Inhibition of Colony-Stimulating Factor 1 Receptor by PLX3397 Prevents Amyloid Beta Pathology and Rescues Dopaminergic Signaling in Aging 5xFAD Mice

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
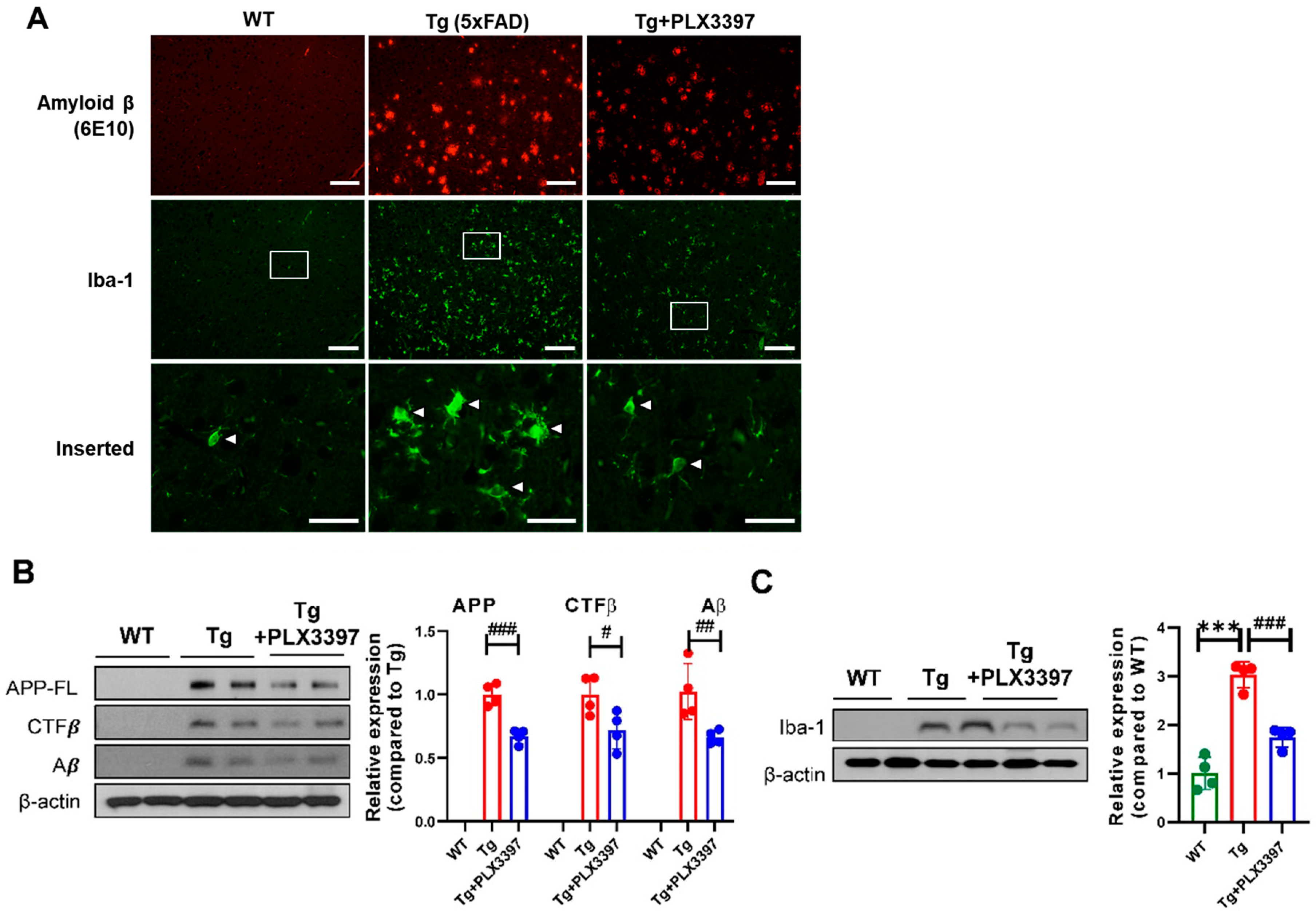
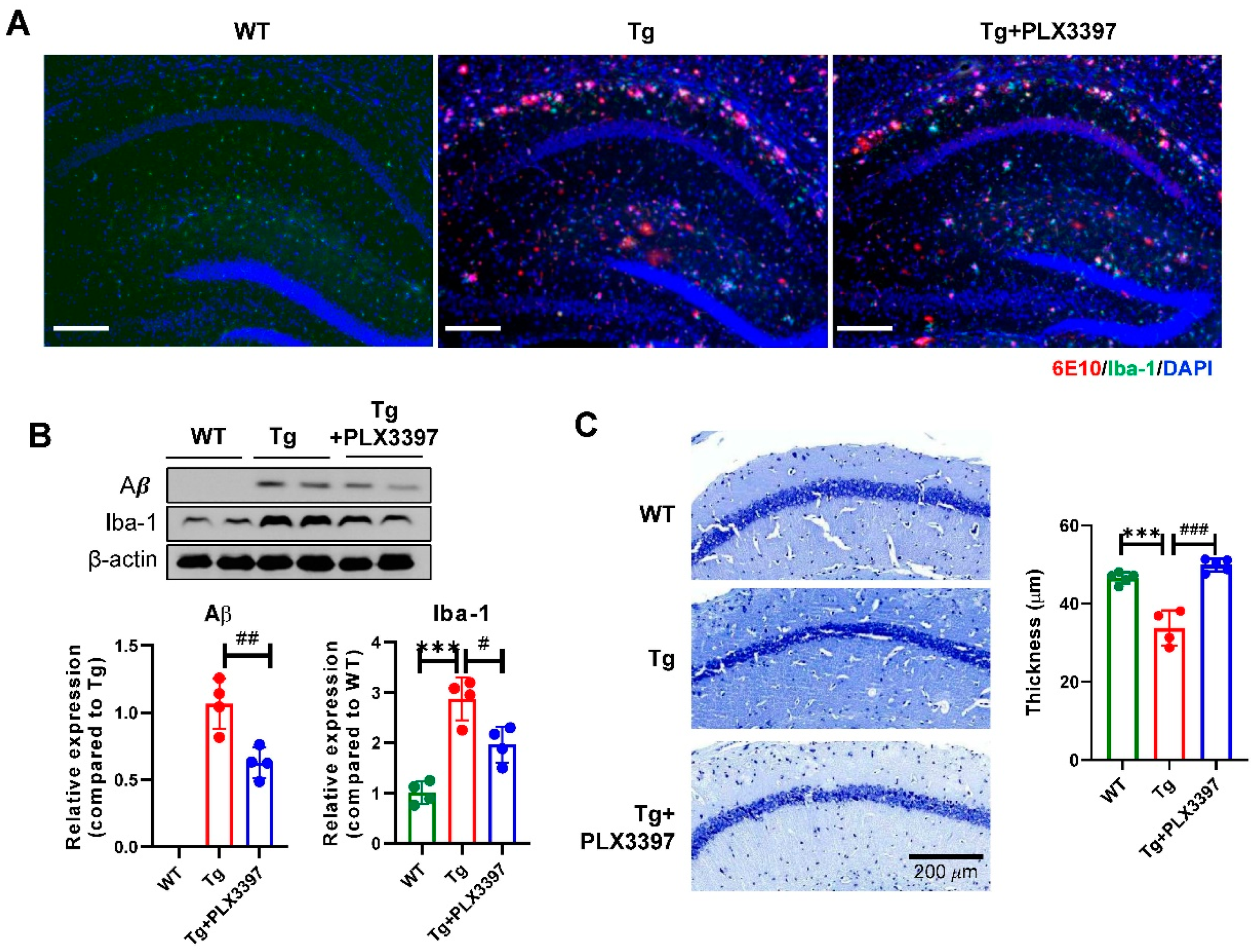
2.1. PLX3397 Alleviated Aβ Pathology in the Cortex and Hippocampus of Aged Tg Mice
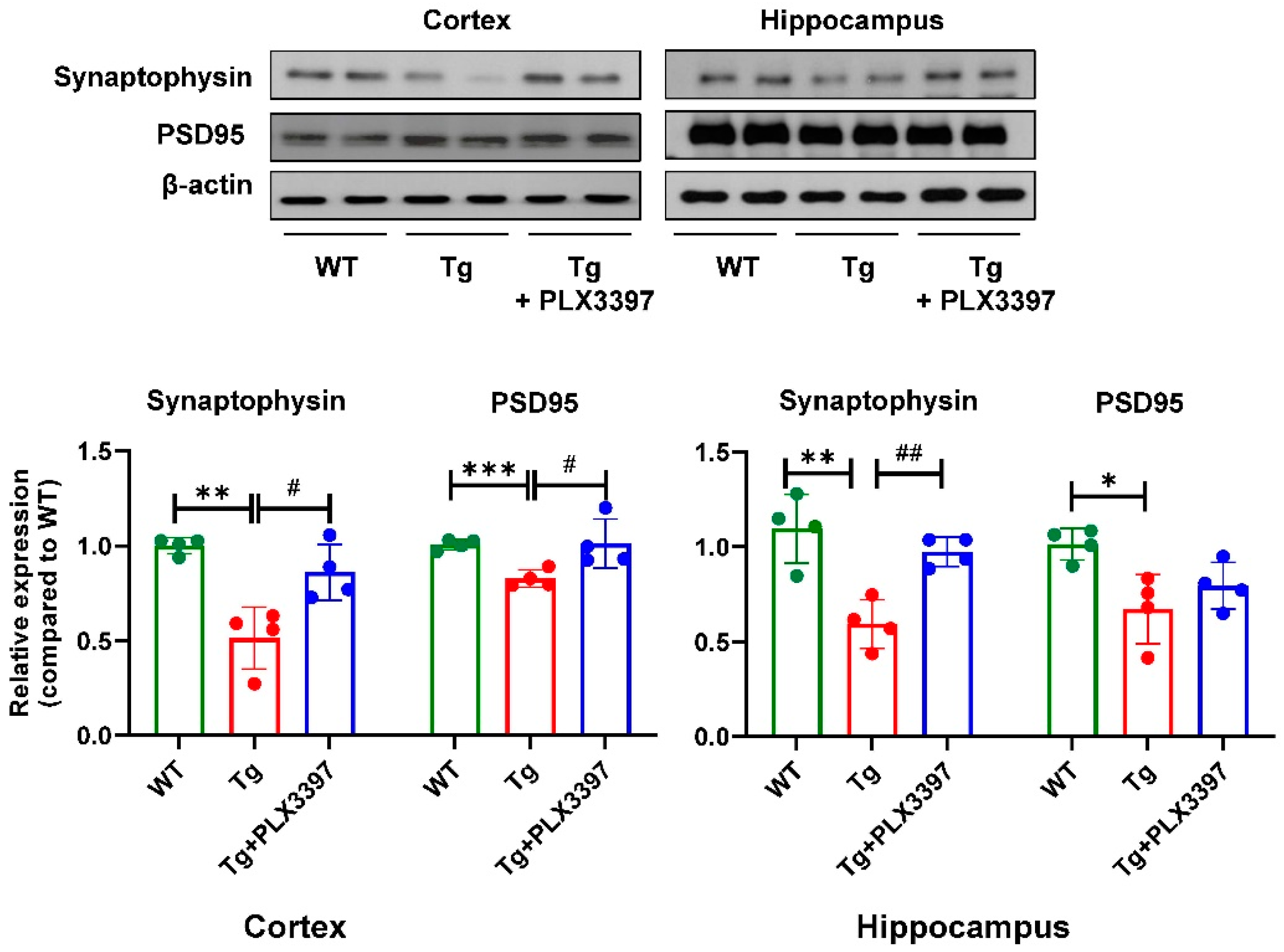
2.2. PLX3397 Increased Synapse-Related Protein Levels in 5xFAD Mice
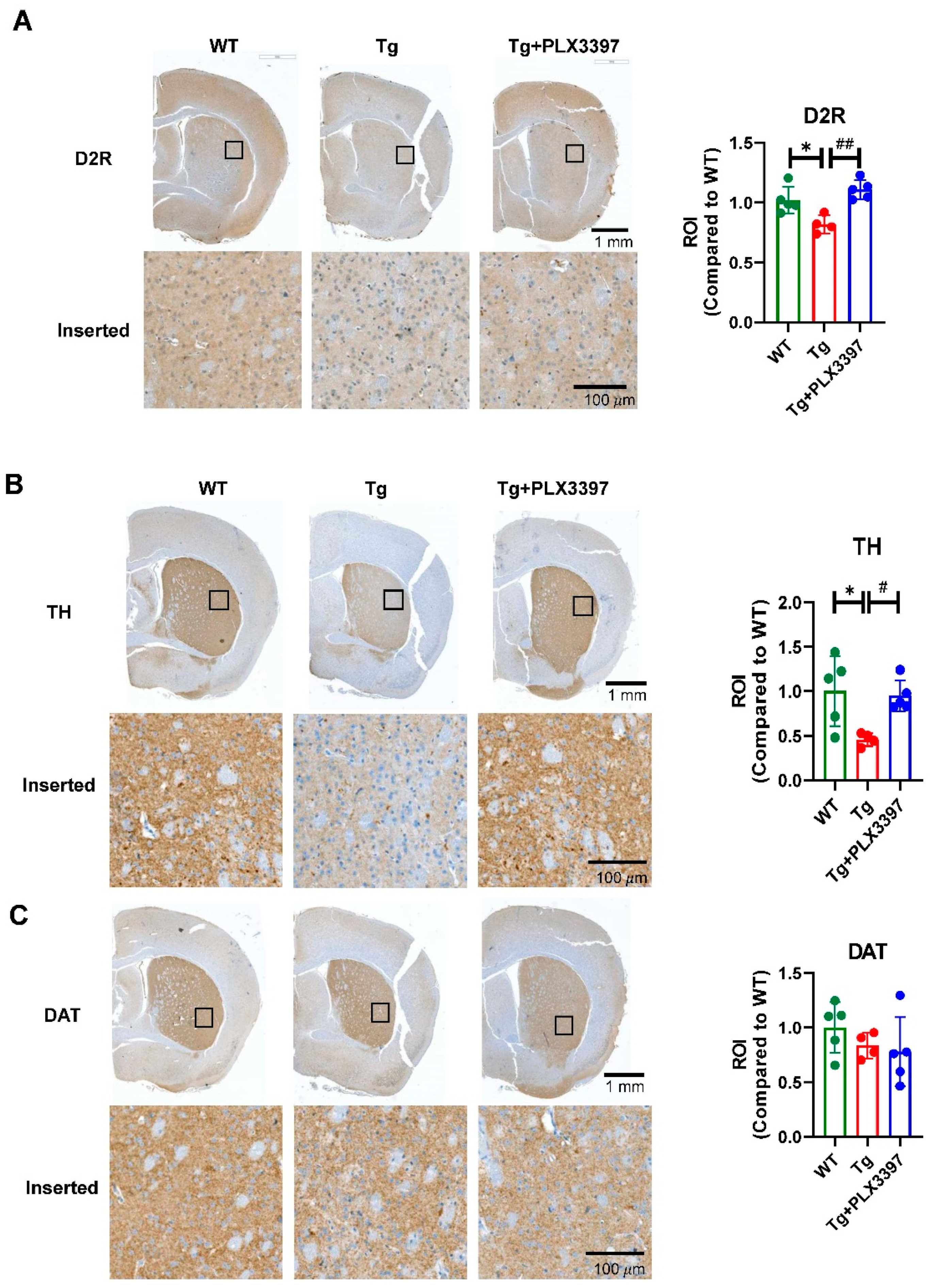
2.3. PLX3397 Protected the dopamine (DA) System in Aged 5xFAD Mice
3. Discussion
4. Materials and Methods
4.1. Animals and Drug Administration
4.2. Immunofluorescence Staining
4.3. Immunohistochemistry
4.4. Cresyl Violet Staining
4.5. Western Blotting
4.6. PET Scans
4.7. PET Image Analysis
4.8. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- A DeTure, M.; Dickson, D.W. The Neuropathological Diagnosis of Alzheimer’s Disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsia, A.Y.; Masliah, E.; McConlogue, L.; Yu, G.Q.; Tatsuno, G.; Hu, K.; Kholodenko, D.; Malenka, R.C.; Nicoll, R.A.; Mucke, L. Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc. Natl. Acad. Sci. USA 1999, 96, 3228–3233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folch, J.; Petrov, D.; Ettcheto, M.; Abad, S.; Sanchez-Lopez, E.; Garcia, M.L.; Olloquequi, J.; Beas-Zarate, C.; Auladell, C.; Camins, A. Current Research Therapeutic Strategies for Alzheimer’s Disease Treatment. Neural Plast. 2016, 2016, 8501693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.; Corbett, A.; Ballard, C. Emerging treatments for Alzheimer’s disease for non-amyloid and non-tau targets. Expert Rev. Neurother. 2017, 17, 683–695. [Google Scholar] [CrossRef]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef]
- Dani, M.; Wood, M.; Mizoguchi, R.; Fan, Z.; Walker, Z.; Morgan, R.; Hinz, R.; Biju, M.; Kuruvilla, T.; Brooks, D.J.; et al. Microglial activation correlates in vivo with both tau and amyloid in Alzheimer’s disease. Brain J. Neurol. 2018, 141, 2740–2754. [Google Scholar] [CrossRef] [PubMed]
- Majerova, P.; Zilkova, M.; Kazmerova, Z.; Kovac, A.; Paholikova, K.; Kovacech, B.; Zilka, N.; Novak, M. Microglia display modest phagocytic capacity for extracellular tau oligomers. J. Neuroinflammation 2014, 11, 161. [Google Scholar] [CrossRef] [Green Version]
- Perlmutter, L.S.; Barron, E.; Chui, H.C. Morphologic association between microglia and senile plaque amyloid in Alzheimer’s disease. Neurosci. Lett. 1990, 119, 32–36. [Google Scholar] [CrossRef]
- Sarlus, H.; Heneka, M.T. Microglia in Alzheimer’s disease. J. Clin. Invest. 2017, 127, 3240–3249. [Google Scholar] [CrossRef]
- Rivera-Escalera, F.; Pinney, J.J.; Owlett, L.; Ahmed, H.; Thakar, J.; Olschowka, J.A.; Elliott, M.R.; O’Banion, M.K. IL-1beta-driven amyloid plaque clearance is associated with an expansion of transcriptionally reprogrammed microglia. J. Neuroinflammation 2019, 16, 261. [Google Scholar] [CrossRef]
- Sosna, J.; Philipp, S.; Albay, R., 3rd; Reyes-Ruiz, J.M.; Baglietto-Vargas, D.; LaFerla, F.M.; Glabe, C.G. Early long-term administration of the CSF1R inhibitor PLX3397 ablates microglia and reduces accumulation of intraneuronal amyloid, neuritic plaque deposition and pre-fibrillar oligomers in 5XFAD mouse model of Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 11. [Google Scholar] [CrossRef] [PubMed]
- Spangenberg, E.E.; Lee, R.J.; Najafi, A.R.; Rice, R.A.; Elmore, M.R.; Blurton-Jones, M.; West, B.L.; Green, K.N. Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-beta pathology. Brain J. Neurol. 2016, 139, 1265–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmore, M.R.; Najafi, A.R.; Koike, M.A.; Dagher, N.N.; Spangenberg, E.E.; Rice, R.A.; Kitazawa, M.; Matusow, B.; Nguyen, H.; West, B.L.; et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 2014, 82, 380–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannarile, M.A.; Weisser, M.; Jacob, W.; Jegg, A.M.; Ries, C.H.; Ruttinger, D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J. Immunother. Cancer 2017, 5, 53. [Google Scholar] [CrossRef] [PubMed]
- Kaur, D.; Sharma, V.; Deshmukh, R. Activation of microglia and astrocytes: A roadway to neuroinflammation and Alzheimer’s disease. Inflammopharmacology 2019, 27, 663–677. [Google Scholar] [CrossRef]
- Li, J.W.; Zong, Y.; Cao, X.P.; Tan, L.; Tan, L. Microglial priming in Alzheimer’s disease. Annu. Transl. Med. 2018, 6, 176. [Google Scholar] [CrossRef]
- Spangenberg, E.; Severson, P.L.; Hohsfield, L.A.; Crapser, J.; Zhang, J.; Burton, E.A.; Zhang, Y.; Spevak, W.; Lin, J.; Phan, N.Y.; et al. Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat. Commun. 2019, 10, 3758. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef] [Green Version]
- Almeida, C.G.; Tampellini, D.; Takahashi, R.H.; Greengard, P.; Lin, M.T.; Snyder, E.M.; Gouras, G.K. Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol. Dis. 2005, 20, 187–198. [Google Scholar] [CrossRef]
- Revilla, S.; Sunol, C.; Garcia-Mesa, Y.; Gimenez-Llort, L.; Sanfeliu, C.; Cristofol, R. Physical exercise improves synaptic dysfunction and recovers the loss of survival factors in 3xTg-AD mouse brain. Neuropharmacology 2014, 81, 55–63. [Google Scholar] [CrossRef]
- Collingridge, G.L.; Singer, W. Excitatory amino acid receptors and synaptic plasticity. Trends Pharmacol. Sci. 1990, 11, 290–296. [Google Scholar] [CrossRef]
- Ferreira, S.T.; Klein, W.L. The Abeta oligomer hypothesis for synapse failure and memory loss in Alzheimer’s disease. Neurobiol. Learn. Mem. 2011, 96, 529–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Revett, T.J.; Baker, G.B.; Jhamandas, J.; Kar, S. Glutamate system, amyloid ss peptides and tau protein: Functional interrelationships and relevance to Alzheimer disease pathology. J. Psychiatry Neurosci. 2013, 38, 6–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donthamsetti, P.; Gallo, E.F.; Buck, D.C.; Stahl, E.L.; Zhu, Y.; Lane, J.R.; Bohn, L.M.; Neve, K.A.; Kellendonk, C.; Javitch, J.A. Arrestin recruitment to dopamine D2 receptor mediates locomotion but not incentive motivation. Mol. Psychiatry 2018. [Google Scholar] [CrossRef] [PubMed]
- Vorobyov, V.; Bakharev, B.; Medvinskaya, N.; Nesterova, I.; Samokhin, A.; Deev, A.; Tatarnikova, O.; Ustyugov, A.; Sengpiel, F.; Bobkova, N. Loss of Midbrain Dopamine Neurons and Altered Apomorphine EEG Effects in the 5xFAD Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2019, 70, 241–256. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.; Lee, H.J.; Jeong, Y.J.; Oh, S.J.; Kang, K.J.; Han, S.J.; Nam, K.R.; Lee, Y.J.; Lee, K.C.; Ryu, Y.H.; et al. Age dependency of mGluR5 availability in 5xFAD mice measured by PET. Neurobiol. Aging 2019, 84, 208–216. [Google Scholar] [CrossRef]
- Michel, P.P.; Hirsch, E.C.; Hunot, S. Understanding Dopaminergic Cell Death Pathways in Parkinson Disease. Neuron 2016, 90, 675–691. [Google Scholar] [CrossRef] [Green Version]
- Nardone, R.; Höller, Y.; Thomschewski, A.; Kunz, A.B.; Lochner, P.; Golaszewski, S.; Trinka, E.; Brigo, F. Dopamine differently modulates central cholinergic circuits in patients with Alzheimer disease and CADASIL. J. Neural. Transm. (Vienna) 2014, 121, 1313–1320. [Google Scholar]
- Nam, E.; Derrick, J.S.; Lee, S.; Kang, J.; Han, J.; Lee, S.J.C.; Chung, S.W.; Lim, M.H. Regulatory Activities of Dopamine and Its Derivatives toward Metal-Free and Metal-Induced Amyloid-beta Aggregation, Oxidative Stress, and Inflammation in Alzheimer’s Disease. ACS Chem. Neurosci. 2018, 9, 2655–2666. [Google Scholar] [CrossRef]
- Son, Y.; Kim, J.S.; Jeong, Y.J.; Jeong, Y.K.; Kwon, J.H.; Choi, H.D.; Pack, J.K.; Kim, N.; Lee, Y.S.; Lee, H.J. Long-term RF exposure on behavior and cerebral glucose metabolism in 5xFAD mice. Neurosci. Lett. 2018, 666, 64–69. [Google Scholar] [CrossRef]
- Manji, Z.; Rojas, A.; Wang, W.; Dingledine, R.; Varvel, N.H.; Ganesh, T. 5xFAD Mice Display Sex-Dependent Inflammatory Gene Induction During the Prodromal Stage of Alzheimer’s Disease. J. Alzheimer’s Dis. 2019, 70, 1259–1274. [Google Scholar] [CrossRef] [PubMed]
- Sadleir, K.R.; Popovic, J.; Vassar, R. ER stress is not elevated in the 5XFAD mouse model of Alzheimer’s disease. J. Biol. Chem. 2018, 293, 18434–18443. [Google Scholar] [CrossRef] [Green Version]
- Moon, B.S.; Park, J.H.; Lee, H.J.; Kim, J.S.; Kil, H.S.; Lee, B.S.; Chi, D.Y.; Lee, B.C.; Kim, Y.K.; Kim, S.E. Highly efficient production of [(18)F]fallypride using small amounts of base concentration. Appl. Radiat. Isot. 2010, 68, 2279–2284. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Lee, H.J.; Park, I.S.; Park, J.A.; Kwon, Y.J.; Ryu, Y.H.; Kim, C.H.; Kang, J.H.; Hyun, I.Y.; Lee, K.C.; et al. Abeta pathology downregulates brain mGluR5 density in a mouse model of Alzheimer. Neuropharmacology 2018, 133, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Boja, J.W.; Mitchell, W.M.; Patel, A.; Kopajtic, T.A.; Carroll, F.I.; Lewin, A.H.; Abraham, P.; Kuhar, M.J. High-affinity binding of [125I]RTI-55 to dopamine and serotonin transporters in rat brain. Synapse 1992, 12, 27–36. [Google Scholar] [CrossRef]
- Cline, E.J.; Scheffel, U.; Boja, J.W.; Mitchell, W.M.; Carroll, F.I.; Abraham, P.; Lewin, A.H.; Kuhar, M.J. In vivo binding of [125I]RTI-55 to dopamine transporters: Pharmacology and regional distribution with autoradiography. Synapse 1992, 12, 37–46. [Google Scholar] [CrossRef]
- Elmenhorst, D.; Minuzzi, L.; Aliaga, A.; Rowley, J.; Massarweh, G.; Diksic, M.; Bauer, A.; Rosa-Neto, P. In vivo and in vitro validation of reference tissue models for the mGluR(5) ligand [(11)C]ABP688. J. Cereb. Blood Flow Metab. 2010, 30, 1538–1549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shigemoto, R.; Nomura, S.; Ohishi, H.; Sugihara, H.; Nakanishi, S.; Mizuno, N. Immunohistochemical localization of a metabotropic glutamate receptor, mGluR5, in the rat brain. Neurosci. Lett. 1993, 163, 53–57. [Google Scholar] [CrossRef]
- Logan, J.; Fowler, J.S.; Volkow, N.D.; Wolf, A.P.; Dewey, S.L.; Schlyer, D.J.; MacGregor, R.R.; Hitzemann, R.; Bendriem, B.; Gatley, S.J.; et al. Graphical analysis of reversible radioligand binding from time-activity measurements applied to [N-11C-methyl]-(-)-cocaine PET studies in human subjects. J. Cereb. Blood Flow Metab. 1990, 10, 740–747. [Google Scholar] [CrossRef] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Son, Y.; Jeong, Y.J.; Shin, N.-R.; Oh, S.J.; Nam, K.R.; Choi, H.-D.; Choi, J.Y.; Lee, H.-J. Inhibition of Colony-Stimulating Factor 1 Receptor by PLX3397 Prevents Amyloid Beta Pathology and Rescues Dopaminergic Signaling in Aging 5xFAD Mice. Int. J. Mol. Sci. 2020, 21, 5553. https://doi.org/10.3390/ijms21155553
Son Y, Jeong YJ, Shin N-R, Oh SJ, Nam KR, Choi H-D, Choi JY, Lee H-J. Inhibition of Colony-Stimulating Factor 1 Receptor by PLX3397 Prevents Amyloid Beta Pathology and Rescues Dopaminergic Signaling in Aging 5xFAD Mice. International Journal of Molecular Sciences. 2020; 21(15):5553. https://doi.org/10.3390/ijms21155553
Chicago/Turabian StyleSon, Yeonghoon, Ye Ji Jeong, Na-Rae Shin, Se Jong Oh, Kyung Rok Nam, Hyung-Do Choi, Jae Yong Choi, and Hae-June Lee. 2020. "Inhibition of Colony-Stimulating Factor 1 Receptor by PLX3397 Prevents Amyloid Beta Pathology and Rescues Dopaminergic Signaling in Aging 5xFAD Mice" International Journal of Molecular Sciences 21, no. 15: 5553. https://doi.org/10.3390/ijms21155553
APA StyleSon, Y., Jeong, Y. J., Shin, N. -R., Oh, S. J., Nam, K. R., Choi, H. -D., Choi, J. Y., & Lee, H. -J. (2020). Inhibition of Colony-Stimulating Factor 1 Receptor by PLX3397 Prevents Amyloid Beta Pathology and Rescues Dopaminergic Signaling in Aging 5xFAD Mice. International Journal of Molecular Sciences, 21(15), 5553. https://doi.org/10.3390/ijms21155553