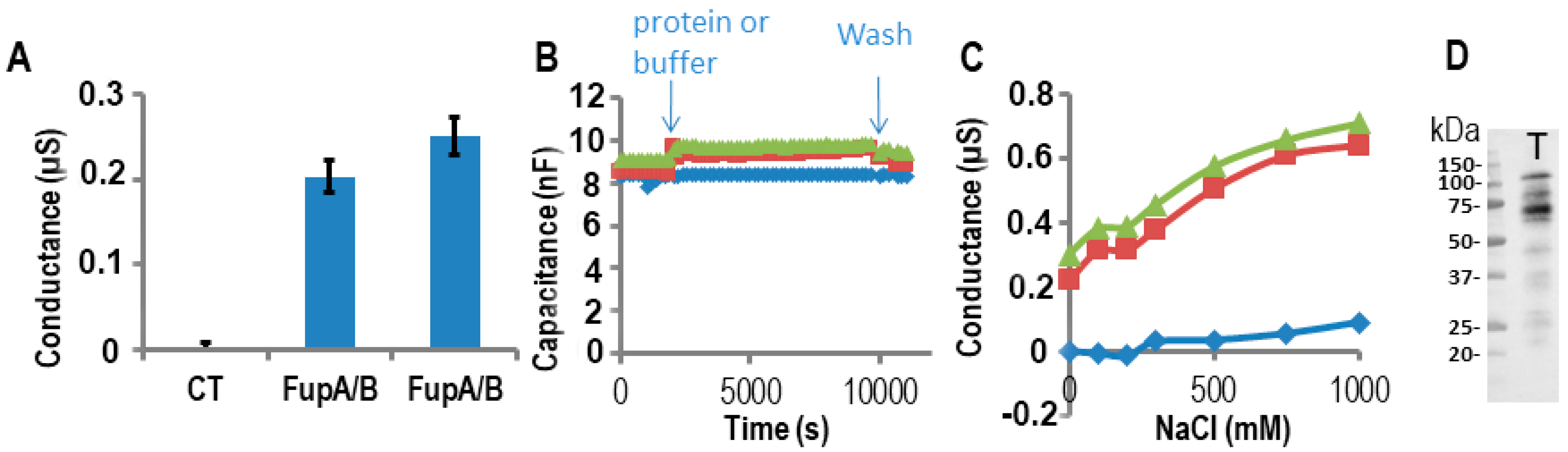
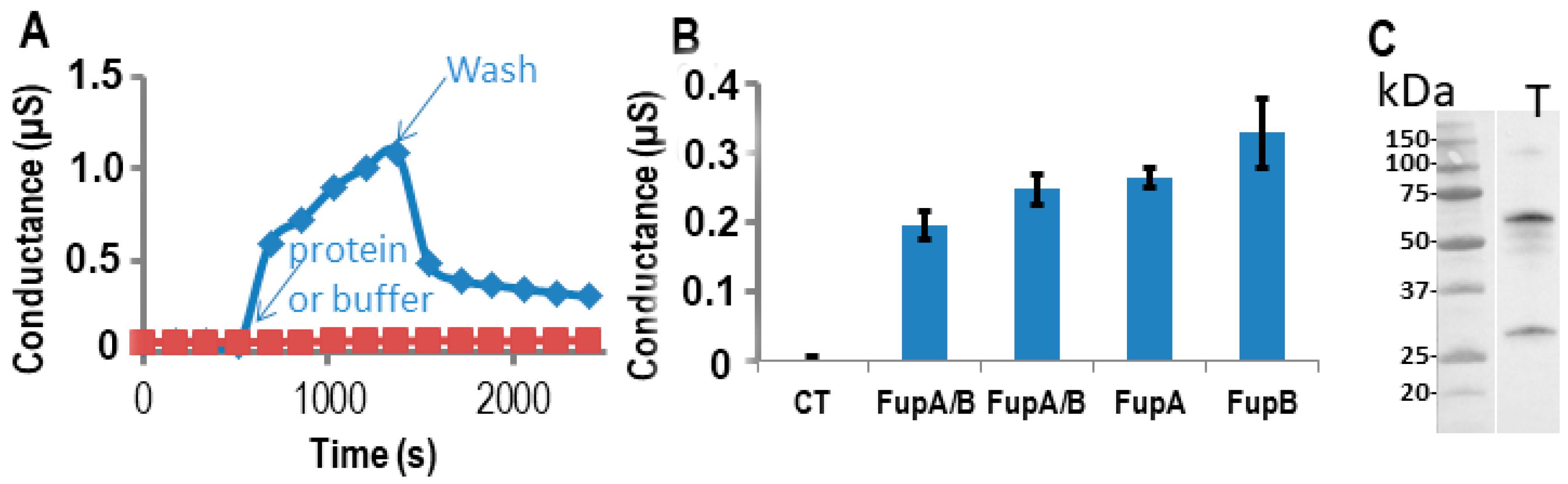
2.3. Impedance Spectroscopy Experiments Using Tethered Lipid Bilayer Membranes Confirmed the Porin Activity of FupA/B
3. Material and Methods
3.1. Protein Purification
3.2. Protein Insertion in Liposomes
3.3. Porin Activity by Fluorescent Uptake Assay in Proteo-Liposomes
3.4. Electrophysiological Measurement of Porin Ion Conductance in Tethered Lipid Bilayer Membranes
3.5. Evaluation of the Amount of Protein Inserted in the Tethered Lipid Bilayer Membrane
3.6. Statistical Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Oyston, P.C.F.; Sjostedt, A.; Titball, R.W. Tularaemia: Bioterrorism defence renews interest in Francisella tularensis. Nat. Rev. Microbiol. 2004, 2, 967–978. [Google Scholar] [CrossRef]
- Eigelsbach, H.T.; Downs, C.M. Prophylactic effectiveness of live and killed tularemia vaccines. I. Production of vaccine and evaluation in the white mouse and guinea pig. J. Immunol. 1961, 87, 415–425. [Google Scholar]
- Oyston, P.C.F. Francisella tularensis vaccines. Vaccine 2009, 27 (Suppl. S4), D48–D51. [Google Scholar] [CrossRef]
- Twine, S.; Byström, M.; Chen, W.; Forsman, M.; Golovliov, I.; Johansson, A.; Kelly, J.; Lindgren, H.; Svensson, K.; Zingmark, C.; et al. A mutant of Francisella tularensis strain SCHU S4 lacking the ability to express a 58-kilodalton protein is attenuated for virulence and is an effective live vaccine. Infect. Immun. 2005, 73, 8345–8352. [Google Scholar] [CrossRef] [Green Version]
- Salomonsson, E.; Kuoppa, K.; Forslund, A.L.; Zingmark, C.; Golovliov, I.; Sjöstedt, A.; Noppa, L.; Forsberg, Å. Reintroduction of two deleted virulence loci restores full virulence to the live vaccine strain of Francisella tularensis. Infect. Immun. 2009, 77, 3424–3431. [Google Scholar] [CrossRef] [Green Version]
- Lindgren, H.; Honn, M.; Golovlev, I.; Kadzhaev, K.; Conlan, W.; Sjöstedt, A. The 58-kilodalton major virulence factor of Francisella tularensis is required for efficient utilization of iron. Infect. Immun. 2009, 77, 4429–4436. [Google Scholar] [CrossRef] [Green Version]
- Ramakrishnan, G.; Sen, B.; Johnson, R. Paralogous outer membrane proteins mediate uptake of different forms of iron and synergistically govern virulence in Francisella tularensis tularensis. J. Biol. Chem. 2012, 287, 25191–25202. [Google Scholar] [CrossRef] [Green Version]
- Sen, B.; Meeker, A.; Ramakrishnan, G. The fslE homolog, FTL-0439 (fupA/B), mediates siderophore-dependent iron uptake in Francisella tularensis LVS. Infect. Immun. 2010, 78, 4276–4285. [Google Scholar] [CrossRef] [Green Version]
- Siebert, C.; Lindgren, H.; Ferré, S.; Villers, C.; Boisset, S.; Perard, J.; Sjöstedt, A.; Maurin, M.; Brochier-Armanet, C.; Couté, Y.; et al. Francisella tularensis: FupA mutation contributes to fluoroquinolone resistance by increasing vesicle secretion and biofilm formation. Emerg. Microbes Infect. 2019, 8, 808–822. [Google Scholar] [CrossRef]
- Ramakrishnan, G. Iron and Virulence in Francisella tularensis. Front. Cell. Infect. Microbiol. 2017, 7, 107. [Google Scholar] [CrossRef] [Green Version]
- BLAST Proteins. Available online: https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins (accessed on 30 July 2020).
- Gabaldón, T.; Koonin, E.V. Functional and evolutionary implications of gene orthology. Nat. Rev. Genet. 2013, 14, 360–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramakrishnan, G.; Sen, B. The FupA/B protein uniquely facilitates transport of ferrous iron and siderophore-associated ferric iron across the outer membrane of Francisella tularensis live vaccine strain. Microbiology 2014, 160, 446–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez, N.; Johnson, R.; Sen, B.; Ramakrishnan, G. Two parallel pathways for ferric and ferrous iron acquisition support growth and virulence of the intracellular pathogen Francisella tularensis Schu S4. Microbiologyopen 2016, 5, 453–468. [Google Scholar] [CrossRef] [PubMed]
- Henderson, J.C.; Zimmerman, S.M.; Crofts, A.A.; Boll, J.M.; Kuhns, L.G.; Herrera, C.M.; Trent, M.S. The Power of Asymmetry: Architecture and Assembly of the Gram-Negative Outer Membrane Lipid Bilayer. Annu. Rev. Microbiol. 2016, 70, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [Green Version]
- Pongprayoon, P.; Beckstein, O.; Wee, C.L.; Sansom, M.S.P. Simulations of anion transport through OprP reveal the molecular basis for high affinity and selectivity for phosphate. Proc. Natl. Acad. Sci. USA 2009, 106, 21614–21618. [Google Scholar] [CrossRef] [Green Version]
- Cortes, S.; Barette, C.; Beroud, R.; De Waard, M.; Schaack, B. Functional characterization of cell-free expressed Kv1.3 channel using a voltage-sensitive fluorescent dye. Protein Expr. Purif. 2018, 145, 94–99. [Google Scholar] [CrossRef]
- Crouch, C.H.; Bost, M.H.; Kim, T.H.; Green, B.M.; Arbuckle, D.S.; Grossman, C.H.; Howard, K.P. Optimization of detergent-mediated reconstitution of influenza A M2 protein into proteoliposomes. Membranes 2018, 8, 103. [Google Scholar] [CrossRef] [Green Version]
- Ruysschaert, T.; Marque, A.; Duteyrat, J.L.; Lesieur, S.; Winterhalter, M.; Fournier, D. Liposome retention in size exclusion chromatography. BMC Biotechnol. 2005, 5, 1–13. [Google Scholar]
- Vemuri, S.; Rhodes, C.T. Separation of liposomes by a gel filtration chromatographic technique: A preliminary evaluation. Pharm. Acta Helv. 1994, 69, 107–113. [Google Scholar] [CrossRef]
- Maccarini, M.; Gayet, L.; Alcaraz, J.P.; Liguori, L.; Stidder, B.; Watkins, E.B.; Lenormand, J.L.; Martin, D.K. Functional Characterization of Cell-Free Expressed OprF Porin from Pseudomonas aeruginosa Stably Incorporated in Tethered Lipid Bilayers. Langmuir 2017, 33, 9988–9996. [Google Scholar] [CrossRef] [PubMed]
- Geertsma, E.R.; Nik Mahmood, N.A.B.; Schuurman-Wolters, G.K.; Poolman, B. Membrane reconstitution of ABC transporters and assays of translocator function. Nat. Protoc. 2008, 3, 256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigaud, J.-L. Membrane proteins: Functional and structural studies using reconstituted proteoliposomes and 2-D crystals. Braz. J. Med. Biol. Res. 2002, 35, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Stockbridge, R.B.; Tsai, M.F. Lipid reconstitution and recording of recombinant ion channels. Methods Enzymol. 2015, 556, 385–404. [Google Scholar] [PubMed]
- Subrini, O.; Sotomayor-Pérez, A.C.; Hessel, A.; Spiaczka-Karst, J.; Selwa, E.; Sapay, N.; Veneziano, R.; Pansieri, J.; Chopineau, J.; Ladant, D.; et al. Characterization of a membrane-active peptide from the bordetella pertussis CyaA toxin. J. Biol. Chem. 2013, 288, 32585–32598. [Google Scholar] [CrossRef] [Green Version]
- Cranfield, C.G.; Cornell, B.A.; Grage, S.L.; Duckworth, P.; Carne, S.; Ulrich, A.S.; Martinac, B. Transient potential gradients and impedance measures of tethered bilayer lipid membranes: Pore-forming peptide insertion and the effect of electroporation. Biophys J. 2014, 106, 182–189. [Google Scholar] [CrossRef] [Green Version]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siebert, C.; Mercier, C.; Martin, D.K.; Renesto, P.; Schaack, B. Physicochemical Evidence that Francisella FupA and FupB Proteins Are Porins. Int. J. Mol. Sci. 2020, 21, 5496. https://doi.org/10.3390/ijms21155496
Siebert C, Mercier C, Martin DK, Renesto P, Schaack B. Physicochemical Evidence that Francisella FupA and FupB Proteins Are Porins. International Journal of Molecular Sciences. 2020; 21(15):5496. https://doi.org/10.3390/ijms21155496
Chicago/Turabian StyleSiebert, Claire, Corinne Mercier, Donald K. Martin, Patricia Renesto, and Beatrice Schaack. 2020. "Physicochemical Evidence that Francisella FupA and FupB Proteins Are Porins" International Journal of Molecular Sciences 21, no. 15: 5496. https://doi.org/10.3390/ijms21155496
APA StyleSiebert, C., Mercier, C., Martin, D. K., Renesto, P., & Schaack, B. (2020). Physicochemical Evidence that Francisella FupA and FupB Proteins Are Porins. International Journal of Molecular Sciences, 21(15), 5496. https://doi.org/10.3390/ijms21155496