Pyrazoline Hybrids as Promising Anticancer Agents: An Up-to-Date Overview



Abstract
: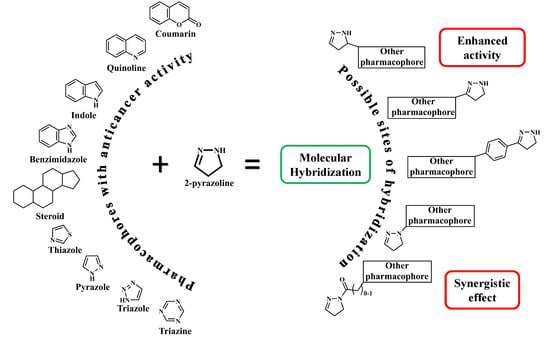
1. Introduction, Overview, and Classification
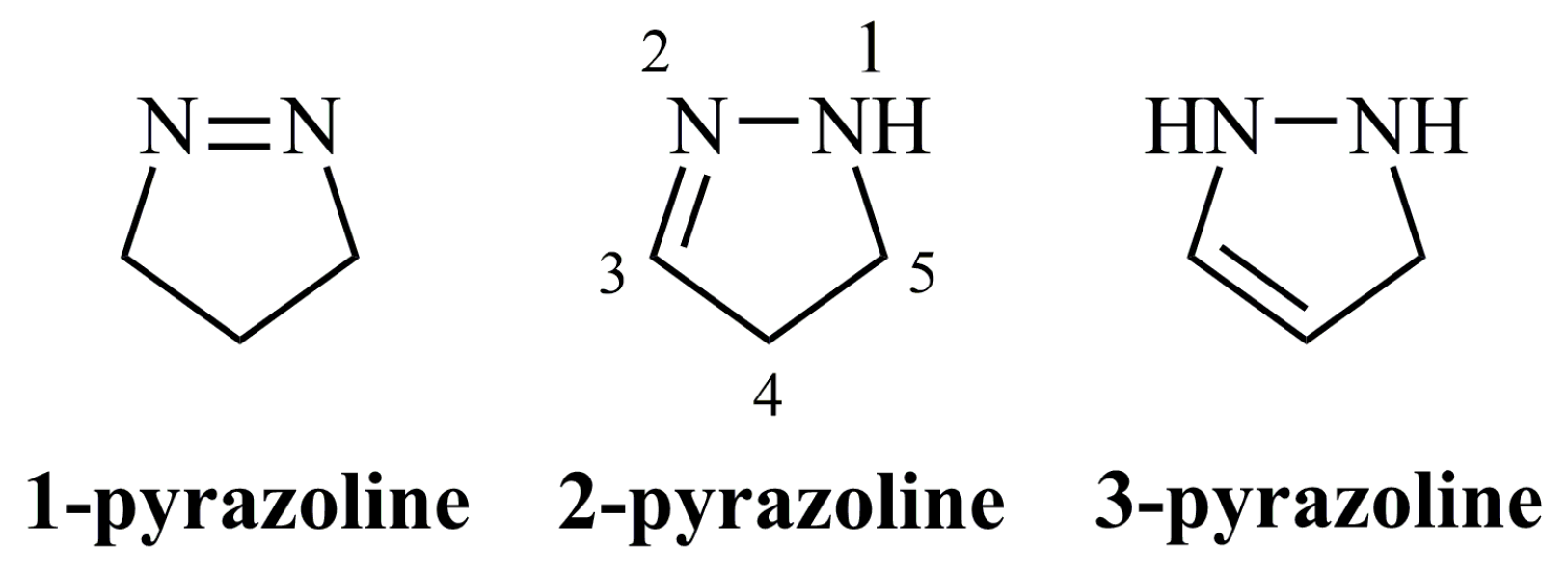
- 1-Pyrazolines;
- 3-Pyrazolines;
- 2-Pyrazolines.
- ○
- Clinical drug-pyrazoline hybrids.
- ○
- Coumarin, quinoline, and other benzene-fused six-membered heterocyclic pyrazoline hybrids.
- ▪
- Coumarin-pyrazoline hybrids.
- ▪
- Quinoline and quinolinone pyrazoline hybrids.
- ▪
- Other benzene-fused six membered heterocyclic pyrazoline hybrids.
- ○
- Indole-pyrazoline hybrids and other benzene-fused five-membered heterocyclic pyrazoline hybrids.
- ▪
- Indole-pyrazoline hybrids.
- ▪
- Isatin-pyrazoline hybrids.
- ▪
- Other benzene-fused five-membered heterocyclic pyrazoline hybrids.
- ○
- Thiazole and thiazolidinone pyrazoline hybrids.
- ▪
- Thiazole and benzothiazole pyrazoline hybrids.
- ▪
- Thiazolidinone-pyrazoline hybrids.
- ○
- Two nitrogen containing heterocyclic–pyrazoline hybrids and analogues.
- ▪
- Pyrazole-pyrazoline hybrids.
- ▪
- Imidazole and benzimidazole-pyrazoline hybrids.
- ▪
- Other two heteroatom heterocyclic pyrazoline hybrids.
- ○
- Three or more nitrogen heterocyclic–pyrazoline hybrids and analogues.
- ▪
- Triazole-pyrazoline hybrids.
- ▪
- Triazine-pyrazoline hybrids.
- ▪
- Other three or more heteroatom heterocyclic-pyrazoline hybrids.
- ○
- Steroidal pyrazoline hybrids.
- ○
- Other non-classified pyrazoline based hybrids.
2. Literature Survey
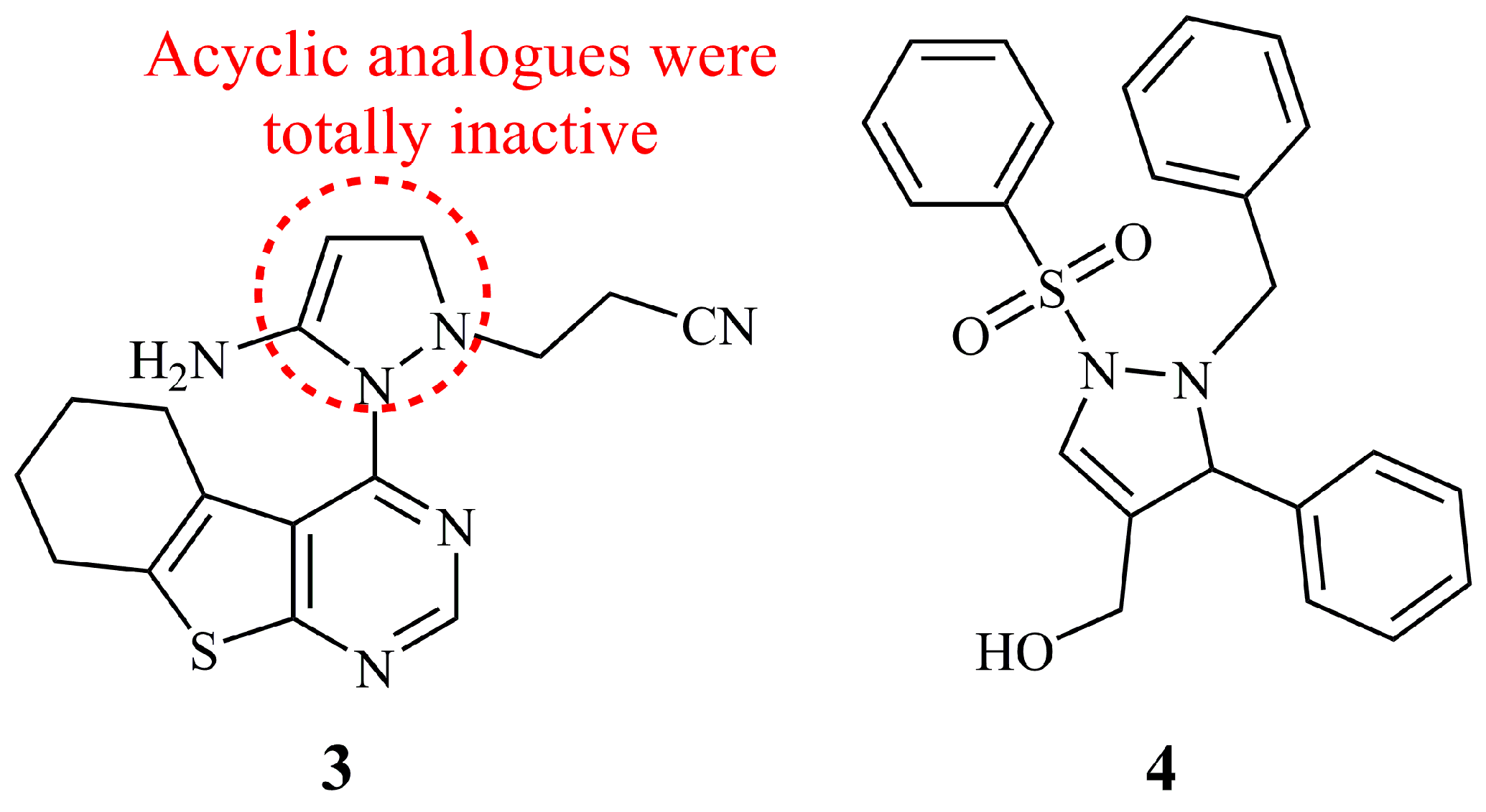
3. 1-Pyrazolines
4. 3-Pyrazolines
5. 2-Pyrazolines
5.1. Clinical Drug-Pyrazoline Hybrids
5.2. Coumarin, Quinoline, and Other Benzene-Fused Six-Membered Heterocyclic Pyrazoline Hybrids
5.2.1. Coumarin-Pyrazoline Hybrids
5.2.2. Quinoline-Pyrazoline Hybrids
5.2.3. Other Benzene-Fused Six Membered Heterocyclic Pyrazoline Hybrids
5.3. Indole-Pyrazoline Hybrids and Other Benzene-Fused Five-Membered Heterocyclic Pyrazoline Hybrids
5.3.1. Indole-Pyrazoline Hybrids
5.3.2. Isatin-Pyrazoline Hybrids
5.3.3. Other Benzene-Fused Five-Membered Heterocyclic Pyrazoline Hybrids
5.4. Thiazole and Thiazolidinone Pyrazoline Hybrids
5.4.1. Thiazole and Benzothiazole Pyrazoline Hybrids
5.4.2. Thiazolidinone-Pyrazoline Hybrids
5.5. Two Nitrogen Containing Heterocyclic–Pyrazoline Hybrids and Analogues
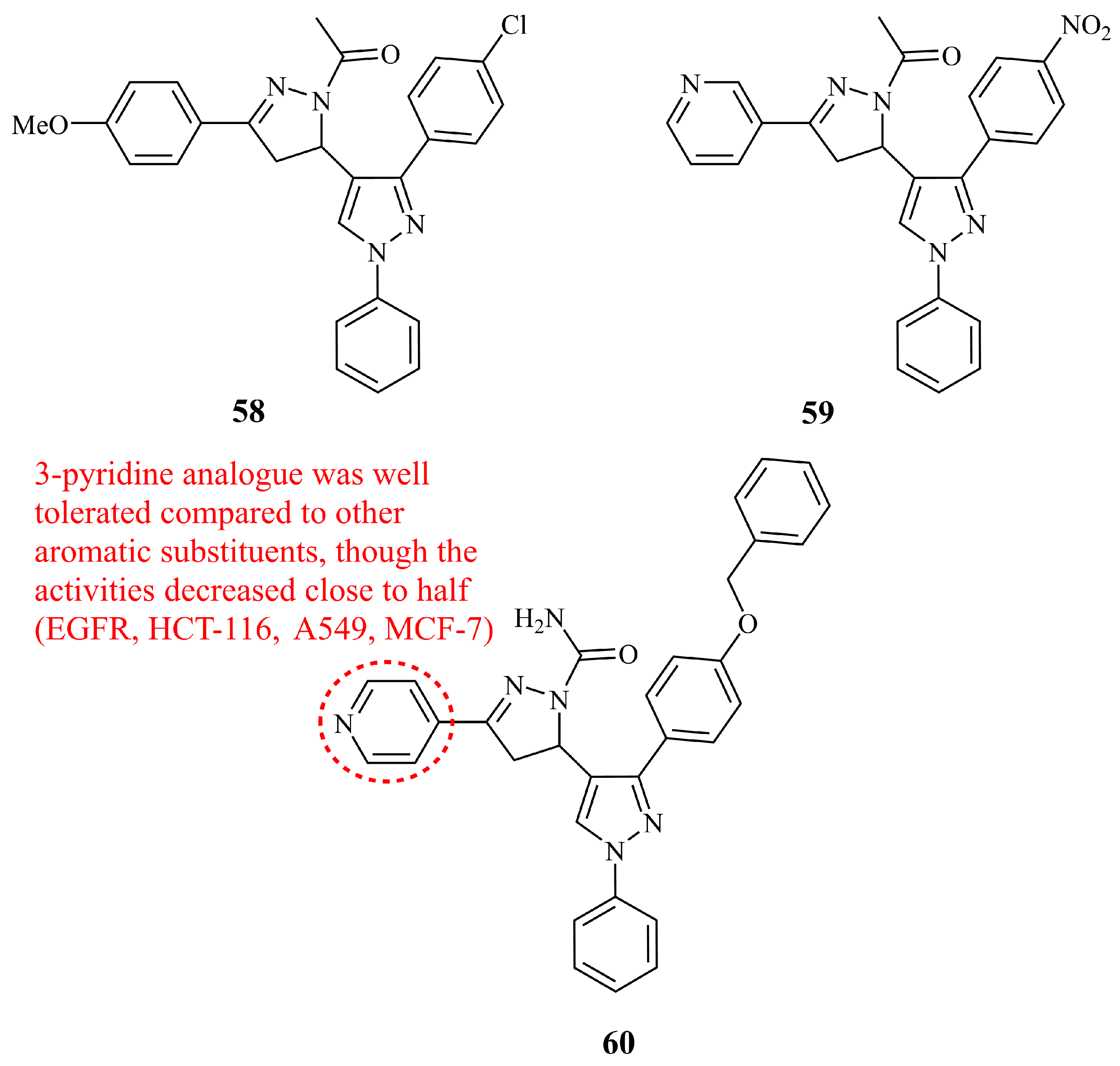
5.5.1. Pyrazole-Pyrazoline Hybrids
5.5.2. Imidazole and Benzimidazole Pyrazoline Hybrids
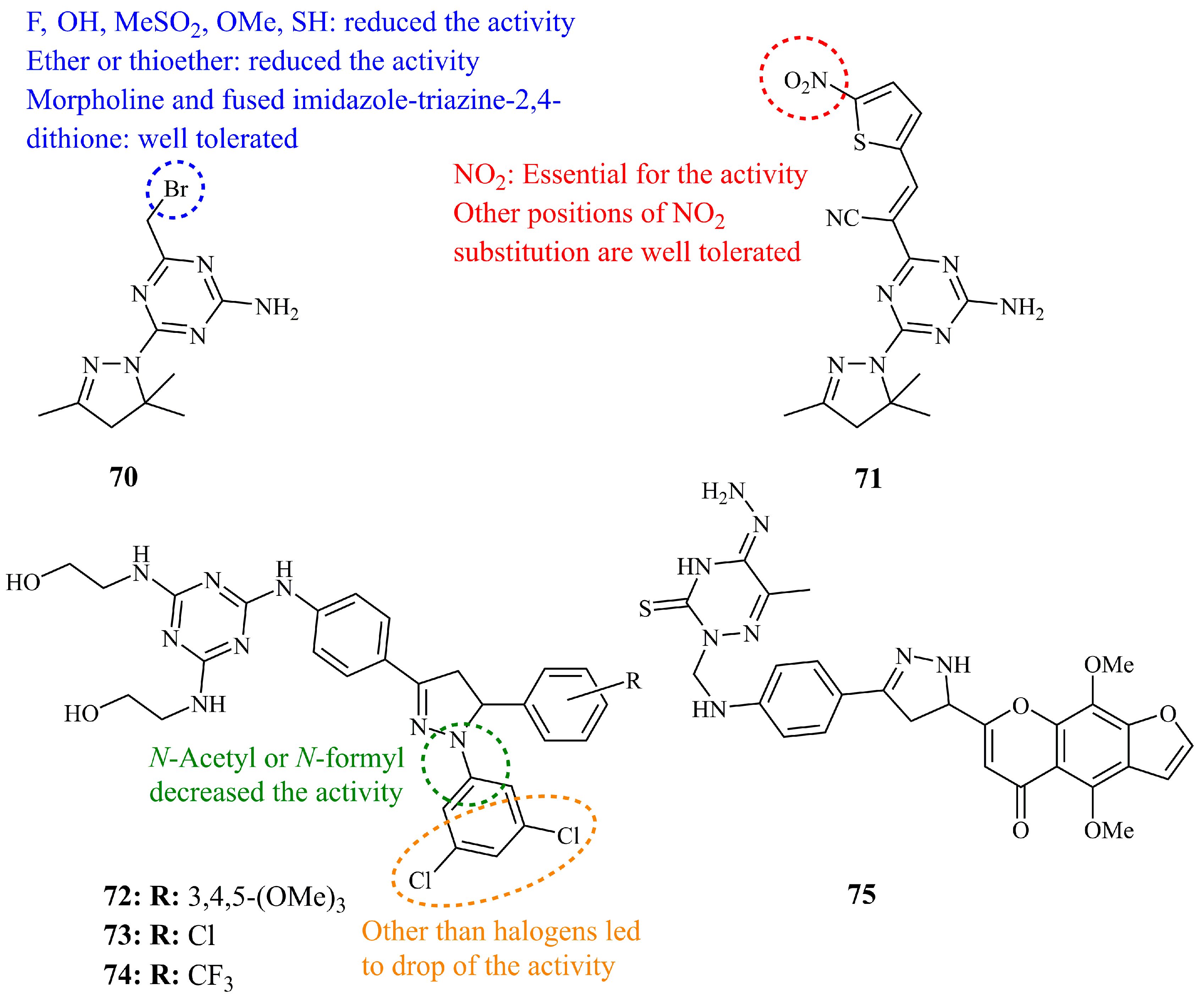
5.5.3. Other Two Heteroatom Heterocyclic Pyrazoline Hybrids
5.6. Three or More Nitrogen Heterocyclic–Pyrazoline Hybrids and Analogues
5.6.1. Triazole-Pyrazoline Hybrids
5.6.2. Triazine-Pyrazoline Hybrids
5.6.3. Additional Three or More Heteroatom Heterocyclic–Pyrazoline Hybrids and Analogues
5.7. Steroidal Pyrazoline Hybrids
5.8. Other Non-Classified Pyrazoline Based Conjugates
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organization. Cancer: Key Facts. Available online: http://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 15 March 2020).
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N. Engl. J. Med. 2012, 366, 1382–1392. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol. Sci. 2015, 36, 422–439. [Google Scholar] [CrossRef] [Green Version]
- Byrd, J.C.; Furman, R.R.; Coutre, S.E.; Flinn, I.W.; Burger, J.A.; Blum, K.A.; Grant, B.; Sharman, J.P.; Coleman, M.; Wierda, W.G.; et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 2013, 369, 32–42. [Google Scholar] [CrossRef]
- Yusuf, M.; Jain, P. Synthetic and biological studies of pyrazolines and related heterocyclic compounds. Arab. J. Chem. 2014, 7, 553–596. [Google Scholar] [CrossRef] [Green Version]
- Farooq, S.; Ngaini, Z. One-pot and two-pot synthesis of chalcone based mono and bis-pyrazolines. Tetrah. Lett. 2020, 61, 151416. [Google Scholar] [CrossRef]
- Marella, A.; Ali, R.; Alam, T.; Saha, R.; Tanwar, O.; Akhter, M.; Shaquiquzzaman; Alam, M. Pyrazolines: A biological review. Mini-Rev. Med. Chem. 2013, 13, 921–931. [CrossRef] [PubMed]
- Alex, J.M.; Kumar, R. 4,5-Dihydro-1H-pyrazole: An indispensable scaffold. J. Enzyme Inhib. Med. Chem. 2014, 29, 427–442. [Google Scholar] [CrossRef] [PubMed]
- Shaaban, M.R.; Mayhoub, A.S.; Farag, A.M. Recent advances in the therapeutic applications of pyrazolines. Expert Opin. Ther. Patents 2012, 22, 253–291. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Bawa, S.; Drabu, S.; Kumar, R.; Gupta, H. Biological activities of pyrazoline derivatives–A recent development. Recent Pat. Antiinfect. Drug Discov. 2009, 4, 154–163. [Google Scholar] [CrossRef]
- Varghese, B.; Al-Busafi, S.N.; Suliman, F.O.; Al-Kindy, S.M.Z. Unveiling a versatile heterocycle: Pyrazoline–A review. RSC Adv. 2017, 7, 46999–47016. [Google Scholar] [CrossRef] [Green Version]
- Bansal, R.; Singh, R. Steroidal pyrazolines as a promising scaffold in drug discovery. Future Med. Chem. 2020, 12, 949–959. [Google Scholar] [CrossRef] [PubMed]
- Nepali, K.; Sharma, S.; Sharma, M.; Bedi, P.M.S.; Dhar, K.L. Rational approaches, design strategies, structure activity relationship and mechanistic insights for anticancer hybrids. Eur. J. Med. Chem. 2014, 77, 422–487. [Google Scholar] [CrossRef] [PubMed]
- Appendino, G.; Jakupovic, J.; Varese, M.; Belloro, E.; Danieli, B.; Bombardelli, E. Synthesis of 7,9-nitrogen-substituted paclitaxel derivatives. Tetrah. Lett. 1996, 37, 7837–7840. [Google Scholar] [CrossRef]
- García Ruano, J.L.; Alonso de Diego, S.A.; Blanco, D.; Martín Castro, A.M.; Rosario Martín, M.; Rodríguez Ramos, J.H. (Z)-3-p-Tolylsulfinylacrylonitriles as chiral dipolarophiles: Reactions with diazoalkanes. Org. Lett. 2001, 3, 3173–3176. [Google Scholar] [CrossRef]
- Louhichi, N.; Houas, A.; Hamadi, N.B.; Msaddek, M. Synthesis and chemistry of new spiro-Δ1-pyrazoline. J. Heterocycl. Chem. 2012, 49, 267–271. [Google Scholar] [CrossRef]
- Schneider, M.P.; Goldbach, M. Facile synthesis of fucoserratene and the (±)-dictyopterenes B, D, and D’ (= ectocarpene): Constituents of marine brown algae. J. Am. Chem. Soc. 1980, 102, 6114–6116. [Google Scholar] [CrossRef]
- Andrews, S.D.; Day, A.C. Photochemistry of organic nitrogen compounds. Part IV. The photolysis of 4-alkylidene-1-pyrazolines: A possible route to trismethylenemethyl diradicals. J. Chem. Soc. (B) 1968, 1271–1279. [Google Scholar] [CrossRef]
- Quast, H.; Fuss, A. Tetramethylcyclopropanone and isopropylidene(dimethyl)thiirane via photolysis of reluctant 3,3,5,5-tetramethyl-1-pyrazolines; influence of solvent and temperature on the competition between [3+2]- and [4 + 1]- photocycloelimination. Angew. Chem. Int. Ed. Engl. 1981, 20, 291–292. [Google Scholar] [CrossRef]
- Muray, E.; Illa, O.; Castillo, J.A.; Álvarez-Larena, Á.; Bourdelande, J.L.; Branchadell, V.; Ortuño, R.M. Photolysis of chiral 1-pyrazolines to cyclopropanes: Mechanism and stereospecificity. J. Org. Chem. 2003, 68, 4906–4911. [Google Scholar] [CrossRef]
- Crawford, R.J.; Mishra, A. The mechanism of the thermal decomposition of 1-pyrazolines and its relationship to cyclopropane isomerizations. J. Am. Chem. Soc. 1966, 87, 3963–3969. [Google Scholar] [CrossRef]
- Tóth, G.; Lévai, A.; Dinya, Z.; Snatzke, G. Thermal decomposition of some new spiro-1-pyrazolines. Tetrahedron 1991, 47, 8119–8132. [Google Scholar] [CrossRef]
- Hamaguchi, M.; Nakaishi, M.; Nagai, T.; Tamura, H. Effects of an electron-withdrawing group on thermal decomposition of 4-alkylidene-1-pyrazolines: A novel stereoselective formation of alkylidenecyclopropane due to participation of π-electrons on the methylene carbon in decomposition. J. Org. Chem. 2003, 68, 9711–9722. [Google Scholar] [CrossRef] [PubMed]
- Kosemura, S.; Yamamura, S. Citreoazopyrone, a novel metabolite of a hybrid strain KO 0011 derived from Penicillium citreo-viride B. IFO 6200 and 4691. Tetrah. Lett. 1997, 38, 3025–3026. [Google Scholar] [CrossRef]
- Cui, B.C.; Yoon, I.; Li, J.Z.; Shim, Y.K. Novel cationic purpurinimides as potential photosensitizers: Design, synthesis and biological evaluation. J. Chem. Pharm. Res. 2013, 5, 818–823. [Google Scholar]
- Adamus-Grabicka, A.A.; Markowicz-Piasecka, M.; Cieślak, M.; Królewska-Golińska, K.; Hikisz, P.; Kusz, J.; Małecka, M.; Budzisz, E. Biological evaluation of 3-benzylidenechromanones and their spiropyrazolines-based analogues. Molecules 2020, 25, 1613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okitsu, T.; Sato, K.; Wada, A. Reagent-controlled oxidative aromatization in iodocyclization: Switchable access to dihydropyrazoles and pyrazoles. Org. Lett. 2010, 12, 3506–3509. [Google Scholar] [CrossRef]
- Shintani, R.; Fu, G.C. A new copper-catalyzed [3 + 2] cycloaddition: Enantioselective coupling of terminal alkynes with azomethine imines to generate five-membered nitrogen heterocycles. J. Am. Chem. Soc. 2003, 125, 10778–10779. [Google Scholar] [CrossRef]
- Shapiro, N.D.; Shi, Y.; Toste, F.D. Gold-catalyzed [3+3]-annulation of azomethine imines with propargyl esters. J. Am. Chem. Soc. 2009, 131, 11654–11655. [Google Scholar] [CrossRef] [Green Version]
- Capretto, D.A.; Brouwer, C.; Poor, C.B.; He, C. Gold(I)-catalyzed formation of 3-pyrazolines through cycloaddition of diaziridine to alkynes. Org. Lett. 2011, 13, 5842–5845. [Google Scholar] [CrossRef]
- El-Metwally, S.A.; Khalil, A.K.; El-Sayed, W.M. Design, molecular modeling and anticancer evaluation of thieno[2,3-d]pyrimidine derivatives as inhibitors of topoisomerase II. Bioorg. Chem. 2020, 94, 103492. [Google Scholar] [CrossRef] [PubMed]
- Mamedova, G.; Mahmudova, A.; Mamedov, S.; Erden, Y.; Taslimi, P.; Tüzün, B.; Tas, R.; Farzaliyev, V.; Sujayev, A.; Alwasel, S.H.; et al. Novel tribenzylaminobenzolsulphonylimine based on their pyrazine and pyridazines: Synthesis, characterization, antidiabetic, anticancer, anticholinergic, and molecular docking studies. Bioorg. Chem. 2019, 93, 103313. [Google Scholar] [CrossRef] [PubMed]
- Siegrist, A.E.; Eckhardt, C.; Kaschig, J.; Schmidt, E. Optical Brighteners; Ullmann’s Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2000. [Google Scholar]
- Wang, S.-Q.; Wu, Q.-H.; Wang, H.-Y.; Zheng, X.-X.; Shen, S.-L.; Zhang, Y.-R.; Miao, J.-Y.; Zhao, B.-X. Novel pyrazoline-based fluorescent probe for detecting glutathione and its application in cells. Biosens. Bioelectron. 2014, 55, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, F.-W.; Wang, S.-Q.; Ge, F.; Zhao, B.-X.; Miao, J.-Y. A highly sensitive fluorescent probe based on simple pyrazoline for Zn2+ in living neuron cells. Org. Biomol. Chem. 2012, 10, 8640–8644. [Google Scholar] [CrossRef]
- Borsenberger, P.M.; Schein, L.B. Hole transport in 1-phenyl-3-((diethylamino)styryl)-5-(p-(diethylamino)phenyl)pyrazoline-doped polymers. J. Phys. Chem. 1994, 98, 233–239. [Google Scholar] [CrossRef]
- Sano, T.; Nishio, Y.; Hamada, Y.; Takahasi, H.; Usuki, T.; Shibata, K. Design of conjugated molecular materials for optoelectronics. J. Mater. Chem. 2000, 10, 157–161. [Google Scholar] [CrossRef]
- Rathish, I.G.; Javed, K.; Ahmad, S.; Bano, S.; Alam, M.S.; Pillai, K.K.; Singh, S.; Bagchi, V. Synthesis and antiinflammatory activity of some new 1,3,5-trisubstituted pyrazolines bearing benzene sulfonamide. Bioorg. Med. Chem. Lett. 2009, 19, 255–258. [Google Scholar] [CrossRef]
- Bansal, E.; Srivastava, V.K.; Kumar, A. Synthesis and anti-inflammatory activity of 1-acetyl-5-substitute daryl-3-(β-aminonaphthyl)-2-pyrazolines and β-(substituted aminoethyl) amidonaphthalenes. Eur. J. Med. Chem. 2001, 36, 81–92. [Google Scholar] [CrossRef]
- Karthikeyan, M.S.; Holla, B.S.; Kumari, N.S. Synthesis and antimicrobial studies on novel chloro-fluorine containing hydroxy pyrazolines. Eur. J. Med. Chem. 2007, 42, 30–36. [Google Scholar] [CrossRef]
- Siddiqui, Z.N.; Musthafa, T.N.M.; Ahmad, A.; Khan, A.U. Thermal solvent-free synthesis of novel pyrazolyl chalcones and pyrazolines as potential antimicrobial agents. Bioorg. Med. Chem. Lett. 2011, 21, 2860–2865. [Google Scholar] [CrossRef]
- Mishra, V.K.; Mishra, M.; Kashaw, V.; Kashaw, S.K. Synthesis of 1,3,5-trisubstituted pyrazolines as potential antimalarial and antimicrobial agents. Bioorg. Med. Chem. 2017, 25, 1949–1962. [Google Scholar] [CrossRef] [PubMed]
- Shahar Yar, M.; Afroz Bakht, M.; Siddiqui, A.A.; Abdullah, M.M.; de Clercq, E. Synthesis and evaluation of in vitro antiviral activity of novel phenoxy acetic acid derivatives. J. Enzyme Inhib. Med. Chem. 2009, 24, 876–882. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, S.; Paliwal, D.; Kaushik, D.; Gupta, G.K.; Kumar, A. Synthesis, antimalarial evaluation and SAR study of some 1,3,5-trisubstituted pyrazoline derivatives. Lett. Org. Chem. 2019, 16, 807–817. [Google Scholar] [CrossRef]
- Havrylyuk, D.; Zimenkovsky, B.; Karpenko, O.; Grellier, P.; Lesyk, R. Synthesis of pyrazoline-thiazolidinone hybrids with trypanocidal activity. Eur. J. Med. Chem. 2014, 85, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.-L.; Wang, F.; Zhang, M.-Z.; Liu, Z.-M.; Huang, W.; Yang, G.-F. Synthesis, fungicidal, and insecticidal activities of β-methoxyacrylate-containing N-acetyl pyrazoline derivatives. J. Agric. Food Chem. 2008, 56, 10767–10773. [Google Scholar] [CrossRef] [PubMed]
- Luan, S.; Zhong, H.; Zhao, X.; Yang, J.; Jing, Y.; Liu, D.; Zhao, L. Synthesis, anticancer evaluation and pharmacokinetic study of novel 10-O-phenyl ethers of dihydroartemisinin. Eur. J. Med. Chem. 2017, 141, 584–595. [Google Scholar] [CrossRef] [PubMed]
- Gordaliza, M.; Castro, M.A.; Miguel del Corral, J.M.; San Feliciano, A. Antitumor properties of podophyllotoxin and related compounds. Curr. Pharm. Des. 2000, 6, 1811–1839. [Google Scholar] [CrossRef]
- Gordaliza, M.; Miguel del Corral, J.M.; Castro, M.A.; López-Vásquez, M.L.; San Feliciano, A.; García-Grávalos, M.D.; Carpy, A. Synthesis and evaluation of pyrazolignans. A new class of cytotoxic agents. Bioorg. Med. Chem. 1995, 3, 1203–1210. [Google Scholar] [CrossRef]
- Gordaliza, M.; Miguel del Corral, J.M.; Castro, M.A.; García- García, P.A.; San Feliciano, A. Cytotoxic cyclolignans related to podophyllotoxin. Il Pharmaco. 2001, 56, 297–304. [Google Scholar] [CrossRef]
- Thakur, A.; Singla, R.; Jaitak, V. Coumarins as anticancer agents: A review on synthetic strategies, mechanism of action and SAR studies. Eur. J. Med. Chem. 2015, 101, 476–495. [Google Scholar] [CrossRef]
- Liu, X.-H.; Liu, H.-F.; Chen, J.; Yang, Y.; Song, B.-A.; Bai, L.-S.; Liu, J.-X.; Zhu, H.-L.; Qi, X.-B. Synthesis and molecular docking study of novel coumarin derivatives containing 4,5-dihydropyrazole moiety as potential antitumor agents. Bioorg. Med. Chem. Lett. 2010, 20, 5705–5708. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.-Q.; Huang, C.; Jia, Y.-M.; Song, B.A.; Liu, X.-H. Novel coumarin-dihydropyrazole thio-ethanone derivatives: Design, synthesis and anticancer activity. Eur. J. Med. Chem. 2014, 74, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Qiang, D.Z.; Shi, J.B.; Song, B.A.; Liu, X.H. Novel 2H-chromen derivatives: Design, synthesis and anticancer activity. RSC Adv. 2014, 4, 5607–5617. [Google Scholar] [CrossRef]
- Chen, Y.Y.; Wu, X.Q.; Tang, W.J.; Shi, J.B.; Li, J.; Liu, X.H. Novel dihydropyrazole-chromen: Design and modulates hTERT inhibition proliferation of MGC-803. Eur. J. Med. Chem. 2016, 110, 65–75. [Google Scholar] [CrossRef]
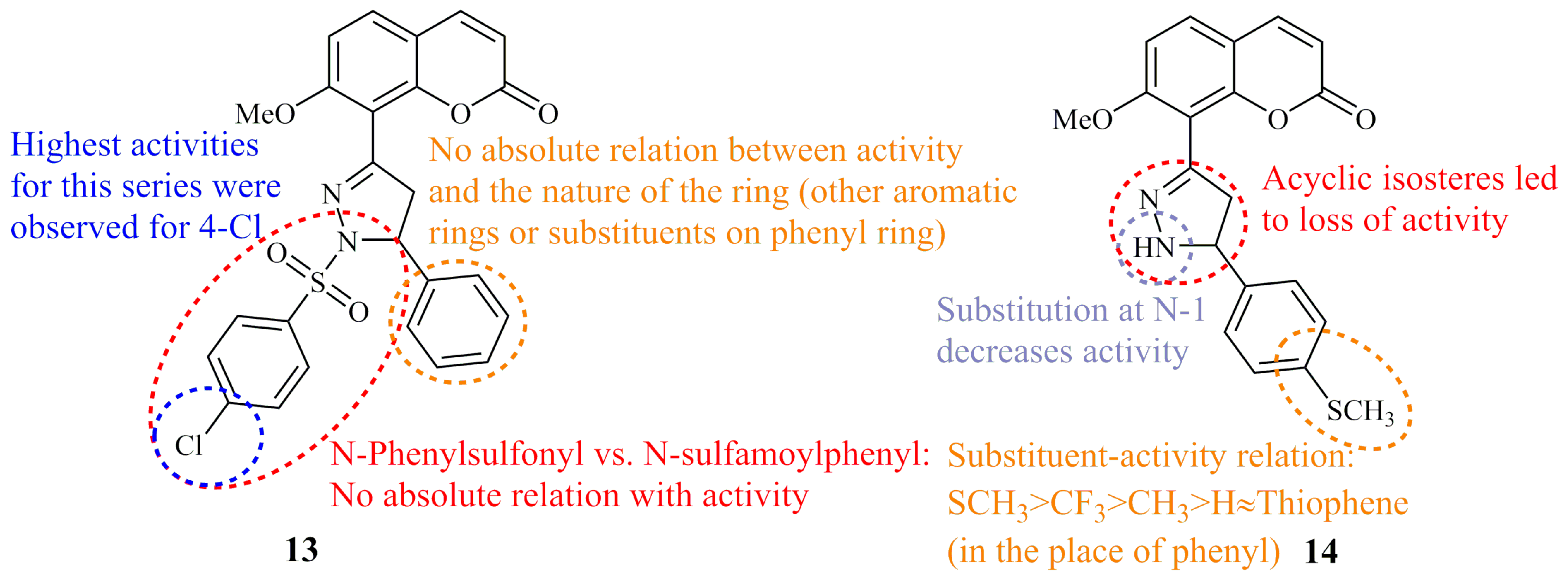
- Amin, K.M.; Eissa, A.A.M.; Abou-Seri, S.M.; Awadallah, F.M.; Hassan, G.S. Synthesis and biological evaluation of novel coumarine-pyrazoline hybrids endowed with phenylsulfonyl moiety as antitumor agents. Eur. J. Med. Chem. 2013, 60, 187–198. [Google Scholar] [CrossRef]
- Amin, K.M.; Abou-Seri, S.M.; Awadallah, F.M.; Eissa, A.A.M.; Hassan, G.S.; Abdulla, M.M. Synthesis and anticancer activity of some 8-substituted-7-methoxy-2H-chromen-2-one derivatives toward hepatocellular carcinoma HepG2 cells. Eur. J. Med. Chem. 2015, 90, 221–231. [Google Scholar] [CrossRef]
- Latif, N.A.A.; Batran, R.Z.; Khedr, M.A.; Abdalla, M.M. 3-Substituted-4-hydroxycoumarin as a new scaffold with potent CDK inhibition and promising anticancer effect: Synthesis, molecular modeling and QSAR studies. Bioorg. Chem. 2016, 67, 116–129. [Google Scholar] [CrossRef]
- Wei, Q.; Ning, J.-Y.; Dai, X.; Gao, Y.-D.; Su, L.; Zhao, B.-Z.; Miao, J.-Y. Discovery of novel HSP90 inhibitors that induced apoptosis and impaired autophagic flux in A549 lung cancer cells. Eur. J. Med. Chem. 2018, 145, 551–558. [Google Scholar] [CrossRef]
- Bai, S.-Y.; Dai, X.; Zhao, B.-X.; Miao, J.-Y. Discovery of a novel fluorescent HSP90 inhibitor and its anti-lung cancer effect. RSC Adv. 2014, 4, 19887–19890. [Google Scholar] [CrossRef]
- Kumar, N.; Bhatnagar, A.; Dudhe, R. Synthesis of 3-(4,5-dihydro-1-phenyl-5-substituted-1-phenyl-1H-pyrazol-3-yl)-2H-chromen-2-one derivatives and evaluation of their anticancer activity. Arab. J. Chem. 2017, 10, S2443–S2452. [Google Scholar] [CrossRef] [Green Version]
- Garazd, Y.; Garazd, M.; Lesyk, R. Synthesis and evaluation of anticancer activity of 6-pyrazolinylcoumarin derivatives. Saudi Pharm. J. 2017, 25, 214–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, T.D.M.; Ziora, Z.M.; Blaskovich, M.A.T. Quinolone antibiotics. Med. Chem. Comm. 2019, 10, 1719–1739. [Google Scholar] [CrossRef] [PubMed]
- Afzal, O.; Kumar, S.; Haider, R.; Ali, R.; Kumar, R.; Jaggi, M.; Bawa, S. A review on anticancer potential of bioactive heterocycle quinoline. Eur. J. Med. Chem. 2015, 97, 871–910. [Google Scholar] [CrossRef]
- Wissner, A.; Overbeek, E.; Reich, M.F.; Floyd, M.B.; Johnson, B.D.; Mamuya, N.; Rosfjord, E.C.; Discafani, C.; Davis, R.; Shi, X.; et al. Synthesis and structure-activity relationships of 6,7-disubstituted 4-anilinoquinoline-3-carbonitriles. The design of an orally active, irreversible inhibitor of the tyrosine kinase activity of the epidermal growth factor receptor (EGFR) and the human epidermal growth factor receptor-2 (HER-2). J. Med. Chem. 2003, 46, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Cherian, M.A.; Ma, C.X. The role of neratinib in HER2-driven breast cancer. Future Oncol. 2017, 13, 1931–1943. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.E.; Kim, D.-W.; Kantarjian, H.M.; Brümmendorf, T.H.; Dyagil, I.; Griskevicius, L.; Malhotra, H.; Powell, C.; Gogat, K.; Countouriotis, A.M.; et al. Bosutinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia: Results from the BELA trial. J. Clin. Oncol. 2012, 30, 3486–3492. [Google Scholar] [CrossRef]
- Ramírez-Prada, J.; Robledo, S.M.; Vélez, I.D.; del Pilar Crespo, M.; Quiroga, J.; Abonia, R.; Montoya, A.; Svetaz, L.; Zacchino, S.; Insuasty, B. Synthesis of novel quinoline-based 4,5-dihydro-1H-pyrazoles as potential anticancer, antifungal, antibacterial and antiprotozoal agents. Eur. J. Med. Chem. 2017, 131, 237–254. [Google Scholar] [CrossRef]
- Insuasty, B.; Montoya, A.; Becerra, D.; Quiroga, J.; Abonia, R.; Robledo, S.; Vélez, I.D.; Upequi, Y.; Nogueras, M.; Cobo, J. Synthesis of novel analogs of 2-pyrazoline obtained from [(7-chloroquinolin-4-yl)amino]chalcones and hydrazine as potential antitumor and antimalarial agents. Eur. J. Med. Chem. 2013, 67, 252–262. [Google Scholar] [CrossRef]
- Charris, J.E.; Monasterios, M.C.; Acosta, M.E.; Rodríquez, M.A.; Gamboa, N.D.; Martinez, G.P.; Rojas, V.; Mijares, M.R.; De Sanctis, J.B. Antimalarial, antiproliferative, and apoptotic activity of quinoline-chalcone and quinoline-pyrazoline hybrids. A dual action. Med. Chem. Res. 2019, 28, 2050–2066. [Google Scholar] [CrossRef]
- George, R.F.; Samir, E.M.; Abdelhamed, M.N.; Abdel-Aziz, H.A.; Abbas, S.E.-S. Synthesis and anti-proliferative activity of some new quinoline based 4,5-dihydropyrazoles and their thiazole hybrids as EGFR inhibitors. Bioorg. Chem. 2019, 83, 186–197. [Google Scholar] [CrossRef]
- Bagnolini, G.; Milano, D.; Manerba, M.; Schipani, F.; Ortega, J.A.; Gioia, D.; Falchi, F.; Balboni, A.; Farabegoli, F.; De Franco, F.; et al. Synthetic lethality in pancreatic cancer: Discovery of a new RAD51-BRCA2 small molecule disruptor that inhibits homologous recombination and synergizes with olaparib. J. Med. Chem. 2020, 63, 2588–2619. [Google Scholar] [CrossRef] [PubMed]
- Omar, M.T. Synthesis of 1-(heterocyclic substituted aniline)-9H-thioxanthon-9-ones and their antitumor activity. Arch. Pharm. Res. 1997, 20, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Thabit, M.G.; El Bialy, S.A.A.; Nasr, M.N.A. Synthesis and biological evaluation of new 3-(4-substituted phenyl)aminoquinoxaline derivatives as anticancer agents. Heterocycl. Commun. 2015, 21, 25–35. [Google Scholar] [CrossRef]
- El-Sakka, S.; Soliman, M.; El-Shalakany, E. Synthesis, cytotoxicity and molecular docking of some Schiff bases derived quinazolinone bearing pyrazoline. Egypt. J. Chem. 2019, 62, 197–209. [Google Scholar] [CrossRef] [Green Version]
- Gouhar, R.S.; Haneen, D.S.A.; El-Hallouty, S.M. Synthesis and anticancer evaluation of some novel quinazolin-4(3H)-one derivatives. J. Heterocyclic Chem. 2019, 56, 1651–1660. [Google Scholar] [CrossRef]
- Kaushik, N.K.; Kaushik, N.; Attri, P.; Kumar, N.; Kim, C.H.; Verma, A.K.; Choi, E.H. Biomedical importance of indoles. Molecules 2013, 18, 6620–6662. [Google Scholar] [CrossRef]
- Patil, S.A.; Patil, S.; Miller, D.D. Indole molecules as inhibitors of tubulin polymerization: Potential new anticancer agents. Future Med. Chem. 2012, 4, 2085–2115. [Google Scholar] [CrossRef]
- Johnson, I.S.; Armstrong, J.G.; Gorman, M.; Burnett, J.P. The Vinca alcaloids: A new class of oncolytic agents. Cancer Res. 1963, 23, 1390–1427. [Google Scholar]
- Zhang, Y.-L.; Qin, Y.-J.; Tang, D.-J.; Yang, M.-R.; Li, B.-Y.; Wang, Y.-T.; Cai, H.-Y.; Wang, B.-Z.; Zhu, H.-L. Synthesis and biological evaluation of 1-methyl-1H-indole-pyrazoline hybrids as potential tubulin polymerization inhibitors. ChemMedChem 2016, 11, 1446–1458. [Google Scholar] [CrossRef]
- Fan, A.; Zhang, Y.; Zhang, Q.; Wei, J.; Lu, X.; Ren, G.; Zhao, D.; Li, N.; Zhu, H.; Chen, X. Evaluation of the pharmacokinetics, tissue distribution and excretion studies of YMR-65, a tubulin polymerization inhibitor with potential anticancer activity, in rats using UPLC-MS/MS. Xenobiotica 2018, 48, 920–926. [Google Scholar] [CrossRef]
- Fan, A.; Wei, J.; Yang, M.; Zhang, Q.; Zhang, Y.; Liu, Q.; Li, N.; Zhao, D.; Lu, Y.; Li, J.; et al. Pharmacodynamic and pharmacokinetic characteristics of YMR-65, a tubulin inhibitor, in tumor-bearing mice. Eur. J. Pharm. Sci. 2018, 121, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Zhang, Y.-L.; Fan, J.; Ma, X.; Qin, Y.-J.; Zhu, H.-L. Novel nicotinoyl pyrazoline derivatives bearing N-methyl indole moiety as antitumor agents: Design, synthesis and evaluation. Eur. J. Med. Chem. 2018, 156, 722–737. [Google Scholar] [CrossRef] [PubMed]
- Qi, P.-F.; Fang, L.; Li, H.; Li, S.-K.; Yang, Y.-S.; Qi, J.-L.; Xu, C.; Zhu, H.-L. Discovery of novel pyrazoline derivatives containing methyl-1H-indole moiety as potential inhibitors for blocking APC-Asef interactions. Bioorg. Chem. 2020, 99, 103838. [Google Scholar] [CrossRef] [PubMed]
- Fodde, R.; Smits, R.; Clevers, H. APC, signal transduction and genetic instability in colorectal cancer. Nat. Rev. Cancer 2001, 1, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, Â.; Gonçalves, L.M.; Santos, M.M.M. Synthesis of novel spiropyrazoline oxindoles and evaluation of cytotoxicity in cancer cell lines. Eur. J. Med. Chem. 2014, 79, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Nunes, R.C.; Ribeiro, C.J.A.; Monteiro, Â.; Rodriques, C.M.P.; Amaral, J.D.; Santos, M.M.M. In vitro targeting of colon cancer cells using spiropyrazoline oxindoles. Eur. J. Med. Chem. 2017, 139, 168–179. [Google Scholar] [CrossRef]
- Ribeiro, C.J.A.; Amaral, J.D.; Rodriques, C.M.P.; Moreira, R.; Santos, M.M.M. Synthesis and evaluation of spiroisoxazoline oxindoles as anticancer agents. Bioorg. Med. Chem. 2014, 22, 577–584. [Google Scholar] [CrossRef]
- Gangarapu, K.; Thumma, G.; Manda, S.; Jallapally, A.; Jarapula, R.; Rekulapally, S. Design, synthesis and molecular docking of novel structural hybrids of substituted isatin based pyrazoline and thiadiazoline as antitumor agents. Med. Chem. Res. 2017, 26, 819–829. [Google Scholar] [CrossRef]
- Kocyigit, U.M.; Budak, Y.; Gürdere, M.B.; Dürü, N.; Taslimi, P.; Gülçin, İ.; Ceylan, M. Synthesis and investigation of anticancer, antibacterial activities and carbonic anhydrase, acetylcholinesterase inhibition profiles of novel (3aR,4S,7R,7aS)-2-[4-[1-acetyl-5-(aryl/heteroaryl)-4,5-dihydro-1H-pyrazol-3-yl]phenyl]-3a,4,7,7a-tetrahydro-1H-4,7-methanoisoindole-1,3(2H)-diones. Monats. Chem. 2019, 150, 721–731. [Google Scholar] [CrossRef]
- Kumari, P.; Mishra, V.S.; Narayana, C.; Khanna, A.; Chakrabarty, A.; Sagar, R. Design and efficient synthesis of pyrazoline and isoxazole bridged indole C-glycoside hybrids as potential anticancer agents. Sci. Rep. 2020, 10, 6660. [Google Scholar] [CrossRef] [Green Version]
- Sonam, V.; Kakkar, R. Isatin and its derivatives: A survey of recent syntheses, reactions, and applications. Med. Chem. Commun. 2019, 10, 351–368. [Google Scholar] [CrossRef]
- Havrylyuk, D.; Kovach, N.; Zimenkovsky, B.; Vasylenko, O.; Lesyk, R. Synthesis and anticancer activity of isatin-based pyrazolines and thiazolidines conjugates. Arch. Pharm. Med. Chem. 2011, 344, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Harkov, S.; Havrylyuk, D.; Atamanyuk, V.; Zimenkovsky, B.; Lesyk, R. Synthesis and biological activity of isatines bearing thiazolidinone and pyrazoline moieties. Pharmacia 2013, 60, 8–18. [Google Scholar]
- Reddy, R.B.; Radhika, T.; Tejaswini, B.; Madhava, R.B.; Harinadha, B.V. Synthesis, characterization, anti-cancer and anti-bacterial study of isatin conjugated 4-azidopyrazoline derivatives. Der Pharma Chem. 2016, 8, 411–418. [Google Scholar]
- Parekh, S.; Bhavsar, D.; Savant, M.; Thakrar, S.; Bavishi, A.; Parmar, M.; Vala, H.; Radadiya, A.; Pandya, N.; Serly, J.; et al. Synthesis of some novel benzofuran-2-yl(4,5-dihyro-3,5-substituted diphenylpyrazol-1-yl) methanones and studies on the antiproliferative effects and reversal of multidrug resistance of human MDR1-gene transfected mouse lymphoma cells in vitro. Eur. J. Med. Chem. 2011, 46, 1942–1948. [Google Scholar] [CrossRef]
- Xu, W.; Pan, Y.; Wang, H.; Li, H.; Peng, Q.; Wei, D.; Chen, C.; Zheng, J. Synthesis and evaluation of new pyrazoline derivatives as potential anticancer agents in HepG-2 cell line. Molecules 2017, 22, 467. [Google Scholar] [CrossRef]
- Taj, T.; Kamble, R.R.; Gireesh, T.M.; Hunnur, R.K.; Margankop, S.B. One-pot synthesis of pyrazoline derivatised carbazoles as antitubercular, anticancer agents, their DNA cleavage and antioxidant activities. Eur. J. Med. Chem. 2011, 46, 4366–4373. [Google Scholar] [CrossRef]
- Höfle, G.; Bedorf, N.; Steinmetz, H.; Schomburg, D.; Gerth, K.; Reichenbach, H. Epothilone A and B–novel 16-membered macrolides with cytotoxic activity: Isolation, crystal structure, and conformation in solution. Angew. Chem. Int. Ed. Engl. 1996, 35, 1567–1569. [Google Scholar] [CrossRef]
- Matsuo, Y.; Kanoh, K.; Yamori, T.; Kasai, H.; Katsuta, A.; Adachi, K.; Shin-ya, K.; Shizuri, Y. Urukthapelstatin A, a novel cytotoxic substance from marine-derived Mechercharimyces asporophorigenens YM11-542. J. Antibiot. 2007, 60, 251–255. [Google Scholar] [CrossRef] [Green Version]
- Portmann, C.; Blom, J.F.; Gademann, K.; Jüttner, F. Aerucyclamides A and B: Isolation and synthesis of toxic ribosomal heterocyclic peptides from the cyanobacterium Microcystis aeruginosa PCC 7806. J. Nat. Prod. 2008, 71, 1193–1196. [Google Scholar] [CrossRef]
- Teruya, T.; Sasaki, H.; Suenaga, K. Hexamollamide, a hexapeptide from an Okinawan ascidian Didemnum molle. Tetrah. Lett. 2008, 49, 5297–5299. [Google Scholar] [CrossRef]
- Cvetkovic, R.S.; Goa, K.L. Lopinavir/Ritonavir: A review of its use in the management of HIV infection. Drugs 2003, 63, 769–802. [Google Scholar] [CrossRef] [PubMed]
- Borelli, C.; Schaller, M.; Niewerth, M.; Nocker, K.; Baasner, B.; Berg, D.; Tiemann, R.; Tietjen, K.; Furgmann, B.; Lang-Fugmann, S.; et al. Modes of action of the new arylguanidine abafungin beyond interference with ergosterol biosynthesis and in vitro activity against medically important fungi. Chemotherapy 2008, 54, 245–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenwood, D. Chapter 29–Sulfonamides. In Antibiotic and Chemotherapy, 9th ed.; Finch, R.G., Greenwood, D., Norrby, S.R., Whitley, R.J., Eds.; W.B. Saunders: Philadelphia, PA, USA, 2010. [Google Scholar]
- Morigi, R.; Locatelli, A.; Leoni, A.; Rambaldi, M. Recent patents in Thiazole derivatives endowed with antitumor activity. Recent Pat. Anticancer Drug Discov. 2015, 10, 280–297. [Google Scholar] [CrossRef]
- Lu, Y.; Li, C.-M.; Wang, Z.; Ross, C.R., 2nd; Chen, J.; Dalton, J.T.; Li, W.; Miller, D.D. Discovery of 4-substituted methoxybenzoyl-aryl-thiazole as novel anticancer agents: Synthesis, biological evaluation, and structure-activity relationships. J. Med. Chem. 2009, 52, 1701–1711. [Google Scholar] [CrossRef] [Green Version]
- Gümüş, M.; Yakan, M.; Koca, İ. Recent advances of thiazole hybrids in biological applications. Future Med. Chem. 2019, 11, 1979–1998. [Google Scholar] [CrossRef]
- Lv, P.-C.; Li, D.-D.; Li, Q.-S.; Lu, X.; Xiao, Z.-P.; Zhu, H.-L. Synthesis, molecular docking and evaluation of thiazolyl-pyrazoline derivatives as EGFR TK inhibitors and potential anticancer agents. Bioorg. Med. Chem. Lett. 2011, 21, 5374–5377. [Google Scholar] [CrossRef]
- Wang, H.-H.; Qiu, K.-M.; Cui, H.-E.; Yang, Y.-S.; Luo, Y.; Xing, M.; Qiu, X.-Y.; Bai, L.-F.; Zhu, H.-L. Synthesis, molecular docking and evaluation of thiazolyl-pyrazoline derivatives containing benzodioxole as potential anticancer agents. Bioorg. Med. Chem. 2013, 21, 448–455. [Google Scholar] [CrossRef]
- Sever, B.; Altintop, M.D.; Radwan, M.O.; Özdemir, A.; Otsuka, M.; Fujita, M.; Ciftci, H.I. Design, synthesis and biological evaluation of a new series of thiazolyl-pyrazolines as dual EGFR and HER2 inhibitors. Eur. J. Med. Chem. 2019, 182, 111648. [Google Scholar] [CrossRef]
- Altintop, M.D.; Özdemir, A.; Turan-Zitouni, G.; Ilgin, S.; Atli, Ö.; Demirel, R.; Kaplancikli, Z.A. A novel series of thiazolylepyrazoline derivatives: Synthesis and evaluation of antifungal activity, cytotoxicity and genotoxicity. Eur. J. Med. Chem. 2015, 92, 342–352. [Google Scholar] [CrossRef]
- Edrees, M.M.; Abu-Melha, S.; Saad, A.M.; Kheder, N.A.; Gomha, S.M.; Muhammad, Z.A. Eco-friendly synthesis, characterization and biological evaluation of some novel pyrazolines containing thiazole moiety as potential anticancer and antimicrobial agents. Molecules 2018, 23, 2970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravula, P.; Vamaraju, H.B.; Paturi, M.; Bodige, S.; Gulipalli, K.C.; Chandra, J.N.N.S. Design, synthesis, and docking studies of novel dimethyl triazene incorporated thiazolyl pyrazolines for anticancer activity. J. Heterocyclic Chem. 2018, 55, 1313–1323. [Google Scholar] [CrossRef]
- Sadashiva, R.; Naral, D.; Kudva, J.; Kumar, S.M.; Byrappa, K.; Shafeeulla, R.M.; Kumsi, M. Synthesis, structure characterization, in vitro and in silico biological evaluation of a new series of thiazole nucleus integrated with pyrazoline scaffolds. J. Mol. Struct. 2017, 1145, 18–31. [Google Scholar] [CrossRef]
- Santosh, R.; Prabhu, A.; Selvam, M.K.; Krishna, P.M.; Nagaraja, G.K.; Rekha, P.D. Design, synthesis and pharmacology of some oxadiazole and hydroxypyrazoline hybrids bearing thiazolyl scaffold: Antiproliferative activity, molecular docking and DNA binding studies. Heliyon 2019, 5, e01255. [Google Scholar] [CrossRef] [Green Version]
- Amnerkar, N.D.; Bhusari, K.P. Preliminary anticancer activity of some prop-2-eneamido, thiazole and 1-acetyl-pyrazolin derivatives of aminobenzothiazoles. Dig. J. Nano. Biol. 2010, 5, 177–187. [Google Scholar]
- Tugrak, M.; Gul, H.I.; Sakagamu, H.; Gulcin, I. Synthesis, cytotoxic, and carbonic anhydrase inhibitory effects of new 2-(3-(4-methoxyphenyl)-5-(aryl)-4,5-dihydro-1H-pyrazol-1-yl)benzo[d]thiazole derivatives. J. Heterocyclic Chem. 2020, 1–7. [Google Scholar] [CrossRef]
- Nirwan, S.; Chahal, V.; Kakkar, R. Thiazolidinones: Synthesis, reactivity and their biological applications. J. Heterocyclic Chem. 2019, 56, 1239–1253. [Google Scholar] [CrossRef]
- Patel, N.B.; Patel, H.R.; Shaikh, F.M.; Rajani, D. New 4-thiazolidinones from 5-ethyl pyridine-2-ethanol: Their antibacterial, antifungal, and antitubercular activity. Med. Chem. Res. 2014, 23, 1360–1370. [Google Scholar] [CrossRef]
- Patel, D.; Kumari, P.; Patel, N. Synthesis and biological evaluation of some thiazolidinones as antimicrobial agents. Eur. J. Med. Chem. 2012, 48, 354–362. [Google Scholar] [CrossRef]
- Nitsche, C.; Schreier, V.N.; Behnam, M.A.M.; Kumar, A.; Bartenschlager, R.; Klein, C.D. Thiazolidinone-peptide hybrids as dengue virus protease inhibitors with antiviral activity in cell culture. J. Med. Chem. 2013, 56, 8389–8403. [Google Scholar] [CrossRef]
- Szychowski, K.A.; Leja, M.L.; Kaminskyy, D.V.; Binduga, U.E.; Pinyazhko, O.R.; Lesyk, R.B.; Gmiński, J. Study of novel anticancer 4-thiazolidinone derivatives. Chem. Biol. Interact. 2017, 262, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Havrylyuk, D.; Zimenkovsky, B.; Vasylenko, O.; Zaprutko, L.; Gzella, A.; Lesyk, R. Synthesis of novel thiazolone-based compounds containing pyrazoline moiety and evaluation of their anticancer activity. Eur. J. Med. Chem. 2009, 44, 1396–1404. [Google Scholar] [CrossRef] [PubMed]
- Havrylyuk, D.; Zimenkovsky, B.; Vasylenko, O.; Gzella, A.; Lesyk, R. Synthesis of new 4-thiazolidinone-, pyrazoline-, and isatin-based conjugates with promising antitumor activity. J. Med. Chem. 2012, 55, 8630–8641. [Google Scholar] [CrossRef] [PubMed]
- Havrylyuk, D.; Zimenkovsky, B.; Vasylenko, O.; Lesyk, R. Synthesis and anticancer and antiviral activities of new 2-pyrazoline-substituted 4-thiazolidinones. J. Heterocyclic Chem. 2013, 50, E55–E62. [Google Scholar] [CrossRef]
- Havrylyuk, D.; Zimenkovsky, B.; Vasylenko, O.; Day, C.W.; Smee, D.F.; Grellier, P.; Lesyk, R. Synthesis and biological activity evaluation of 5-pyrazoline substituted 4-thiazolidinones. Eur. J. Med. Chem. 2013, 66, 228–237. [Google Scholar] [CrossRef]
- Khalil, N.A.; Ahmed, E.M.; El-Nassan, H.B. Synthesis, characterization, and biological evaluation of certain 1,3-thiazolone derivatives bearing pyrazoline moiety as potential anti-breast cancer agents. Med. Chem. Res. 2013, 22, 1021–1027. [Google Scholar] [CrossRef]
- Abdullah, J.H.; Yahya, T.A.A.; Al-Ghoraphi, M.A.H.; Yassin, S.H. Synthesis and evaluation of new pyrazoline and thiazolidinone derivatives as anticancer activity. Der. Pharm. Chem. 2014, 6, 203–210. [Google Scholar]
- Ansari, A.; Ali, A.; Asif, M. Shamsuzzaman Review: Biologically active pyrazole derivatives. New. J. Chem. 2017, 41, 16–41. [Google Scholar] [CrossRef]
- Fustero, S.; Sánchez-Roselló, M.; Barrio, P.; Simón-Fuentes, A. From 2000 to mid-2010: A fruitful decade for the synthesis of pyrazoles. Chem. Rev. 2011, 111, 6984–7034. [Google Scholar] [CrossRef]
- Casadia, I.; de Albuquerque, D.Y.; do Carmo Capiotto, A.; de Pereira, C.M.P.; Pizzuti, L. Oxidative aromatization of pyrazolines. Curr. Org. Synth. 2017, 14, 691–703. [Google Scholar] [CrossRef]
- Insuasty, B.; Tigreros, A.; Orozco, F.; Quiroga, J.; Abonía, R.; Nogueras, M.; Sanchez, A.; Cobo, J. Synthesis of novel pyrazolic analogues of chalcones and their 3-aryl-4-(3-aryl-4,5-dihydro-1H-pyrazol-5-yl)-1-phenyl-1H-pyrazole derivatives as potential antitumor agents. Bioorg. Med. Chem. 2010, 18, 4965–4974. [Google Scholar] [CrossRef] [PubMed]
- Alam, R.; Alam, A.; Panda, A.K. Rahisuddin, Design, synthesis and cytotoxicity evaluation of pyrazolyl pyrazoline and pyrazolyl aminopyrimidine derivatives as potential anticancer agents. Med. Chem. Res. 2018, 27, 560–570. [Google Scholar] [CrossRef]
- Nawaz, F.; Alam, O.; Perwez, A.; Rizvi, M.A.; Naim, M.J.; Siddiqui, N.; Pottoo, F.H.; Jha, M. 3′-(4-(Benzyloxy)phenyl)-1′-phenyl-5-(heteroaryl/aryl)-3,4-dihydro-1′H,2H-[3,4′-bipyrazole]-2-carboxamides as EGFR kinase inhibitors: Synthesis, anticancer evaluation, and molecular docking studies. Arch. Pharm. Chem. Life Sci. 2020, 353, e1900262. [Google Scholar] [CrossRef] [PubMed]
- Shaharyar, M.; Abdullah, M.M.; Bakht, M.A.; Majeed, J. Pyrazoline bearing benzimidazoles: Search for anticancer agent. Eur. J. Med. Chem. 2010, 45, 114–119. [Google Scholar] [CrossRef]
- Akhtar, M.J.; Khan, A.A.; Ali, Z.; Dewangan, R.P.; Rafi, M.; Hassan, M.Q.; Akhtar, M.S.; Siddiqui, A.A.; Partap, S.; Pasha, S.; et al. Synthesis of stable benzimidazole derivatives bearing pyrazole as anticancer and EGFR receptor inhibitors. Bioorg. Chem. 2018, 78, 158–169. [Google Scholar] [CrossRef]
- Chouiter, M.I.; Boulebd, H.; Pereira, D.M.; Valentão, P.; Andrade, P.B.; Belfaitah, A.; Silva, A.M.S. New chalcone-type compounds and 2-pyrazoline derivatives: Synthesis and caspase-dependent anticancer activity. Future Med. Chem. 2020, 12, 493–509. [Google Scholar] [CrossRef]
- Kuthyala, S.; Hanumanthappa, M.; Kumar, S.M.; Sheik, S.; Karikannar, N.G.; Prabhu, A. Crystal, Hirshfeld, ADMET, drug-like and anticancer study of some newly synthesized imidazopyridine containing pyrazoline derivatives. J. Mol. Struct. 2019, 1197, 65–72. [Google Scholar] [CrossRef]
- Nofal, Z.M.; Soliman, E.A.; El-Karim, S.S.A.; El-Zahar, M.I.; Srour, A.M.; Sethumadhavan, S.; Maher, T.J. Novel benzimidazole derivatives as expected anticancer agents. Acta. Pol. Pharm. 2011, 68, 519–534. [Google Scholar]
- Dinesha; Viveka, S.; Naik, P.; Nagaraja, G.K. Synthesis, characterization of new imidazoquinonyl chalcones and pyrazolines as potential anticancer and antioxidant agents. Med. Chem. Res. 2014, 23, 4189–4197. [Google Scholar] [CrossRef]
- Ahmed, N.M.; Youns, M.; Soltan, M.K.; Said, A.M. Design, synthesis, molecular modelling, and biological evaluation of novel substituted pyrimidine derivatives as potential anticancer agents for hepatocellular carcinoma. J. Enzyme Inhib. Med. Chem. 2019, 34, 1110–1120. [Google Scholar] [CrossRef]
- Shaik, A.; Bhandare, R.R.; Palleapati, K.; Nissankararao, S.; Kancharlapalli, V.; Shaik, S. Antimicrobial, antioxidant, and anticancer activities of some novel isoxazole ring containing chalcone and dihydropyrazole derivatives. Molecules 2020, 25, 1047. [Google Scholar] [CrossRef] [Green Version]
- Bailey, E.M.; Krakowsky, D.J.; Rybak, M.J. The triazole antifungal agents: A review of itraconazole and fluconazole. Pharmacotherapy 1990, 10, 146–153. [Google Scholar] [CrossRef]
- Bozorov, K.; Zhao, J.; Aisa, H.A. 1,2,3-Triazole-containing hybrids as leads in medicinal chemistry: A recent overview. Bioorg. Med. Chem. 2019, 27, 3511–3531. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Zhao, S.-J.; Liu, Y. 1,2,3-Triazole-containing hybrids as potential anticancer agents: Current developments, action mechanisms and structure-activity relationships. Eur. J. Med. Chem. 2019, 183, 111700. [Google Scholar] [CrossRef] [PubMed]
- Hussaini, S.M.A.; Yedla, P.; Babu, K.S.; Shaik, T.B.; Chityal, G.K.; Kamal, A. Synthesis and biological evaluation of 1,2,3-triazole tethered pyrazoline and chalcone derivatives. Chem. Biol. Drug Des. 2016, 88, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Pankaj, S.; Saha, S.T.; Kaur, M.; Oluwakemi, E.; Awolade, P.; Singh, P.; Kumar, V. Synthesis and in vitro anti-proliferative evaluation of naphthalimide–chalcone/pyrazoline conjugates as potential SERMs with computational validation. RSC Adv. 2020, 10, 15836–15845. [Google Scholar] [CrossRef] [Green Version]
- Siliveri, S.; Vamaraju, H.B.; Raj, S. Design, synthesis, molecular docking, ADMET studies, and biological evaluation of isoxazoline and pyrazoline incorporating 1,2,3-triazole benzene sulfonamides. Russ. J. Bioorg. Chem. 2019, 45, 381–390. [Google Scholar] [CrossRef]
- Elgogary, S.R.; Khidre, R.E.; El-Telbani, E.M. Regioselective synthesis and evaluation of novel sulfonamide 1,2,3-triazole derivatives as antitumor agents. J. Iran Chem. Soc. 2020, 17, 765–776. [Google Scholar] [CrossRef]
- Cascioferro, S.; Parrino, B.; Spanò, V.; Carbone, A.; Montalbano, A.; Barraja, P.; Diana, P.; Cirrincione, G. 1,3,4-Triazines: A promising scaffold for anticancer drugs development. Eur. J. Med. Chem. 2017, 142, 523–549. [Google Scholar] [CrossRef]
- Cascioferro, S.; Parrino, B.; Spanò, V.; Carbone, A.; Montalbano, A.; Barraja, P.; Diana, P.; Cirrincione, G. An overview on the recent developments of 1,2,4-triazine derivatives as anticancer compounds. Eur. J. Med. Chem. 2017, 142, 328–375. [Google Scholar] [CrossRef] [PubMed]
- Brzozowski, Z.; Sączewski, F.; Gdaniec, M. Synthesis, structural characterization and antitumor activity of novel 2,4-diamino-1,3,5-triazine derivatives. Eur. J. Med. Chem. 2000, 35, 1053–1064. [Google Scholar] [CrossRef]
- Brzozowski, Z.; Sączewski, F. Synthesis and antitumor activity of novel 2-amino-4-(3,5,5-trimethyl-2-pyrazolino)-1,3,5-triazine derivatives. Eur. J. Med. Chem. 2002, 37, 709–720. [Google Scholar] [CrossRef]
- Moreno, L.M.; Quiroga, J.; Abonia, R.; Ramírez-Prada, J.; Insuasty, B. Synthesis of new 1,3,5-triazine-based 2-pyrazolines as potential anticancer agents. Molecules 2018, 23, 1956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-All, A.S.A.; Osman, S.A.; Roaiah, H.M.F.; Abdalla, M.M.; El Aty, A.A.A.; El-Hady, W.H.A. Potent anticancer and antimicrobial activities of pyrazole, oxazole and pyridine derivatives containing 1,2,4-triazine moiety. Med. Chem. Res. 2015, 24, 4093–4104. [Google Scholar] [CrossRef]
- ElMonaem, H.S.A.; Abdel-Aziz, N.I.; Morsy, M.A.; Badria, F.A.; ElSenduny, F.; El-Ashmawy, M.B.; Moustafa, M.A. Synthesis, in vitro antiproliferative evaluation and molecular docking of new tetrazole-chalcone and tetrazole-pyrazoline hybrids. J. Appl. Pharm. Sci. 2018, 8, 75–87. [Google Scholar] [CrossRef] [Green Version]
- Karabacak, M.; Altintop, M.D.; Çiftçi, H.İ.; Koga, R.; Otsuka, M.; Fujita, M.; Özdemir, A. Synthesis and evaluation of new pyrazoline derivatives as potential anticancer agents. Molecules 2015, 20, 19066–19084. [Google Scholar] [CrossRef]
- Salvador, J.A.R.; Carvalho, J.F.S.; Neves, M.A.C.; Silvestre, S.M.; Leitão, A.J.; Silva, M.M.C.; e Melo, M.L.S. Anticancer steroids: Linking natural and semi-synthetic compounds. Nat. Prod. Rep. 2013, 30, 324–374. [Google Scholar] [CrossRef]
- Banday, A.H.; Mir, B.P.; Lone, I.H.; Suri, K.A.; Kumar, H.M.S. Studies on novel D-ring substituted steroidal pyrazolines as potential anticancer agents. Steroids 2010, 75, 805–809. [Google Scholar] [CrossRef]
- Iványi, Z.; Wölfling, J.; Görbe, T.; Szécsi, M.; Wittmann, T.; Schneider, G. Synthesis of regioisomeric 17β-N-phenylpyrazolyl steroid derivatives and their inhibitory effect on 17α-hydroxylase/C17,20-lyase. Steroids 2010, 75, 450–456. [Google Scholar] [CrossRef]
- Iványi, Z.; Szabó, N.; Huber, J.; Wölfling, J.; Zupkó, I.; Szécsi, M.; Wittmann, T.; Schneider, G. Synthesis of D-ring-substituted (5’R)- and (5’S)-17β-pyrazolinylandrostene epimers and comparison of their potential anticancer activities. Steroids 2012, 77, 566–574. [Google Scholar] [CrossRef]
- Choudhary, M.I.; Alam, M.S.; ur Rahman, A.; Yousuf, S.; Wu, Y.-C.; Lin, A.-S.; Shaheen, F. Pregnenolone derivatives as potential anticancer agents. Steroids 2011, 76, 1554–1559. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.-L.; Wu, Y.; Liu, C.-J.; Wei, C.-Y.; Tao, J.-C.; Liu, H.-M. Design and stereoselective synthesis of novel isosteviol-fused pyrazolines and pyrazoles as potential anticancer agents. Eur. J. Med. Chem. 2013, 65, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wu, H.; Li, A.-J.; Pei, J.; Zhao, L. Synthesis and biological evaluation of hydrazone and pyrazoline derivatives derived from androstenedione. Res. Chem. Intermed. 2018, 44, 7029–7046. [Google Scholar] [CrossRef]
- Kankala, S.; Kankala, R.K.; Kommidi, D.R.; Mudithanapelli, C.; Balaboina, R.; Vadde, R.; Jonnalagadda, S.B.; Vasam, C.S. Synthesis and anti-cancer evaluation of steroidal diglycoside-pyrazoline hybrids. RSC Adv. 2014, 4, 40305–40311. [Google Scholar] [CrossRef]
- Shamsuzzaman; Khanam, H.; Mashrai, A.; Sherwani, A.; Owais, M.; Siddiqui, N. Synthesis and anti-tumor evaluation of B-ring substituted steroidal pyrazoline derivatives. Steroids 2013, 78, 1263–1272. [Google Scholar] [CrossRef]
- Shamsuzzaman; Khanam, H.; Dar, A.M.; Siddiqui, N.; Rehman, S. Synthesis, characterization and anticancer studies of new steroidal pyrazolines. J. Saudi Chem. Soc. 2016, 20, 7–12. [Google Scholar] [CrossRef] [Green Version]
- Bai, X.; Shi, W.Q.; Chen, H.F.; Zhang, P.; Li, Y.; Yin, S.F. Synthesis and antitumor activity of 1-acetyl-3-(4-phenyl)-4,5-dihydro-2-pyrazoline-5-phenylursolate and 4-chalcone ursolate derivatives. Chem. Nat. Comp. 2012, 48, 60–65. [Google Scholar] [CrossRef]
- Wang, R.; Chen, H.; Yan, W.; Zheng, M.; Zhang, T.; Zhang, Y. Ferrocene-containing hybrids as potential anticancer agents: Current developments, mechanisms of action and structure-activity relationships. Eur. J. Med. Chem. 2020, 190, 112109. [Google Scholar] [CrossRef]
- Atmaca, H.; Özkan, A.N.; Zora, M. Novel ferrocenyl pyrazoles inhibit breast cancer cell viability via induction of apoptosis and inhibition of PI3K/Akt and ERK1/2 signaling. Chem. Biol. Interact. 2017, 263, 28–35. [Google Scholar] [CrossRef]
- Shen, S.-L.; Zhu, J.; Li, M.; Zhao, B.-X.; Miao, J.-Y. Synthesis of ferrocenyl pyrazole-containing chiral aminoethanol derivatives and their inhibition against A549 and H322 lung cancer cells. Eur. J. Med. Chem. 2012, 54, 287–294. [Google Scholar] [CrossRef]
- Shen, S.-L.; Shao, J.-H.; Luo, J.-Z.; Liu, J.-T.; Miao, J.-Y.; Zhao, B.-Z. Novel chiral ferrocenylpyrazolo[1,5-a][1,4]diazepin-4-one derivatives—Synthesis, characterization and inhibition against lung cancer cells. Eur. J. Med. Chem. 2013, 63, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Bostancioğlu, R.B.; Demirel, S.; Cin, G.T.; Koparal, A.T. Novel ferrocenyl-containing N-acetyl-2-pyrazolines inhibit in vitro angiogenesis and human lung cancer growth by interfering with F-actin stress fiber polymerization. Drug Chem. Toxixol. 2013, 36, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, M.; Kumar, N.; Baldi, A.; Chandra, R.; Babu, M.A.; Madan, J. 4-Bromo-4’-chloro pyrazoline analog of curcumin augmented anticancer activity against human cervical cancer, HeLa cells: In silico-guided analysis, synthesis, and in vitro cytotoxicity. J. Biomo. Struct. Dyn. 2020, 38, 1335–1353. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
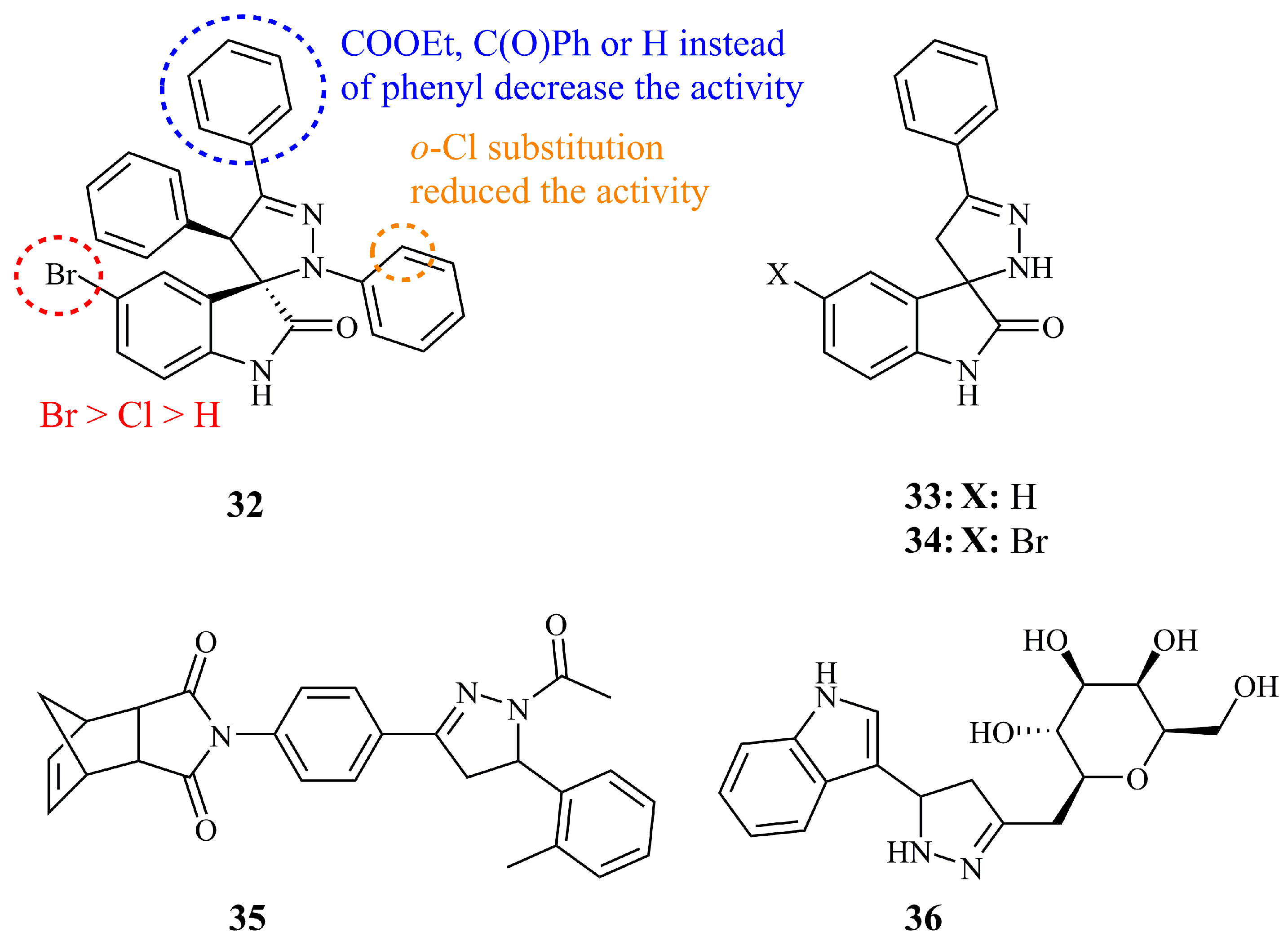{kind=link}
{kind=link}
{kind=link}
{kind=link}
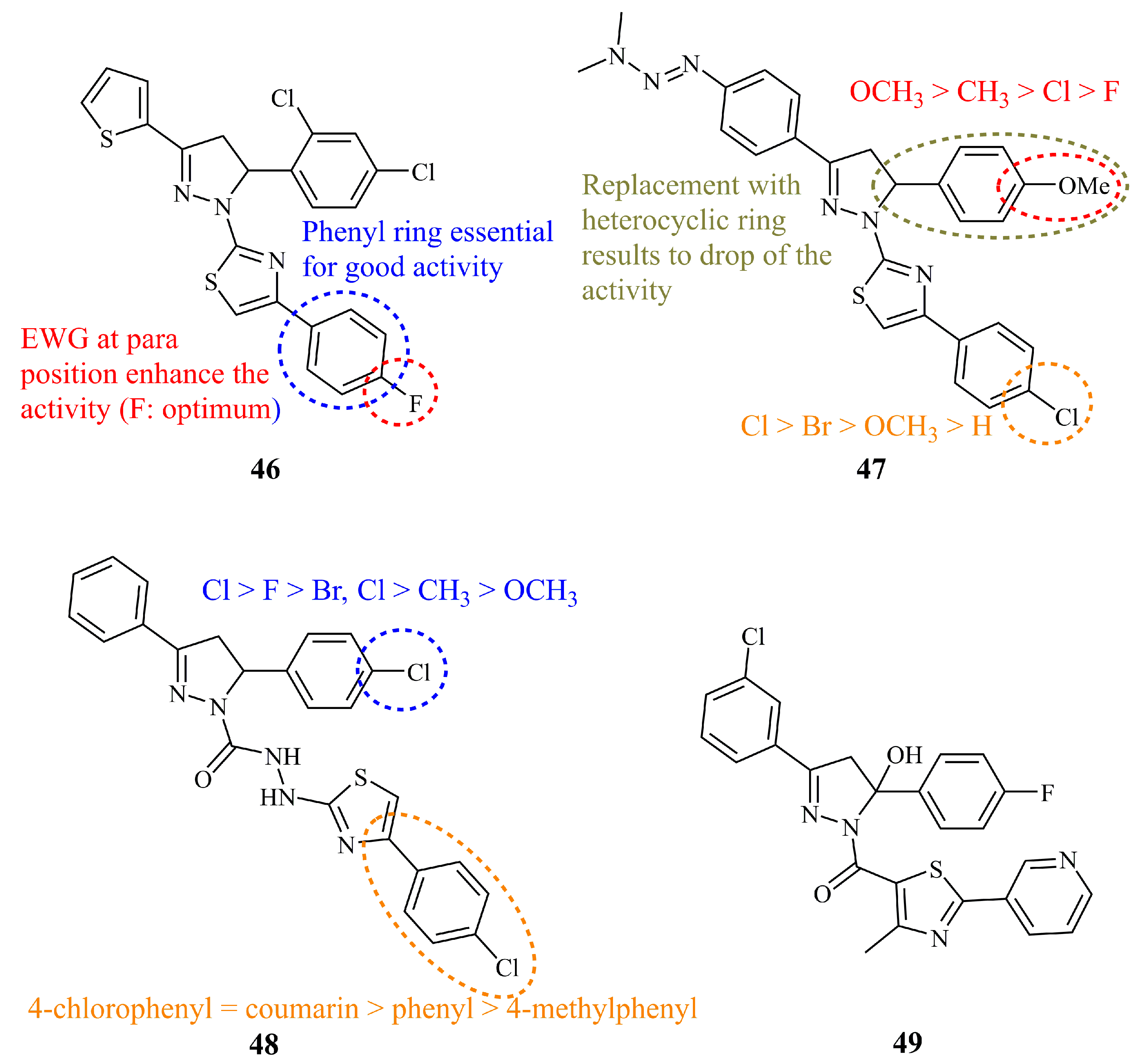{kind=link}
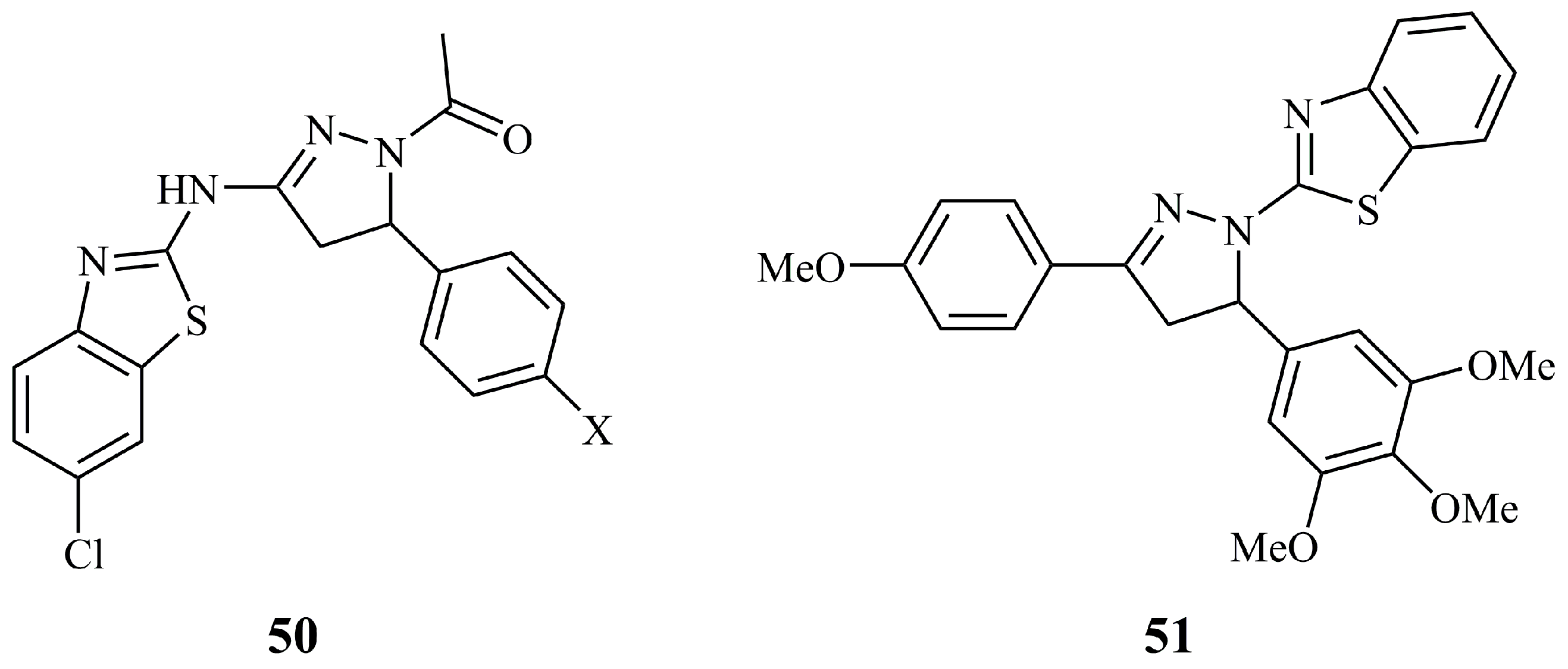{kind=link}
{kind=link}
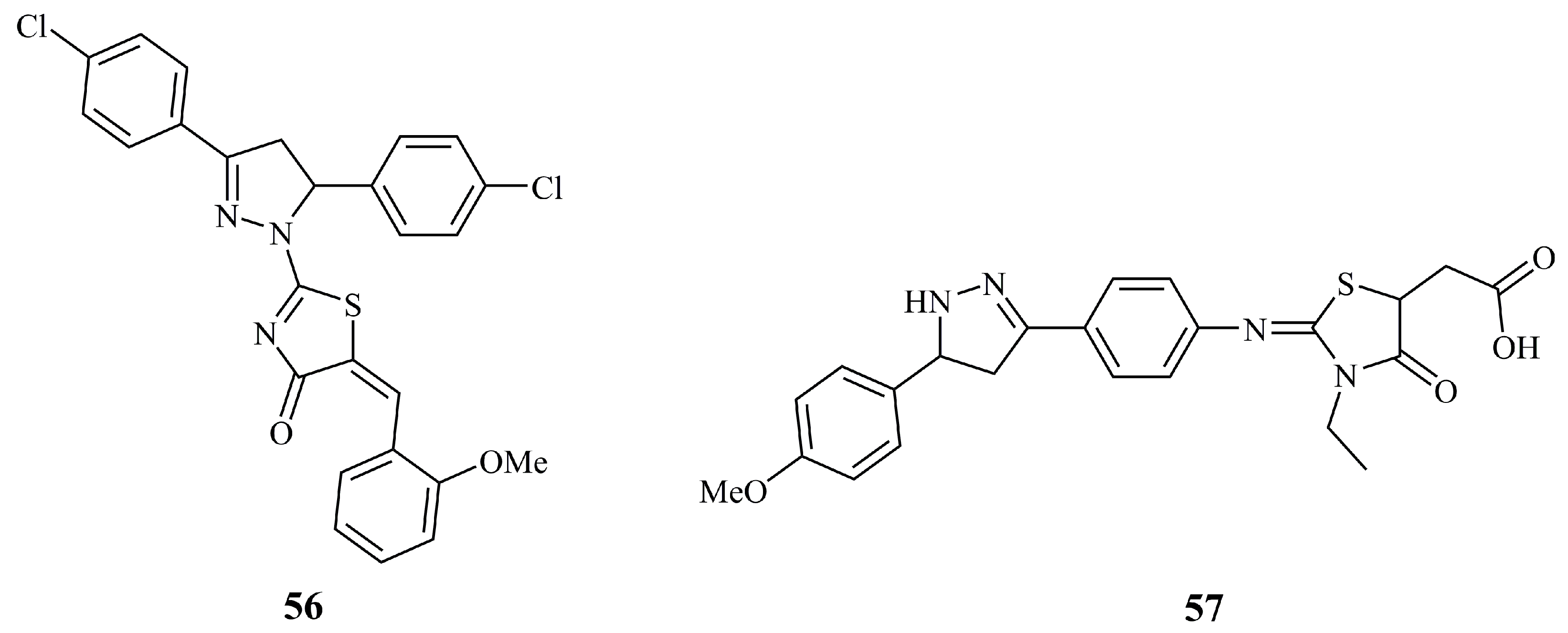{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





















© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matiadis, D.; Sagnou, M. Pyrazoline Hybrids as Promising Anticancer Agents: An Up-to-Date Overview. Int. J. Mol. Sci. 2020, 21, 5507. https://doi.org/10.3390/ijms21155507
Matiadis D, Sagnou M. Pyrazoline Hybrids as Promising Anticancer Agents: An Up-to-Date Overview. International Journal of Molecular Sciences. 2020; 21(15):5507. https://doi.org/10.3390/ijms21155507
Chicago/Turabian StyleMatiadis, Dimitris, and Marina Sagnou. 2020. "Pyrazoline Hybrids as Promising Anticancer Agents: An Up-to-Date Overview" International Journal of Molecular Sciences 21, no. 15: 5507. https://doi.org/10.3390/ijms21155507
APA StyleMatiadis, D., & Sagnou, M. (2020). Pyrazoline Hybrids as Promising Anticancer Agents: An Up-to-Date Overview. International Journal of Molecular Sciences, 21(15), 5507. https://doi.org/10.3390/ijms21155507