α1AMP-Activated Protein Kinase Protects against Lipopolysaccharide-Induced Endothelial Barrier Disruption via Junctional Reinforcement and Activation of the p38 MAPK/HSP27 Pathway

 , , ,
, , ,  ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
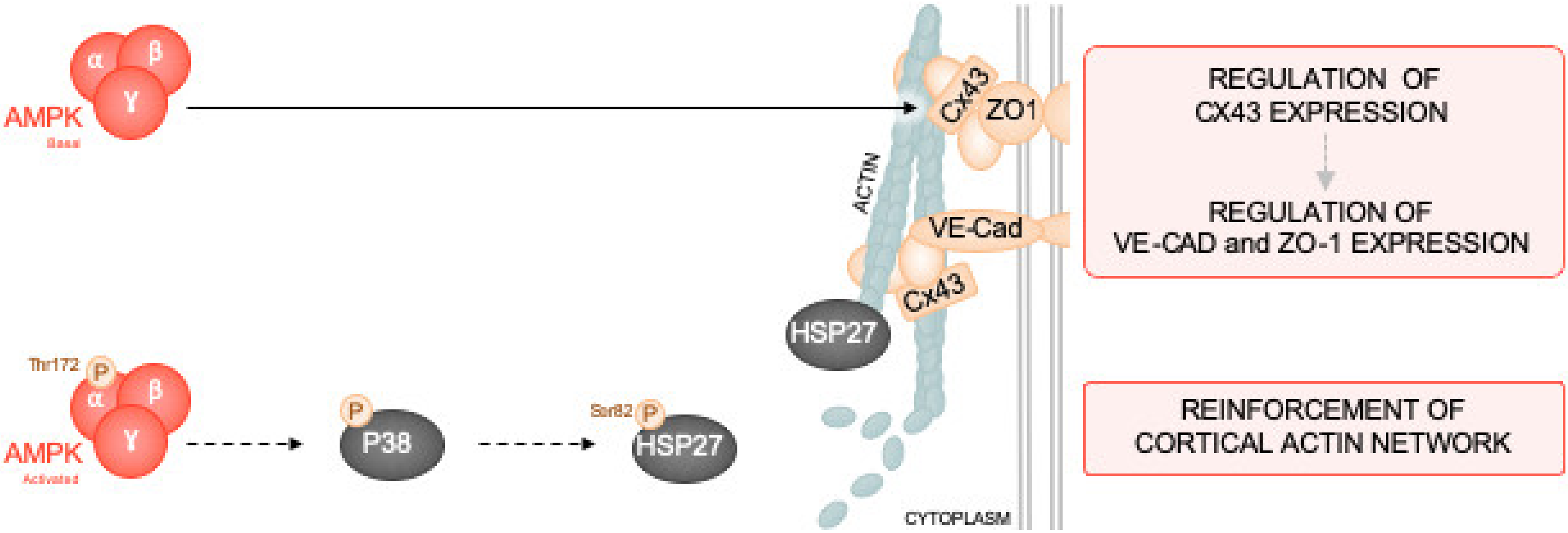
2.1. α1. AMPK Preserves IEJs Integrity
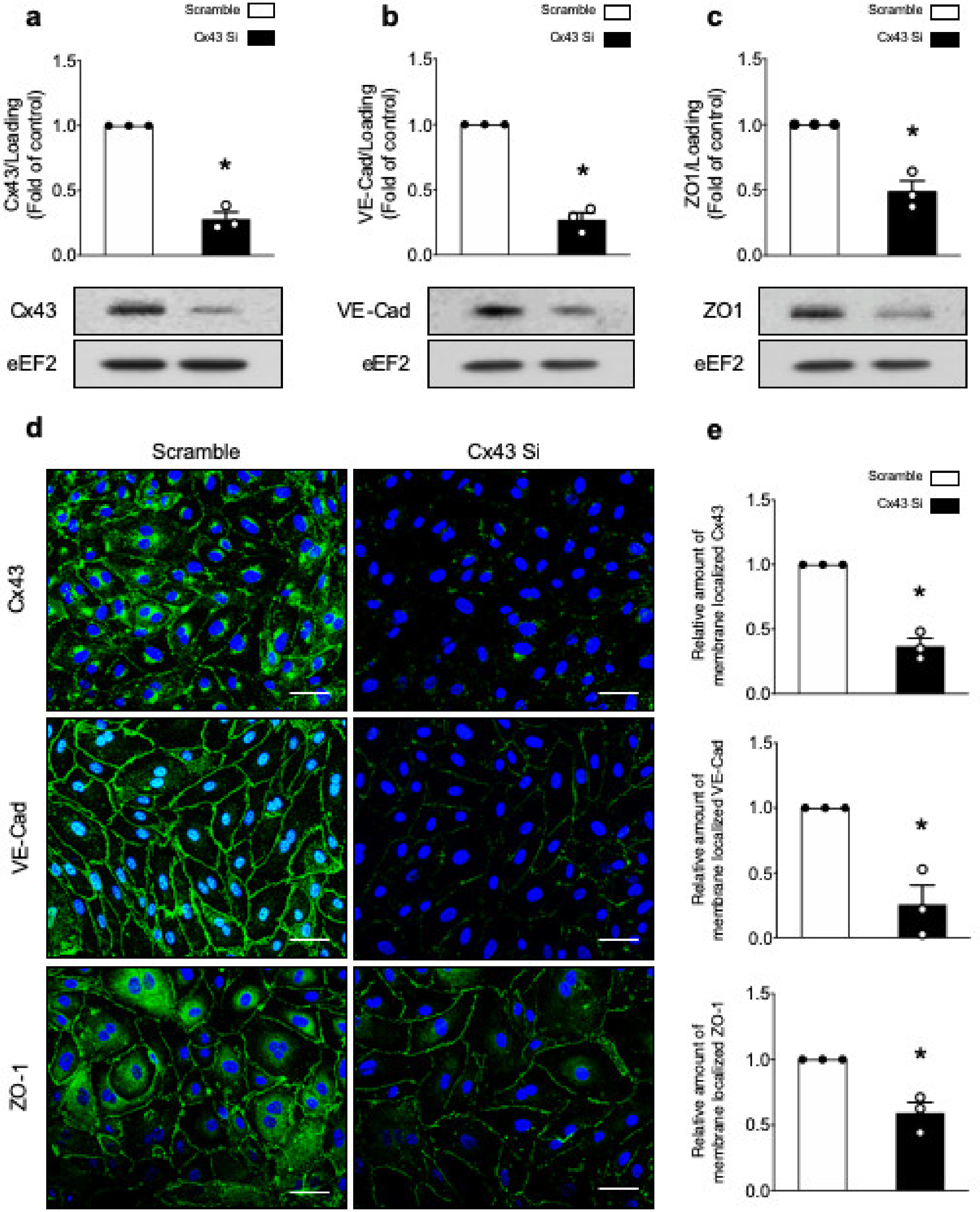
2.1.1. α1. AMPK Deficiency is Associated with Downregulation of VE-Cad, ZO-1, and Cx43 Expression
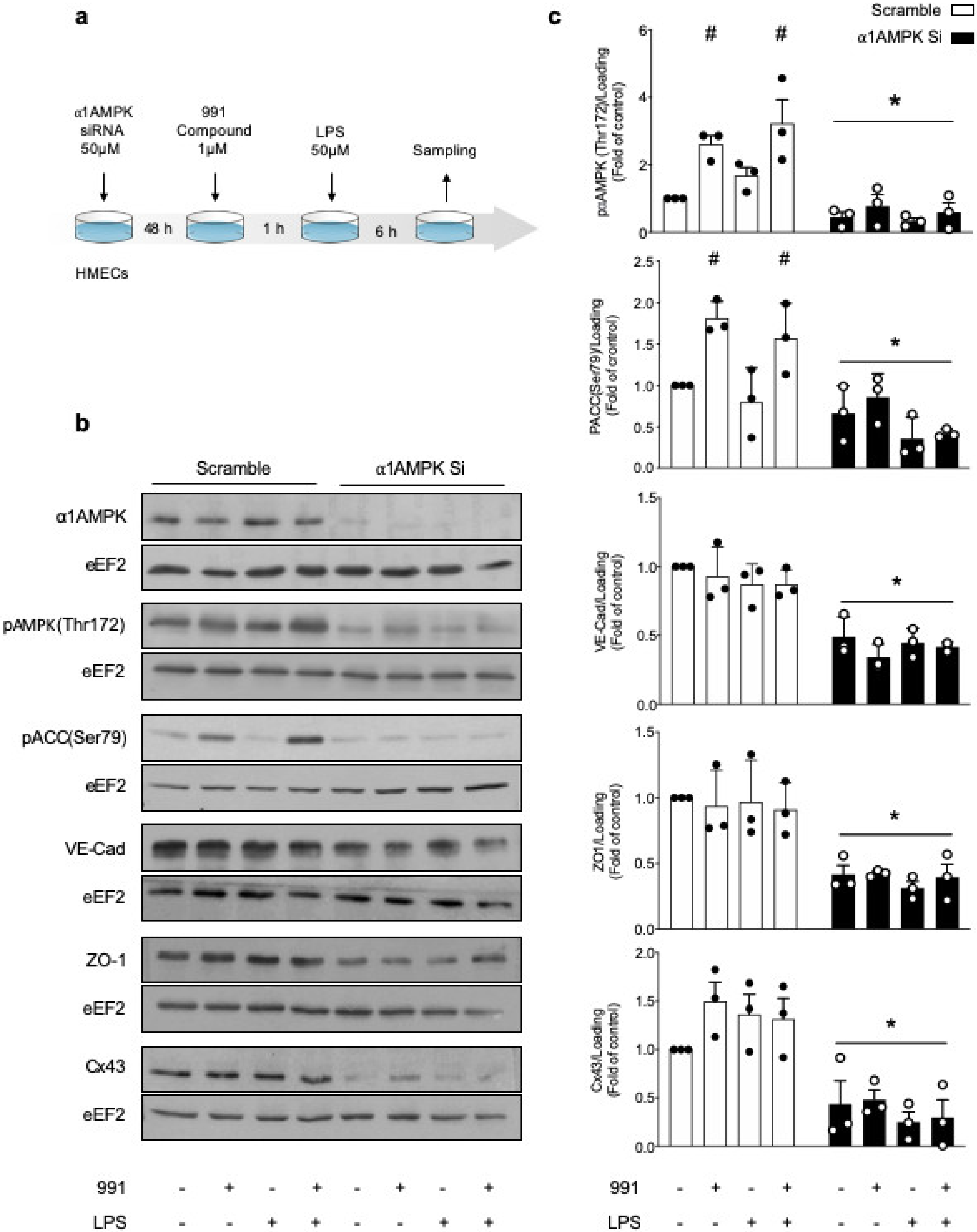
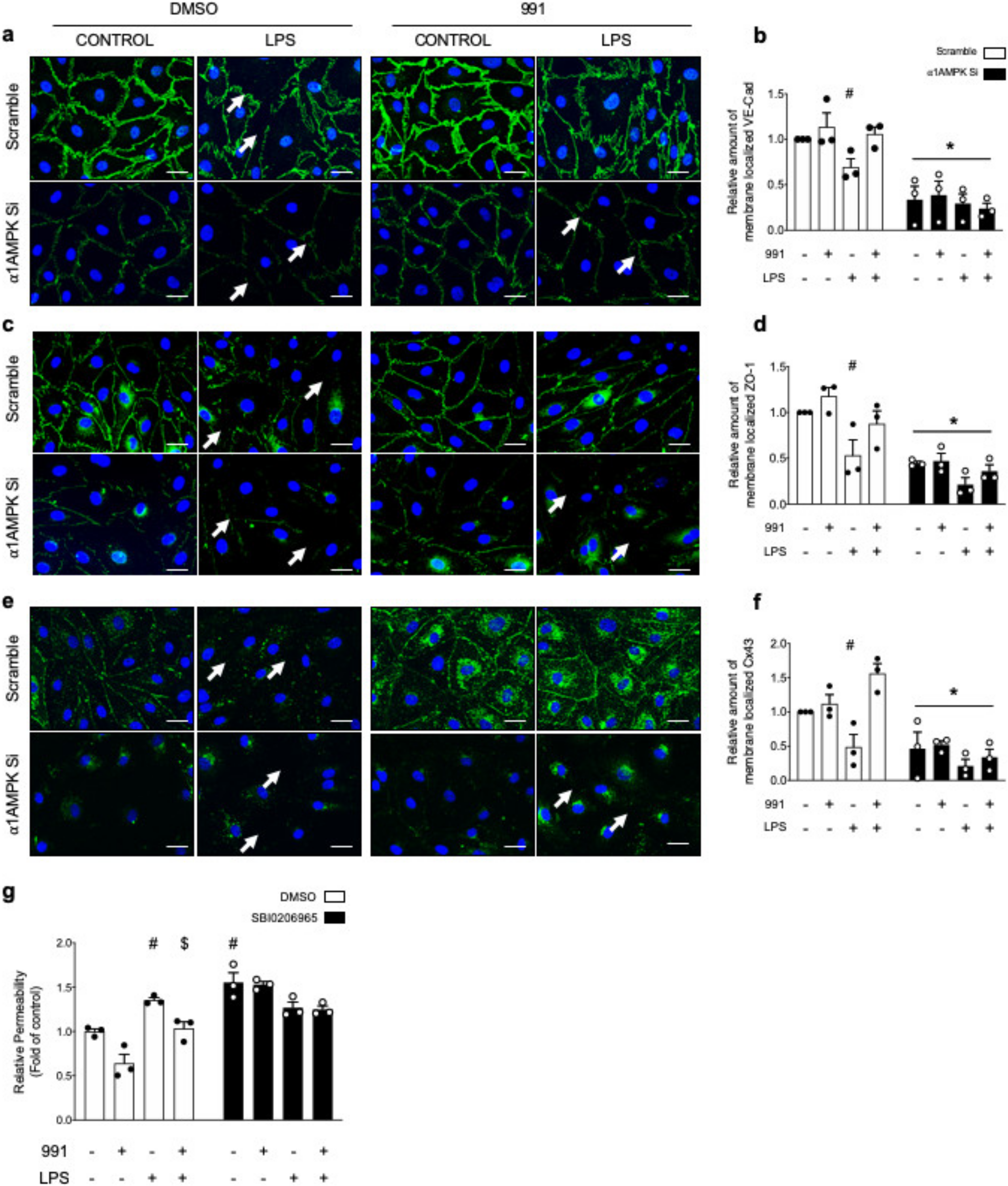
2.1.2. AMPK Activation Prevents LPS-Induced Disruption of IEJs and Endothelial Barrier Dysfunction
2.2. α1. AMPK Induces Actin Cytoskeleton Remodeling
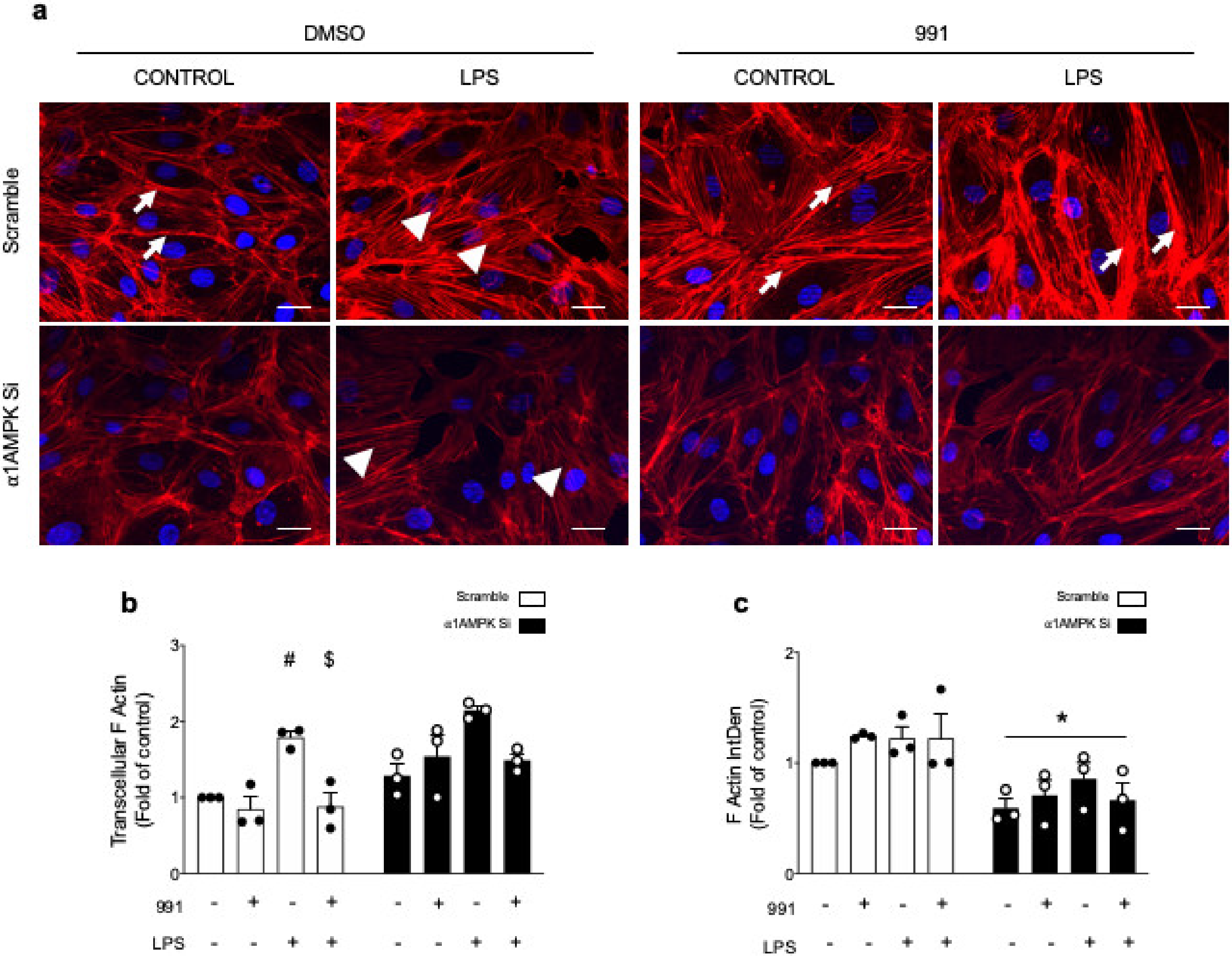
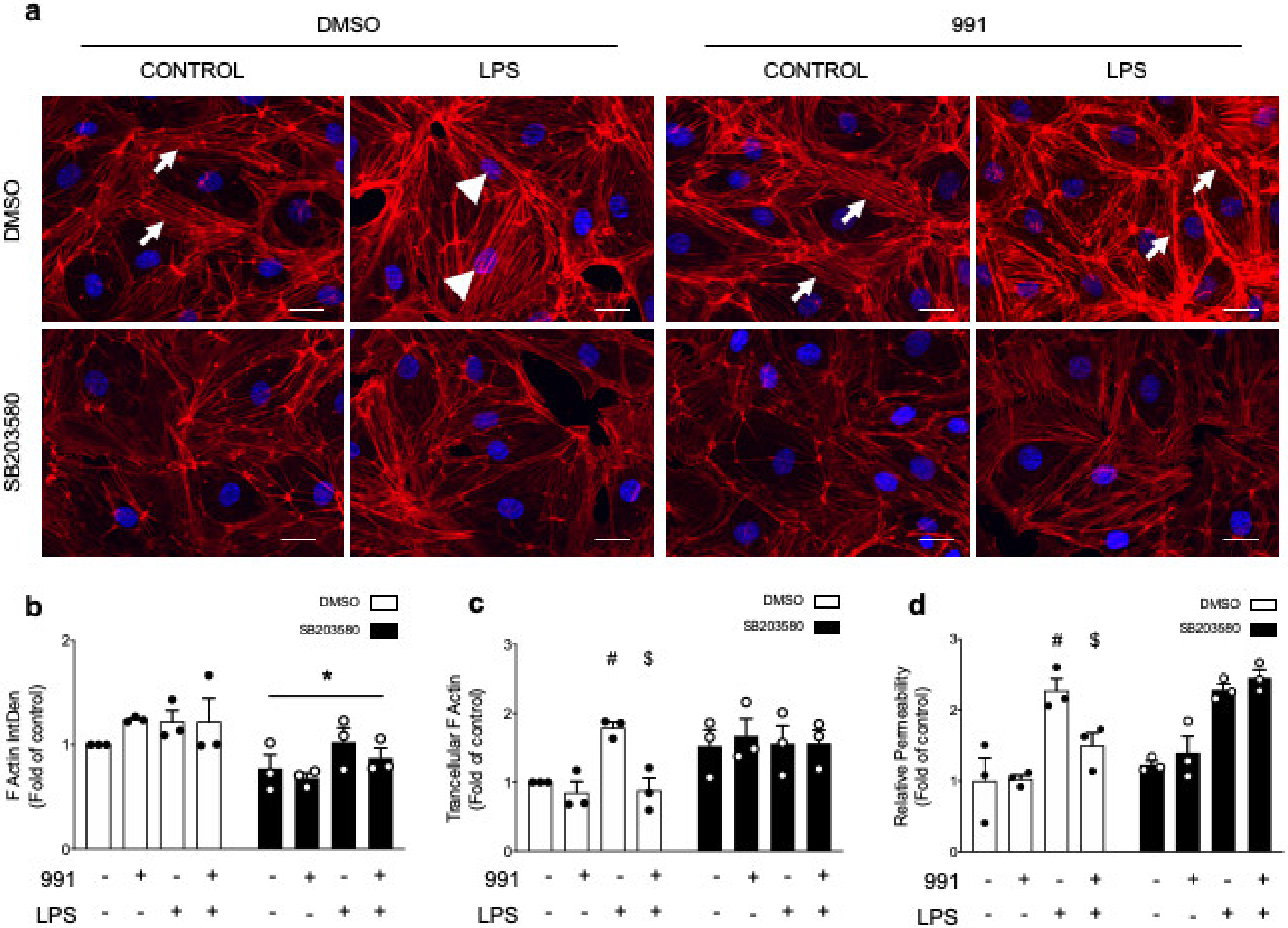
2.2.1. AMPK Activation Modulates Actin Organization and Polymerization
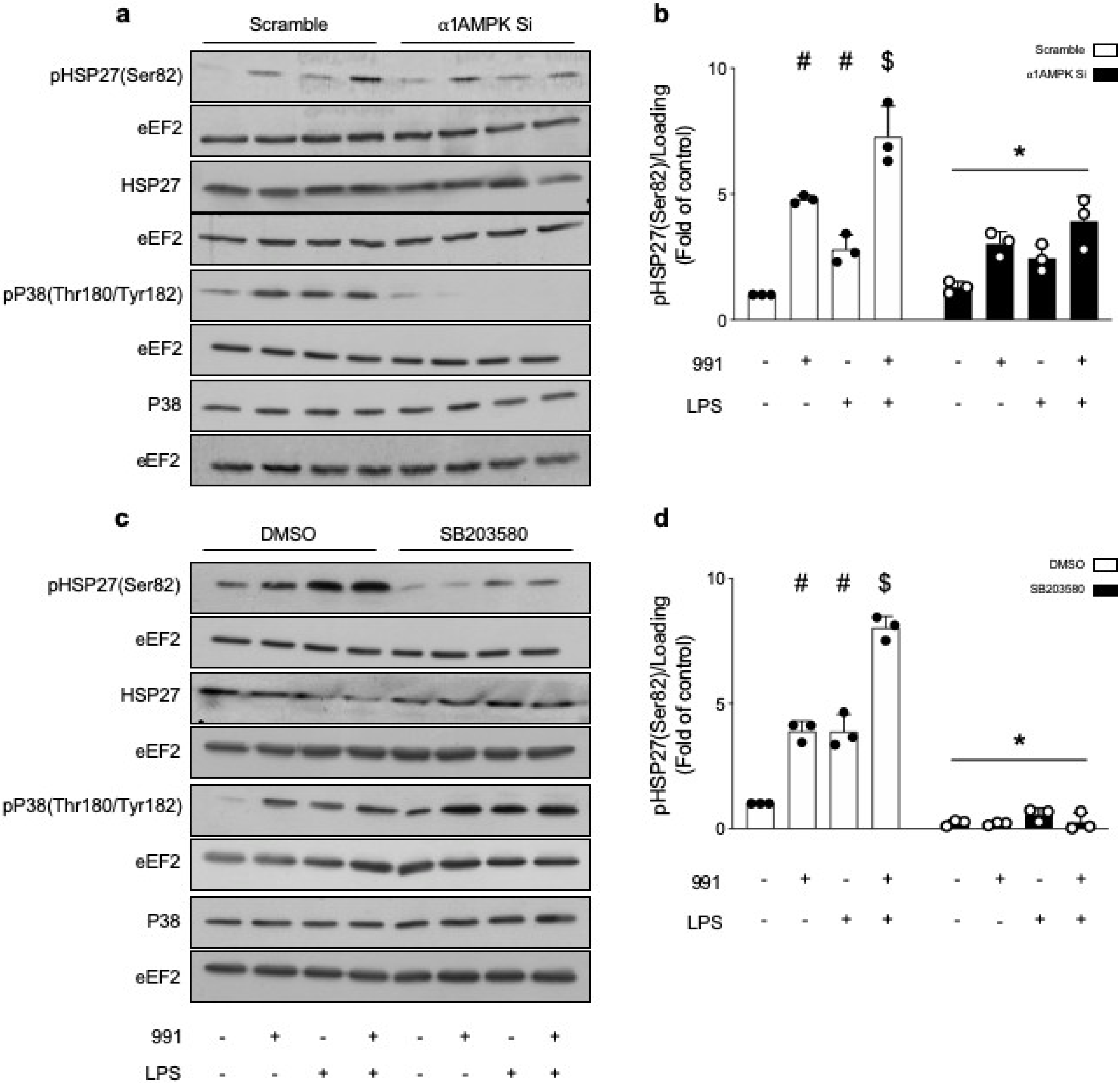
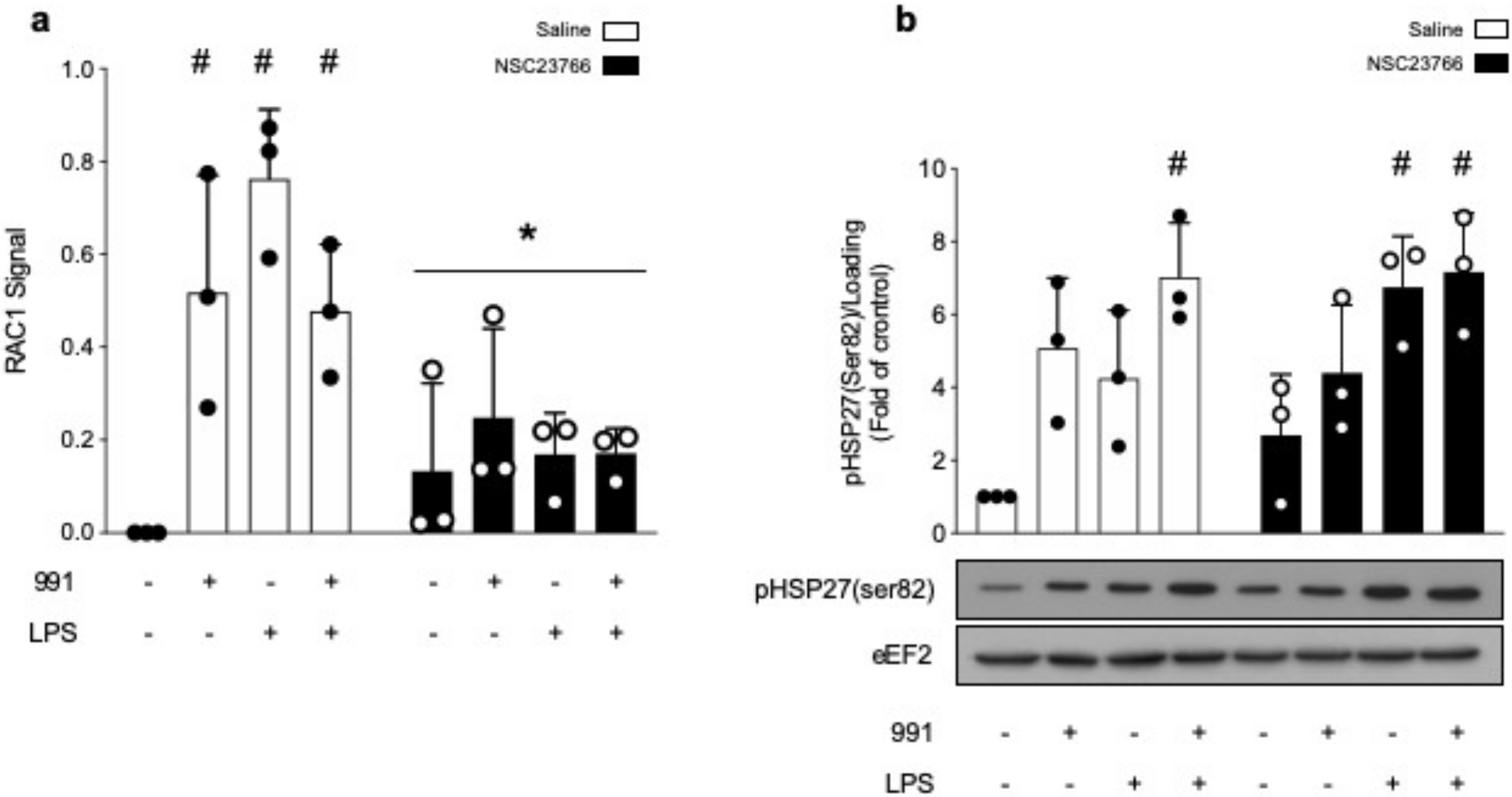
2.2.2. α1. AMPK Regulates Actin Polymerization via Activation of the p38 MAPK/HSP27 Pathway
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Culture and Treatments
4.3. siRNA Transfections
4.4. Western Blotting
4.5. Immunofluorescence Microscopy
4.6. Image Analysis
4.7. Endothelial Permeability Assessment
4.8. Small GTPases Activity Assay
4.9. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACC | Acetyl-CoA carboxylase |
| AICAr | 5-Aminoimidazole-4-carboxamide ribonucleotide |
| AJs | Adherens junctions |
| AMPK | AMP-activated protein kinase |
| Cx43 | Connexin 43 |
| eEF2 | Eukaryotic elongation factor 2 |
| GIV | G-alpha interacting vesicle associated protein |
| HMEC | Human dermal microvascular endothelial cell |
| HSP27 | Heat shock protein 27 |
| IEJs | Inter-endothelial junctions |
| LPS | Lipopolysaccharide |
| p38 MAPK | p38 mitogen-activated protein kinase |
| MAPKAPK | Mitogen-activated protein kinase -activated protein kinase |
| N-Cad | N-cadherin |
| siRNA | Small interfering RNA |
| TAB | Transforming growth factor a-activated kinase 1 binding proteins |
| TJs | Tight junctions |
| VE-Cad | VE-cadherin |
| ZO-1 | Zonula occludens-1 |
References
- Vincent, J.L.; Jones, G.; David, S.; Olariu, E.; Cadwell, K.K. Frequency and mortality of septic shock in Europe and North America: A systematic review and meta-analysis. Crit. Care 2019, 23, 196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincent, J.L.; Marshall, J.C.; Namendys-Silva, S.A.; Francois, B.; Martin-Loeches, I.; Lipman, J.; Reinhart, K.; Antonelli, M.; Pickkers, P.; Njimi, H.; et al. Assessment of the worldwide burden of critical illness: The intensive care over nations (ICON) audit. Lancet Respir. Med. 2014, 2, 380–386. [Google Scholar] [CrossRef]
- Lagu, T.; Rothberg, M.B.; Shieh, M.S.; Pekow, P.S.; Steingrub, J.S.; Lindenauer, P.K. Hospitalizations, costs, and outcomes of severe sepsis in the United States 2003 to 2007. Crit. Care Med. 2012, 40, 754–761. [Google Scholar] [CrossRef]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). Jama 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, F.L.; Bota, D.P.; Bross, A.; Melot, C.; Vincent, J.L. Serial evaluation of the SOFA score to predict outcome in critically ill patients. Jama 2001, 286, 1754–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakr, Y.; Dubois, M.J.; De Backer, D.; Creteur, J.; Vincent, J.L. Persistent microcirculatory alterations are associated with organ failure and death in patients with septic shock. Crit. Care Med. 2004, 32, 1825–1831. [Google Scholar] [CrossRef]
- Hou, P.C.; Filbin, M.R.; Wang, H.; Ngo, L.; Huang, D.T.; Aird, W.C.; Yealy, D.M.; Angus, D.C.; Kellum, J.A.; Shapiro, N.I. Endothelial Permeability and Hemostasis in Septic Shock: Results From the ProCESS Trial. Chest 2017, 152, 22–31. [Google Scholar] [CrossRef]
- Lelubre, C.; Vincent, J.L. Mechanisms and treatment of organ failure in sepsis. Nat. Rev. Nephrol. 2018, 14, 417–427. [Google Scholar] [CrossRef]
- Ratiani, L.; Gamkrelidze, M.; Khuchua, E.; Khutsishvili, T.; Intskirveli, N.; Vardosanidze, K. Altered Microcirculation in Septic Shock. Georgian Med. News 2015, 244–245, 16–24. [Google Scholar]
- Komarova, Y.A.; Kruse, K.; Mehta, D.; Malik, A.B. Protein Interactions at Endothelial Junctions and Signaling Mechanisms Regulating Endothelial Permeability. Circ. Res. 2017, 120, 179–206. [Google Scholar] [CrossRef] [Green Version]
- Garrett, J.P.; Lowery, A.M.; Adam, A.P.; Kowalczyk, A.P.; Vincent, P.A. Regulation of endothelial barrier function by p120-cateninVE-cadherin interaction. Mol. Biol. Cell 2017, 28, 85–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannotta, M.; Trani, M.; Dejana, E. VE-cadherin and endothelial adherens junctions: Active guardians of vascular integrity. Dev. Cell 2013, 26, 441–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trani, M.; Dejana, E. New insights in the control of vascular permeability: Vascular endothelial-cadherin and other players. Curr. Opin. Hematol. 2015, 22, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Dejana, E.; Vestweber, D. The role of VE-cadherin in vascular morphogenesis and permeability control. Prog. Mol. Biol. Transl. Sci. 2013, 116, 119–144. [Google Scholar] [PubMed]
- Soon, A.S.; Chua, J.W.; Becker, D.L. Connexins in endothelial barrier function-novel therapeutic targets countering vascular hyperpermeability. Thromb. Haemost. 2016, 116, 852–867. [Google Scholar] [CrossRef] [Green Version]
- Martins-Marques, T.; Ribeiro-Rodrigues, T.; Batista-Almeida, D.; Aasen, T.; Kwak, B.R.; Girao, H. Biological Functions of Connexin43 Beyond Intercellular Communication. Trends Cell Biol. 2019, 29, 835–847. [Google Scholar] [CrossRef]
- Chen, C.H.; Mayo, J.N.; Gourdie, R.G.; Johnstone, S.R.; Isakson, B.E.; Bearden, S.E. The connexin 43/ZO-1 complex regulates cerebral endothelial F-actin architecture and migration. Am. J. Physiol. Cell Physiol. 2015, 309, C600–C607. [Google Scholar] [CrossRef] [Green Version]
- Herve, J.C.; Derangeon, M.; Sarrouilhe, D.; Giepmans, B.N.; Bourmeyster, N. Gap junctional channels are parts of multiprotein complexes. Biochim. Biophys. Acta 2012, 1818, 1844–1865. [Google Scholar] [CrossRef] [Green Version]
- Leithe, E.; Mesnil, M.; Aasen, T. The connexin 43 C-terminus: A tail of many tales. Biochim. Biophys. Acta Biomembr. 2018, 1860, 48–64. [Google Scholar] [CrossRef]
- Kotini, M.; Barriga, E.H.; Leslie, J.; Gentzel, M.; Rauschenberger, V.; Schambony, A.; Mayor, R. Gap junction protein Connexin-43 is a direct transcriptional regulator of N-cadherin in vivo. Nat. Commun. 2018, 9, 3846. [Google Scholar] [CrossRef]
- Giepmans, B.N.; Moolenaar, W.H. The gap junction protein connexin43 interacts with the second PDZ domain of the zona occludens-1 protein. Curr. Biol. 1998, 8, 931–934. [Google Scholar] [CrossRef] [Green Version]
- Schnoor, M.; Garcia Ponce, A.; Vadillo, E.; Pelayo, R.; Rossaint, J.; Zarbock, A. Actin dynamics in the regulation of endothelial barrier functions and neutrophil recruitment during endotoxemia and sepsis. Cell. Mol. Life Sci. 2017, 74, 1985–1997. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Ponce, A.; Citalan-Madrid, A.F.; Velazquez-Avila, M.; Vargas-Robles, H.; Schnoor, M. The role of actin-binding proteins in the control of endothelial barrier integrity. Thromb. Haemost. 2015, 113, 20–36. [Google Scholar] [CrossRef] [PubMed]
- Claesson-Welsh, L. Vascular permeability—The essentials. Upsala J. Med. Sci. 2015, 120, 135–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Guevara, O.E.; Warburton, R.R.; Hill, N.S.; Gaestel, M.; Kayyali, U.S. Modulation of HSP27 alters hypoxia-induced endothelial permeability and related signaling pathways. J. Cell. Physiol. 2009, 220, 600–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.B.; Ren, X.; Liu, J.; Guo, X.W.; Jiang, X.P.; Zhang, D.X.; Huang, Y.S.; Zhang, J.P. HSP27 phosphorylation protects against endothelial barrier dysfunction under burn serum challenge. Biochem. Biophys. Res. Commun. 2015, 463, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Guay, J.; Lambert, H.; Gingras-Breton, G.; Lavoie, J.N.; Huot, J.; Landry, J. Regulation of actin filament dynamics by p38 map kinase-mediated phosphorylation of heat shock protein 27. J. Cell Sci. 1997, 110, 357–368. [Google Scholar] [PubMed]
- Wang, W.; Weng, J.; Yu, L.; Huang, Q.; Jiang, Y.; Guo, X. Role of TLR4-p38 MAPK-Hsp27 signal pathway in LPS-induced pulmonary epithelial hyperpermeability. BMC Pulm. Med. 2018, 18, 178. [Google Scholar] [CrossRef]
- Chu, Z.G.; Zhang, J.P.; Song, H.P.; Hu, J.Y.; Zhang, Q.; Xiang, F.; Huang, Y.S. p38 MAP kinase mediates burn serum-induced endothelial barrier dysfunction: Involvement of F-actin rearrangement and L-caldesmon phosphorylation. Shock 2010, 34, 222–228. [Google Scholar] [CrossRef]
- Barlow, H.R.; Cleaver, O. Building Blood Vessels-One Rho GTPase at a Time. Cells 2019, 8, 545. [Google Scholar] [CrossRef] [Green Version]
- Timmerman, I.; Heemskerk, N.; Kroon, J.; Schaefer, A.; van Rijssel, J.; Hoogenboezem, M.; van Unen, J.; Goedhart, J.; Gadella, T.W., Jr.; Yin, T.; et al. A local VE-cadherin and Trio-based signaling complex stabilizes endothelial junctions through Rac1. J. Cell Sci. 2015, 128, 3041–3054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daneshjou, N.; Sieracki, N.; van Nieuw Amerongen, G.P.; Conway, D.E.; Schwartz, M.A.; Komarova, Y.A.; Malik, A.B. Rac1 functions as a reversible tension modulator to stabilize VE-cadherin trans-interaction. J. Cell Biol. 2015, 208, 23–32. [Google Scholar] [CrossRef] [Green Version]
- Jian, M.Y.; Liu, Y.; Li, Q.; Wolkowicz, P.; Alexeyev, M.; Zmijewski, J.; Creighton, J. N-cadherin coordinates AMP kinase-mediated lung vascular repair. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 310, L71–L85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivier, S.; Leclerc, J.; Grenier, A.; Foretz, M.; Tamburini, J.; Viollet, B. AMPK Activation Promotes Tight Junction Assembly in Intestinal Epithelial Caco-2 Cells. Int. J. Mol. Sci. 2019, 20, 5171. [Google Scholar] [CrossRef] [Green Version]
- Rowart, P.; Wu, J.; Caplan, M.J.; Jouret, F. Implications of AMPK in the Formation of Epithelial Tight Junctions. Int. J. Mol. Sci. 2018, 19, 2040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miranda, L.; Carpentier, S.; Platek, A.; Hussain, N.; Gueuning, M.A.; Vertommen, D.; Ozkan, Y.; Sid, B.; Hue, L.; Courtoy, P.J.; et al. AMP-activated protein kinase induces actin cytoskeleton reorganization in epithelial cells. Biochem. Biophys. Res. Commun. 2010, 396, 656–661. [Google Scholar] [CrossRef]
- Schubert, K.M.; Qiu, J.; Blodow, S.; Wiedenmann, M.; Lubomirov, L.T.; Pfitzer, G.; Pohl, U.; Schneider, H. The AMP-Related Kinase (AMPK) Induces Ca2+-Independent Dilation of Resistance Arteries by Interfering With Actin Filament Formation. Circ. Res. 2017, 121, 149–161. [Google Scholar] [CrossRef]
- Onselaer, M.B.; Oury, C.; Hunter, R.W.; Eeckhoudt, S.; Barile, N.; Lecut, C.; Morel, N.; Viollet, B.; Jacquet, L.M.; Bertrand, L.; et al. The Ca2+/calmodulin-dependent kinase kinase beta-AMP-activated protein kinase-alpha1 pathway regulates phosphorylation of cytoskeletal targets in thrombin-stimulated human platelets. J. Thromb. Haemost. 2014, 12, 973–986. [Google Scholar] [CrossRef]
- Horman, S.; Morel, N.; Vertommen, D.; Hussain, N.; Neumann, D.; Beauloye, C.; El Najjar, N.; Forcet, C.; Viollet, B.; Walsh, M.P.; et al. AMP-activated protein kinase phosphorylates and desensitizes smooth muscle myosin light chain kinase. J. Biol. Chem. 2008, 283, 18505–18512. [Google Scholar] [CrossRef] [Green Version]
- Tojkander, S.; Ciuba, K.; Lappalainen, P. CaMKK2 Regulates Mechanosensitive Assembly of Contractile Actin Stress Fibers. Cell Rep. 2018, 24, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Thomson, D.M.; Ascione, M.P.; Grange, J.; Nelson, C.; Hansen, M.D. Phosphorylation of VASP by AMPK alters actin binding and occurs at a novel site. Biochem. Biophys. Res. Commun. 2011, 414, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Park, H.Y.; Kunitake, Y.; Hirasaki, N.; Tanaka, M.; Matsui, T. Theaflavins enhance intestinal barrier of Caco-2 Cell monolayers through the expression of AMP-activated protein kinase-mediated Occludin, Claudin-1, and ZO-1. Biosci. Biotechnol. Biochem. 2015, 79, 130–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Wu, Z.; Ji, Y.; Sun, K.; Dai, Z.; Wu, G. L-Glutamine Enhances Tight Junction Integrity by Activating CaMK Kinase 2-AMP-Activated Protein Kinase Signaling in Intestinal Porcine Epithelial Cells. J. Nutr. 2016, 146, 501–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiomi, R.; Shigetomi, K.; Inai, T.; Sakai, M.; Ikenouchi, J. CaMKII regulates the strength of the epithelial barrier. Sci. Rep. 2015, 5, 13262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jian, M.Y.; Alexeyev, M.F.; Wolkowicz, P.E.; Zmijewski, J.W.; Creighton, J.R. Metformin-stimulated AMPK-alpha1 promotes microvascular repair in acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 305, L844–L855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Creighton, J.; Jian, M.; Sayner, S.; Alexeyev, M.; Insel, P.A. Adenosine monophosphate-activated kinase alpha1 promotes endothelial barrier repair. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2011, 25, 3356–3365. [Google Scholar]
- Castanares-Zapatero, D.; Bouleti, C.; Sommereyns, C.; Gerber, B.; Lecut, C.; Mathivet, T.; Horckmans, M.; Communi, D.; Foretz, M.; Vanoverschelde, J.L.; et al. Connection between cardiac vascular permeability, myocardial edema, and inflammation during sepsis: Role of the alpha1AMP-activated protein kinase isoform. Crit. Care Med. 2013, 41, e411–e422. [Google Scholar] [CrossRef]
- Xing, J.; Wang, Q.; Coughlan, K.; Viollet, B.; Moriasi, C.; Zou, M.H. Inhibition of AMP-activated protein kinase accentuates lipopolysaccharide-induced lung endothelial barrier dysfunction and lung injury in vivo. Am. J. Pathol. 2013, 182, 1021–1030. [Google Scholar] [CrossRef] [Green Version]
- Kitzmiller, L.; Ledford, J.R.; Hake, P.W.; O’Connor, M.; Piraino, G.; Zingarelli, B. Activation of AMP-Activated Protein Kinase by A769662 Ameliorates Sepsis-Induced Acute Lung Injury in Adult Mice. Shock 2019, 52, 540–549. [Google Scholar] [CrossRef]
- Fang, M.; Zhong, W.H.; Song, W.L.; Deng, Y.Y.; Yang, D.M.; Xiong, B.; Zeng, H.K.; Wang, H.D. Ulinastatin Ameliorates Pulmonary Capillary Endothelial Permeability Induced by Sepsis Through Protection of Tight Junctions via Inhibition of TNF-alpha and Related Pathways. Front. Pharmacol. 2018, 9, 823. [Google Scholar] [CrossRef]
- Vaez, H.; Najafi, M.; Toutounchi, N.S.; Barar, J.; Barzegari, A.; Garjani, A. Metformin Alleviates Lipopolysaccharide-induced Acute Lung Injury through Suppressing Toll-like Receptor 4 Signaling. Iran. J. Allergy Asthma Immunol. 2016, 15, 498–507. [Google Scholar] [PubMed]
- Chen, F.C.; Kung, C.T.; Cheng, H.H.; Cheng, C.Y.; Tsai, T.C.; Hsiao, S.Y.; Wu, C.H.; Su, C.M. Metformin Affects Serum Lactate Levels in Predicting Mortality of Patients with Sepsis and Bacteremia. J. Clin. Med. 2019, 8, 318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinhart, K.; Daniels, R.; Kissoon, N.; Machado, F.R.; Schachter, R.D.; Finfer, S. Recognizing Sepsis as a Global Health Priority—A WHO Resolution. N. Engl. J. Med. 2017, 377, 414–417. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Sanders, M.J.; Carmena, D.; Bright, N.J.; Haire, L.F.; Underwood, E.; Patel, B.R.; Heath, R.B.; Walker, P.A.; Hallen, S.; et al. Structural basis of AMPK regulation by small molecule activators. Nat. Commun. 2013, 4, 3017. [Google Scholar] [CrossRef] [Green Version]
- Pichon, S.; Bryckaert, M.; Berrou, E. Control of actin dynamics by p38 MAP kinase-Hsp27 distribution in the lamellipodium of smooth muscle cells. J. Cell Sci. 2004, 117, 2569–2577. [Google Scholar] [CrossRef] [Green Version]
- Baumer, Y.; Drenckhahn, D.; Waschke, J. cAMP induced Rac 1-mediated cytoskeletal reorganization in microvascular endothelium. Histochem. Cell Biol. 2008, 129, 765–778. [Google Scholar] [CrossRef]
- Li, X.; Yu, L.; Gao, J.; Bi, X.; Zhang, J.; Xu, S.; Wang, M.; Chen, M.; Qiu, F.; Fu, G. Apelin Ameliorates High Glucose-Induced Downregulation of Connexin 43 via AMPK-Dependent Pathway in Neonatal Rat Cardiomyocytes. Aging Dis. 2018, 9, 66–76. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Zhao, W.T.; Chen, F.X.; Fu, G.S.; Mou, Y.; Hu, S.J. High glucose promotes gap junctional communication in cultured neonatal cardiac fibroblasts via AMPK activation. Mol. Biol. 2014, 48, 687–695. [Google Scholar] [CrossRef]
- Cao, L.; Chen, Y.; Lu, L.; Liu, Y.; Wang, Y.; Fan, J.; Yin, Y. Angiotensin II upregulates fibroblast-myofibroblast transition through Cx43-dependent CaMKII and TGF-β1 signaling in neonatal rat cardiac fibroblasts. Acta Biochim. Biophys. Sin. 2018, 50, 843–852. [Google Scholar] [CrossRef] [Green Version]
- Rauch, J.; Volinsky, N.; Romano, D.; Kolch, W. The secret life of kinases: Functions beyond catalysis. Cell Commun. Signal. 2011, 9, 23. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Pan, L.; Wei, Z.; Zhao, Y.; Zhang, M. Domain-swapped dimerization of ZO-1 PDZ2 generates specific and regulatory connexin43-binding sites. Embo J. 2008, 27, 2113–2123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moorer, M.C.; Hebert, C.; Tomlinson, R.E.; Iyer, S.R.; Chason, M.; Stains, J.P. Defective signaling, osteoblastogenesis and bone remodeling in a mouse model of connexin 43 C-terminal truncation. J. Cell Sci. 2017, 130, 531–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruse, K.; Lee, Q.S.; Sun, Y.; Klomp, J.; Yang, X.; Huang, F.; Sun, M.Y.; Zhao, S.; Hong, Z.; Vogel, S.M.; et al. N-cadherin signaling via Trio assembles adherens junctions to restrict endothelial permeability. J. Cell Biol. 2019, 218, 299–316. [Google Scholar] [CrossRef] [PubMed]
- Meyer, R.A.; Laird, D.W.; Revel, J.P.; Johnson, R.G. Inhibition of gap junction and adherens junction assembly by connexin and A-CAM antibodies. J. Cell Biol. 1992, 119, 179–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, X.; Doble, B.W.; Kardami, E. The carboxy-tail of connexin-43 localizes to the nucleus and inhibits cell growth. Mol. Cell. Biochem. 2003, 242, 35–38. [Google Scholar] [CrossRef] [PubMed]
- Canto, C.; Auwerx, J. AMP-activated protein kinase and its downstream transcriptional pathways. Cell Mol. Life Sci. 2010, 67, 3407–3423. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Zhang, L.; Chen, R.; Lu, H.; Sui, M.; Zhu, Y.; Zeng, L. SIRT3 Protects Against Acute Kidney Injury via AMPK/mTOR-Regulated Autophagy. Front. Physiol. 2018, 9, 1526. [Google Scholar] [CrossRef]
- Li, X.; Jamal, M.; Guo, P.; Jin, Z.; Zheng, F.; Song, X.; Zhan, J.; Wu, H. Irisin alleviates pulmonary epithelial barrier dysfunction in sepsis-induced acute lung injury via activation of AMPK/SIRT1 pathways. Biomed. Pharmacother. 2019, 118, 109363. [Google Scholar] [CrossRef]
- Wang, R.; Xie, Y.; Qiu, J.; Chen, J. The Effects of Dexmedetomidine in a Rat Model of Sepsis-Induced Lung Injury are Mediated Through the Adenosine Monophosphate-Activated Protein Kinase (AMPK)/Silent Information Regulator 1 (SIRT1) Pathway. Med. Sci. Monit. 2020, 26, e919213. [Google Scholar] [CrossRef]
- Sanchez, F.A.; Ehrenfeld, I.P.; Duran, W.N. S-nitrosation of proteins: An emergent regulatory mechanism in microvascular permeability and vascular function. Tissue Barriers 2013, 1, e23896. [Google Scholar] [CrossRef] [Green Version]
- Houssin, E.; Tepass, U.; Laprise, P. Girdin-mediated interactions between cadherin and the actin cytoskeleton are required for epithelial morphogenesis in Drosophila. Development 2015, 142, 1777–1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aznar, N.; Patel, A.; Rohena, C.C.; Dunkel, Y.; Joosen, L.P.; Taupin, V.; Kufareva, I.; Farquhar, M.G.; Ghosh, P. AMP-activated protein kinase fortifies epithelial tight junctions during energetic stress via its effector GIV/Girdin. Elife 2016, 5, e20795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Li, J.; Young, L.H.; Caplan, M.J. AMP-activated protein kinase regulates the assembly of epithelial tight junctions. Proc. Natl. Acad. Sci. USA 2006, 103, 17272–17277. [Google Scholar] [CrossRef] [Green Version]
- Zheng, B.; Cantley, L.C. Regulation of epithelial tight junction assembly and disassembly by AMP-activated protein kinase. Proc. Natl. Acad. Sci. USA 2007, 104, 819–822. [Google Scholar] [CrossRef] [Green Version]
- Xiang, R.L.; Mei, M.; Cong, X.; Li, J.; Zhang, Y.; Ding, C.; Wu, L.L.; Yu, G.Y. Claudin-4 is required for AMPK-modulated paracellular permeability in submandibular gland cells. J. Mol. Cell. Biol. 2014, 6, 486–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Jouret, F.; Rinehart, J.; Sfakianos, J.; Mellman, I.; Lifton, R.P.; Young, L.H.; Caplan, M.J. AMP-activated protein kinase (AMPK) activation and glycogen synthase kinase-3beta (GSK-3beta) inhibition induce Ca2+-independent deposition of tight junction components at the plasma membrane. J. Biol. Chem. 2011, 286, 16879–16890. [Google Scholar] [CrossRef] [Green Version]
- Kameritsch, P.; Kiemer, F.; Beck, H.; Pohl, U.; Pogoda, K. Cx43 increases serum induced filopodia formation via activation of p21-activated protein kinase 1. Biochim. Biophys. Acta 2015, 1853, 2907–2917. [Google Scholar] [CrossRef] [Green Version]
- Garcia, J.G.; Wang, P.; Schaphorst, K.L.; Becker, P.M.; Borbiev, T.; Liu, F.; Birukova, A.; Jacobs, K.; Bogatcheva, N.; Verin, A.D. Critical involvement of p38 MAP kinase in pertussis toxin-induced cytoskeletal reorganization and lung permeability. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2002, 16, 1064–1076. [Google Scholar] [CrossRef]
- Hirano, S.; Rees, R.S.; Yancy, S.L.; Welsh, M.J.; Remick, D.G.; Yamada, T.; Hata, J.; Gilmont, R.R. Endothelial barrier dysfunction caused by LPS correlates with phosphorylation of HSP27 in vivo. Cell Biol. Toxicol. 2004, 20, 1–14. [Google Scholar] [CrossRef]
- Xie, Z.; Zhang, J.; Wu, J.; Viollet, B.; Zou, M.H. Upregulation of mitochondrial uncoupling protein-2 by the AMP-activated protein kinase in endothelial cells attenuates oxidative stress in diabetes. Diabetes 2008, 57, 3222–3230. [Google Scholar] [CrossRef] [Green Version]
- Zarubin, T.; Han, J. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005, 15, 11–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remy, G.; Risco, A.M.; Inesta-Vaquera, F.A.; Gonzalez-Teran, B.; Sabio, G.; Davis, R.J.; Cuenda, A. Differential activation of p38MAPK isoforms by MKK6 and MKK3. Cell. Signal. 2010, 22, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Grimsey, N.J.; Lin, Y.; Narala, R.; Rada, C.C.; Mejia-Pena, H.; Trejo, J. G protein-coupled receptors activate p38 MAPK via a non-canonical TAB1-TAB2- and TAB1-TAB3-dependent pathway in endothelial cells. J. Biol. Chem. 2019, 294, 5867–5878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Miller, E.J.; Ninomiya-Tsuji, J.; Russell, R.R., 3rd; Young, L.H. AMP-activated protein kinase activates p38 mitogen-activated protein kinase by increasing recruitment of p38 MAPK to TAB1 in the ischemic heart. Circ. Res. 2005, 97, 872–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanna, A.; Henson, S.M.; Escors, D.; Akbar, A.N. The kinase p38 activated by the metabolic regulator AMPK and scaffold TAB1 drives the senescence of human T cells. Nat. Immunol. 2014, 15, 965–972. [Google Scholar] [CrossRef] [Green Version]
- Ashwell, J.D. The many paths to p38 mitogen-activated protein kinase activation in the immune system. Nat. Rev. Immunol. 2006, 6, 532–540. [Google Scholar] [CrossRef]
- Jun, J.E.; Kulhanek, K.R.; Chen, H.; Chakraborty, A.; Roose, J.P. Alternative ZAP70-p38 signals prime a classical p38 pathway through LAT and SOS to support regulatory T cell differentiation. Sci. Signal. 2019, 12, eaao0736. [Google Scholar] [CrossRef]
- Tsukita, K.; Yano, T.; Tamura, A.; Tsukita, S. Reciprocal Association between the Apical Junctional Complex and AMPK: A Promising Therapeutic Target for Epithelial/Endothelial Barrier Function? Int. J. Mol. Sci. 2019, 20, 6012. [Google Scholar] [CrossRef] [Green Version]
- Noppe, G.; Dufeys, C.; Buchlin, P.; Marquet, N.; Castanares-Zapatero, D.; Balteau, M.; Hermida, N.; Bouzin, C.; Esfahani, H.; Viollet, B.; et al. Reduced scar maturation and contractility lead to exaggerated left ventricular dilation after myocardial infarction in mice lacking AMPKalpha1. J. Mol. Cell. Cardiol. 2014, 74, 32–43. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angé, M.; Castanares-Zapatero, D.; De Poortere, J.; Dufeys, C.; Courtoy, G.E.; Bouzin, C.; Quarck, R.; Bertrand, L.; Beauloye, C.; Horman, S. α1AMP-Activated Protein Kinase Protects against Lipopolysaccharide-Induced Endothelial Barrier Disruption via Junctional Reinforcement and Activation of the p38 MAPK/HSP27 Pathway. Int. J. Mol. Sci. 2020, 21, 5581. https://doi.org/10.3390/ijms21155581
Angé M, Castanares-Zapatero D, De Poortere J, Dufeys C, Courtoy GE, Bouzin C, Quarck R, Bertrand L, Beauloye C, Horman S. α1AMP-Activated Protein Kinase Protects against Lipopolysaccharide-Induced Endothelial Barrier Disruption via Junctional Reinforcement and Activation of the p38 MAPK/HSP27 Pathway. International Journal of Molecular Sciences. 2020; 21(15):5581. https://doi.org/10.3390/ijms21155581
Chicago/Turabian StyleAngé, Marine, Diego Castanares-Zapatero, Julien De Poortere, Cécile Dufeys, Guillaume E. Courtoy, Caroline Bouzin, Rozenn Quarck, Luc Bertrand, Christophe Beauloye, and Sandrine Horman. 2020. "α1AMP-Activated Protein Kinase Protects against Lipopolysaccharide-Induced Endothelial Barrier Disruption via Junctional Reinforcement and Activation of the p38 MAPK/HSP27 Pathway" International Journal of Molecular Sciences 21, no. 15: 5581. https://doi.org/10.3390/ijms21155581
APA StyleAngé, M., Castanares-Zapatero, D., De Poortere, J., Dufeys, C., Courtoy, G. E., Bouzin, C., Quarck, R., Bertrand, L., Beauloye, C., & Horman, S. (2020). α1AMP-Activated Protein Kinase Protects against Lipopolysaccharide-Induced Endothelial Barrier Disruption via Junctional Reinforcement and Activation of the p38 MAPK/HSP27 Pathway. International Journal of Molecular Sciences, 21(15), 5581. https://doi.org/10.3390/ijms21155581