Involvement of p38 MAPK in Synaptic Function and Dysfunction

 ,
, 
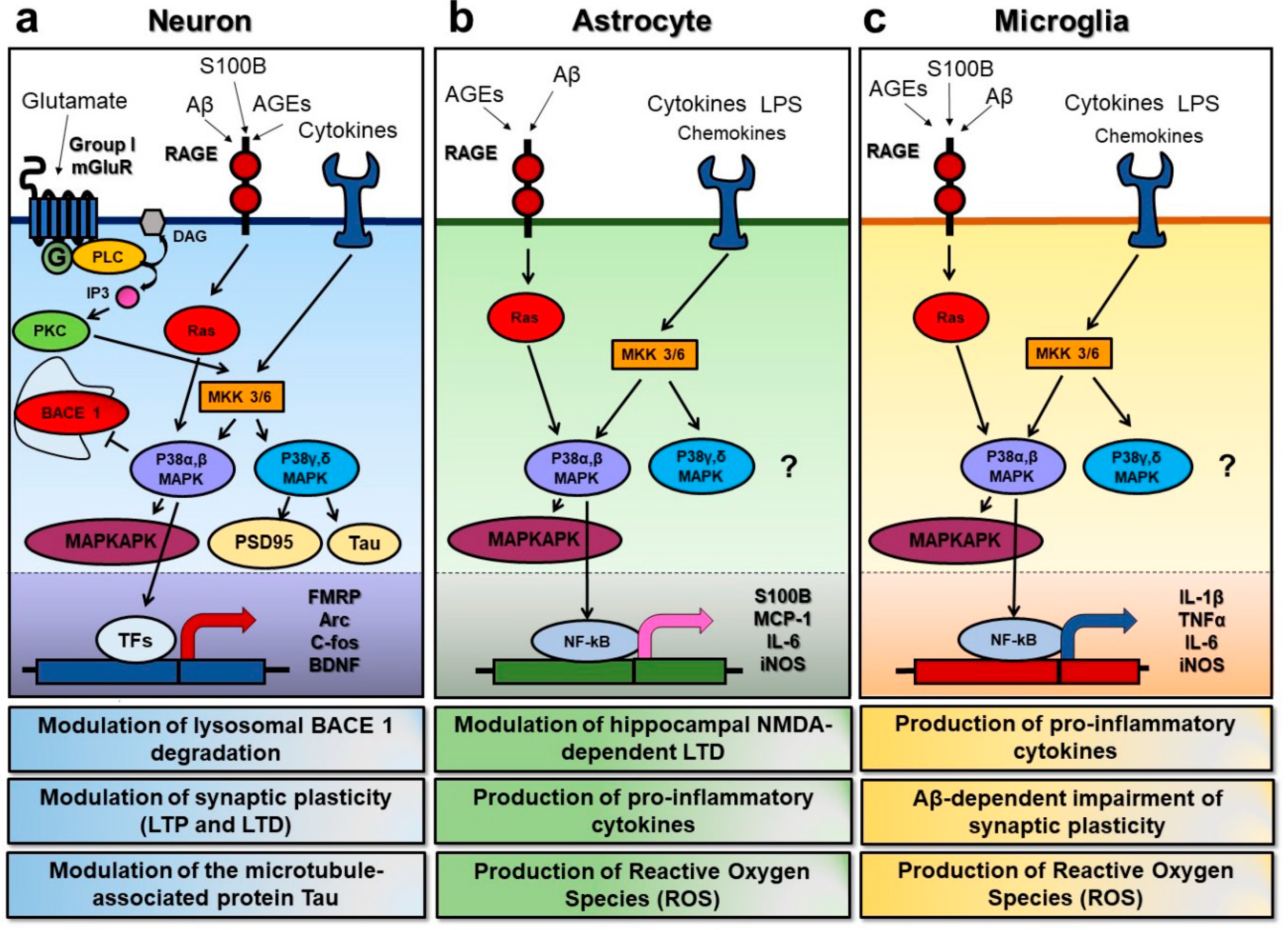{kind=link}
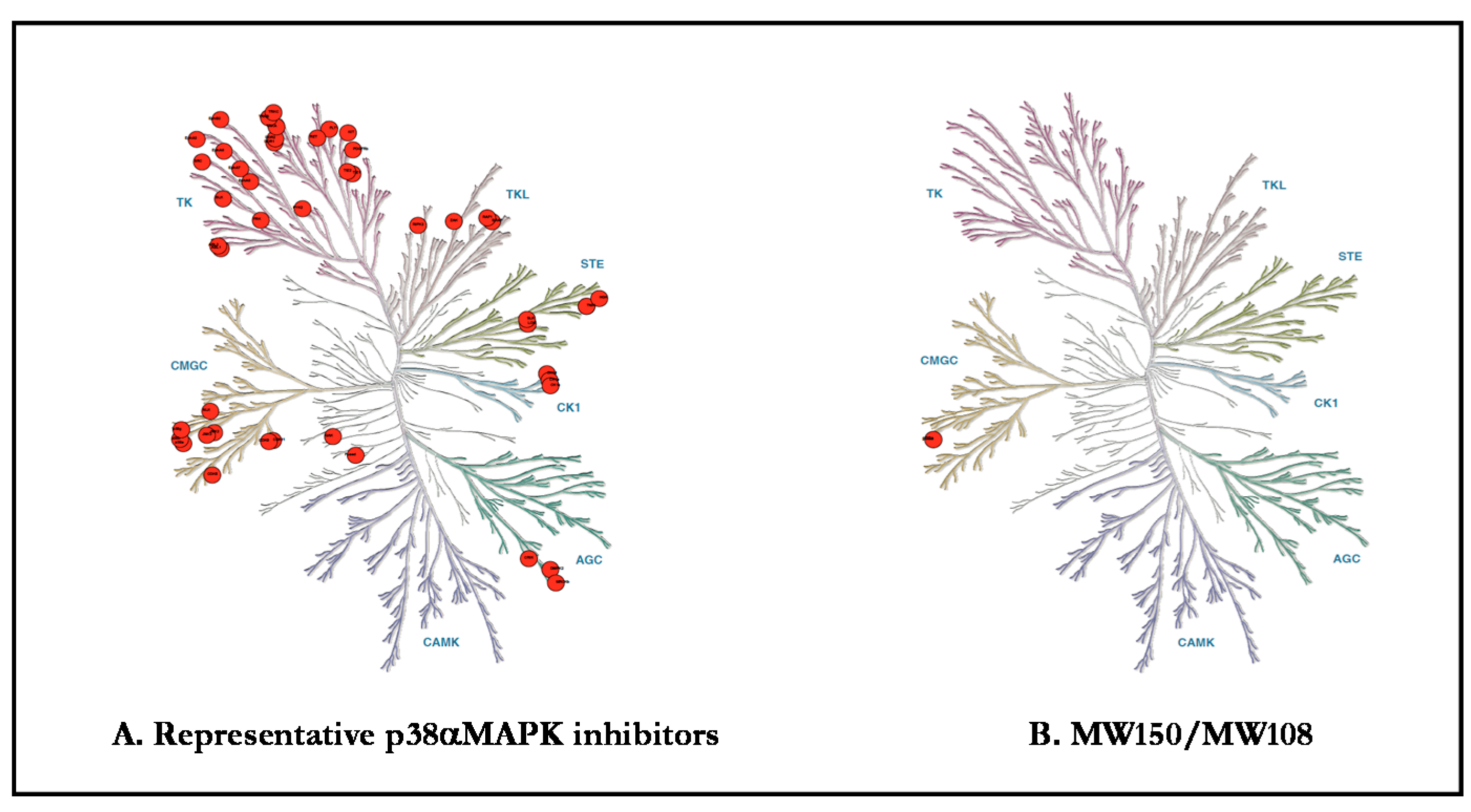{kind=link}
Abstract
:1. P38 Mitogen-Activated Protein Kinases (p38-MAPK)
2. p38 MAPK and Synaptic Function
3. p38 MAPK Neuroinflammation and Synaptic Dysfunction
p38 MAPK, AD Neurodegeneration and Synaptic Dysfunction
4. Small Molecules Targeting p38α MAPK
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| MAPK | mitogen activated protein kinases |
| CNS | central nervous system |
| LTP | long-term potentiation |
| LTD | long-term depression |
| AD | Alzheimer’s disease |
| ERK | extracellular signal-regulated kinases |
| EPSPs | synaptically evoked excitatory postsynaptic potentials |
| JNK | c-Jun amino-terminal kinases |
| ATP | adenosine triphosphate |
| NFAT | nuclear factor of activated T cells |
| ATF | activating transcription factor |
| STAT1 | signal transducer and activator of transcription |
| LPS | lipopolysaccharide |
| IL-1β | interleukin-1β |
| TNFα | tumor necrosis factor |
| AGEs | advanced glycation endproducts |
| mGluRs | metabotropic glutamate receptors |
| TFs | transcription factors |
| FMRP | fragile X mental retardation protein |
| Arc | activity-regulated cytoskeleton |
| BDNF | brain-derived neurotrophic factor |
| PSD95 | postsynaptic density protein 95 |
| RAGE | advanced glycation endproducts receptor |
| MCP-1 | monocyte chemoattractant proten-1 |
| iNOS | inducible nitric oxide synthase |
| NMDA | n-methyl-d-aspartate |
| LFS | low-frequency stimulation |
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| Aβ | amyloid-β |
| EC | entorhinal cortex |
| Tg | transgenic |
| APP | amyloid precursor protein |
| PS1 | mutant human presenilin 1 |
| EP | endophilin |
| NF-KB | nuclear factor-kB |
| ROS | reactive oxygen species |
| MS | multiple sclerosis |
| PD | Parkinson’s disease |
| HD | Huntington’s disease |
| ALS | amyotrophic lateral sclerosis |
| KI | knock in |
| KO | knock out |
| S/T | serine-threonine |
| FPCR | G protein-coupled receptor |
References
- Origlia, N.; Arancio, O.; Domenici, L.; Yan, S.S. MAPK, beta-amyloid and synaptic dysfunction: The role of RAGE. Expert Rev. Neurother. 2009, 9, 1635–1645. [Google Scholar] [CrossRef] [PubMed]
- Correa, S.A.; Eales, K.L. The Role of p38 MAPK and Its Substrates in Neuronal Plasticity and Neurodegenerative Disease. J. Signal Transduct. 2012, 2012, 649079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, G.L.; Lapadat, R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 2002, 298, 1911–1912. [Google Scholar] [CrossRef] [Green Version]
- Jagodzik, P.; Tajdel-Zielinska, M.; Ciesla, A.; Marczak, M.; Ludwikow, A. Mitogen-Activated Protein Kinase Cascades in Plant Hormone Signaling. Front. Plant Sci. 2018, 9, 1387. [Google Scholar] [CrossRef] [PubMed]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. MMBR 2011, 75, 50–83. [Google Scholar] [CrossRef] [Green Version]
- Pei, J.J.; Braak, E.; Braak, H.; Grundke-Iqbal, I.; Iqbal, K.; Winblad, B.; Cowburn, R.F. Localization of active forms of C-jun kinase (JNK) and p38 kinase in Alzheimer’s disease brains at different stages of neurofibrillary degeneration. J. Alzheimer’s Dis. JAD 2001, 3, 41–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Troy, C.M.; Rabacchi, S.A.; Xu, Z.; Maroney, A.C.; Connors, T.J.; Shelanski, M.L.; Greene, L.A. Beta-Amyloid-induced neuronal apoptosis requires c-Jun N-terminal kinase activation. J. Neurochem. 2001, 77, 157–164. [Google Scholar] [CrossRef]
- Zhu, X.; Mei, M.; Lee, H.G.; Wang, Y.; Han, J.; Perry, G.; Smith, M.A. P38 activation mediates amyloid-beta cytotoxicity. Neurochem. Res. 2005, 30, 791–796. [Google Scholar] [CrossRef]
- Hasegawa, Y.; Toyama, K.; Uekawa, K.; Ichijo, H.; Kim-Mitsuyama, S. Role of ASK1/p38 Cascade in a Mouse Model of Alzheimer’s Disease and Brain Aging. J. Alzheimer’s Dis. JAD 2018, 61, 259–263. [Google Scholar] [CrossRef]
- Ono, K.; Han, J. The p38 signal transduction pathway: Activation and function. Cell. Signal. 2000, 12, 1–13. [Google Scholar] [CrossRef]
- Lee, J.K.; Kim, N.J. Recent Advances in the Inhibition of p38 MAPK as a Potential Strategy for the Treatment of Alzheimer’s Disease. Molecules 2017, 22, 1287. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, M.; Cuenda, A.; Spillantini, M.G.; Thomas, G.M.; Buee-Scherrer, V.; Cohen, P.; Goedert, M. Stress-activated protein kinase-3 interacts with the PDZ domain of alpha1-syntrophin. A mechanism for specific substrate recognition. J. Biol. Chem. 1999, 274, 12626–12631. [Google Scholar] [CrossRef] [Green Version]
- Sabio, G.; Arthur, J.S.; Kuma, Y.; Peggie, M.; Carr, J.; Murray-Tait, V.; Centeno, F.; Goedert, M.; Morrice, N.A.; Cuenda, A. p38gamma regulates the localisation of SAP97 in the cytoskeleton by modulating its interaction with GKAP. EMBO J. 2005, 24, 1134–1145. [Google Scholar] [CrossRef] [PubMed]
- Sabio, G.; Reuver, S.; Feijoo, C.; Hasegawa, M.; Thomas, G.M.; Centeno, F.; Kuhlendahl, S.; Leal-Ortiz, S.; Goedert, M.; Garner, C.; et al. Stress- and mitogen-induced phosphorylation of the synapse-associated protein SAP90/PSD-95 by activation of SAPK3/p38gamma and ERK1/ERK2. Biochem. J. 2004, 380, 19–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feijoo, C.; Campbell, D.G.; Jakes, R.; Goedert, M.; Cuenda, A. Evidence that phosphorylation of the microtubule-associated protein Tau by SAPK4/p38delta at Thr50 promotes microtubule assembly. J. Cell Sci. 2005, 118, 397–408. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Park, J.; Che, Y.; Han, P.L.; Lee, J.K. Constitutive activity and differential localization of p38alpha and p38beta MAPKs in adult mouse brain. J. Neurosci. Res. 2000, 60, 623–631. [Google Scholar] [CrossRef]
- Menon, R.; Papaconstantinou, J. p38 Mitogen activated protein kinase (MAPK): A new therapeutic target for reducing the risk of adverse pregnancy outcomes. Expert Opin. Ther. Targets 2016, 20, 1397–1412. [Google Scholar] [CrossRef]
- Schieven, G.L. The biology of p38 kinase: A central role in inflammation. Curr. Top. Med. Chem. 2005, 5, 921–928. [Google Scholar] [CrossRef]
- Bachstetter, A.D.; Xing, B.; de Almeida, L.; Dimayuga, E.R.; Watterson, D.M.; Van Eldik, L.J. Microglial p38alpha MAPK is a key regulator of proinflammatory cytokine up-regulation induced by toll-like receptor (TLR) ligands or beta-amyloid (Abeta). J. Neuroinflamm. 2011, 8, 79. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Li, J.; Bu, X.; Liu, X.; Tankersley, C.G.; Wang, C.; Huang, K. Age-induced augmentation of p38 MAPK phosphorylation in mouse lung. Exp. Gerontol. 2011, 46, 694–702. [Google Scholar] [CrossRef]
- Papaconstantinou, J.; Hsieh, C.C. Activation of senescence and aging characteristics by mitochondrially generated ROS: How are they linked? Cell Cycle 2010, 9, 3831–3833. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, Y.; Gu, Z.; Li, L.; Liu, Y.; Wang, L.; Su, L. p38 MAPK-MK2 pathway regulates the heat-stress-induced accumulation of reactive oxygen species that mediates apoptotic cell death in glial cells. Oncol. Lett. 2018, 15, 775–782. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Zhu, Z.; Tong, B.C.; Iyaswamy, A.; Xie, W.J.; Zhu, Y.; Sreenivasmurthy, S.G.; Senthilkumar, K.; Cheung, K.H.; Song, J.X.; et al. A stress response p38 MAP kinase inhibitor SB202190 promoted TFEB/TFE3-dependent autophagy and lysosomal biogenesis independent of p38. Redox Biol. 2020, 32, 101445. [Google Scholar] [CrossRef] [PubMed]
- Munoz, L.; Ammit, A.J. Targeting p38 MAPK pathway for the treatment of Alzheimer’s disease. Neuropharmacology 2010, 58, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.M.; Grum-Tokars, V.L.; Schavocky, J.P.; Saeed, F.; Staniszewski, A.; Teich, A.F.; Arancio, O.; Bachstetter, A.D.; Webster, S.J.; Van Eldik, L.J.; et al. Targeting human central nervous system protein kinases: An isoform selective p38alphaMAPK inhibitor that attenuates disease progression in Alzheimer’s disease mouse models. Acs Chem. Neurosci. 2015, 6, 666–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Criscuolo, C.; Fontebasso, V.; Middei, S.; Stazi, M.; Ammassari-Teule, M.; Yan, S.S.; Origlia, N. Entorhinal Cortex dysfunction can be rescued by inhibition of microglial RAGE in an Alzheimer’s disease mouse model. Sci. Rep. 2017, 7, 42370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veglianese, P.; Lo Coco, D.; Bao Cutrona, M.; Magnoni, R.; Pennacchini, D.; Pozzi, B.; Gowing, G.; Julien, J.P.; Tortarolo, M.; Bendotti, C. Activation of the p38MAPK cascade is associated with upregulation of TNF alpha receptors in the spinal motor neurons of mouse models of familial ALS. Mol. Cell. Neurosci. 2006, 31, 218–231. [Google Scholar] [CrossRef]
- Guo, W.; Vandoorne, T.; Steyaert, J.; Staats, K.A.; Van Den Bosch, L. The multifaceted role of kinases in amyotrophic lateral sclerosis: Genetic, pathological and therapeutic implications. Brain J. Neurol. 2020, 143, 1651–1673. [Google Scholar] [CrossRef] [PubMed]
- Gui, C.; Ren, Y.; Chen, J.; Wu, X.; Mao, K.; Li, H.; Yu, H.; Zou, F.; Li, W. p38 MAPK-DRP1 signaling is involved in mitochondrial dysfunction and cell death in mutant A53T alpha-synuclein model of Parkinson’s disease. Toxicol. Appl. Pharmacol. 2020, 388, 114874. [Google Scholar] [CrossRef]
- Brobey, R.K.; German, D.; Sonsalla, P.K.; Gurnani, P.; Pastor, J.; Hsieh, C.C.; Papaconstantinou, J.; Foster, P.P.; Kuro-o, M.; Rosenblatt, K.P. Klotho Protects Dopaminergic Neuron Oxidant-Induced Degeneration by Modulating ASK1 and p38 MAPK Signaling Pathways. PLoS ONE 2015, 10, e0139914. [Google Scholar] [CrossRef]
- Kompa, A.R. Do p38 mitogen-activated protein kinase inhibitors have a future for the treatment of cardiovascular disease? J. Thorac. Dis. 2016, 8, E1068–E1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, H.J.; Lee, H.J.; Vuong, T.A.; Choi, K.S.; Choi, D.; Koo, S.H.; Cho, S.C.; Cho, H.; Kang, J.S. Prmt7 Deficiency Causes Reduced Skeletal Muscle Oxidative Metabolism and Age-Related Obesity. Diabetes 2016, 65, 1868–1882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Tu, S.; Yang, S.; Shen, P.; Huang, Y.; Ba, X.; Lin, W.; Huang, Y.; Wang, Y.; Qin, K.; et al. Berberine Modulates LPA Function to Inhibit the Proliferation and Inflammation of FLS-RA via p38/ERK MAPK Pathway Mediated by LPA1. Evid. Based Complementary Altern. Med. Ecam 2019, 2019, 2580207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bredeson, S.; Papaconstantinou, J.; Deford, J.H.; Kechichian, T.; Syed, T.A.; Saade, G.R.; Menon, R. HMGB1 promotes a p38MAPK associated non-infectious inflammatory response pathway in human fetal membranes. PLoS ONE 2014, 9, e113799. [Google Scholar] [CrossRef] [Green Version]
- Bachstetter, A.D.; Van Eldik, L.J. The p38 MAP Kinase Family as Regulators of Proinflammatory Cytokine Production in Degenerative Diseases of the CNS. Aging Dis. 2010, 1, 199–211. [Google Scholar] [PubMed]
- Kim, E.K.; Choi, E.J. Compromised MAPK signaling in human diseases: An update. Arch. Toxicol. 2015, 89, 867–882. [Google Scholar] [CrossRef]
- Li, S.; Tian, X.; Hartley, D.M.; Feig, L.A. The environment versus genetics in controlling the contribution of MAP kinases to synaptic plasticity. Curr. Biol. CB 2006, 16, 2303–2313. [Google Scholar] [CrossRef] [Green Version]
- Bolshakov, V.Y.; Carboni, L.; Cobb, M.H.; Siegelbaum, S.A.; Belardetti, F. Dual MAP kinase pathways mediate opposing forms of long-term plasticity at CA3-CA1 synapses. Nat. Neurosci. 2000, 3, 1107–1112. [Google Scholar] [CrossRef]
- Navarrete, M.; Cuartero, M.I.; Palenzuela, R.; Draffin, J.E.; Konomi, A.; Serra, I.; Colie, S.; Castano-Castano, S.; Hasan, M.T.; Nebreda, A.R.; et al. Astrocytic p38alpha MAPK drives NMDA receptor-dependent long-term depression and modulates long-term memory. Nat. Commun. 2019, 10, 2968. [Google Scholar] [CrossRef] [Green Version]
- Tan, B.; Bitiktas, S.; Kavraal, S.; Dursun, N.; Donmez Altuntas, H.; Suer, C. Low-frequency stimulation induces a durable long-term depression in young adult hyperthyroid rats: The role of p38 mitogen-activated protein kinase and protein phosphatase 1. Neuroreport 2016, 27, 640–646. [Google Scholar] [CrossRef]
- Origlia, N.; Bonadonna, C.; Rosellini, A.; Leznik, E.; Arancio, O.; Yan, S.S.; Domenici, L. Microglial receptor for advanced glycation end product-dependent signal pathway drives beta-amyloid-induced synaptic depression and long-term depression impairment in entorhinal cortex. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 11414–11425. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Kojic, L.Z.; Zhang, L.; Prasad, S.S.; Douglas, R.; Wang, Y.; Cynader, M.S. Anisomycin activates p38 MAP kinase to induce LTD in mouse primary visual cortex. Brain Res. 2006, 1085, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Ittner, A.; Asih, P.R.; Tan, A.R.P.; Prikas, E.; Bertz, J.; Stefanoska, K.; Lin, Y.; Volkerling, A.M.; Ke, Y.D.; Delerue, F.; et al. Reduction of advanced tau-mediated memory deficits by the MAP kinase p38gamma. Acta Neuropathol. 2020. [Google Scholar] [CrossRef]
- Dai, H.L.; Hu, W.Y.; Jiang, L.H.; Li, L.; Gaung, X.F.; Xiao, Z.C. p38 MAPK Inhibition Improves Synaptic Plasticity and Memory in Angiotensin II-dependent Hypertensive Mice. Sci. Rep. 2016, 6, 27600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Origlia, N.; Righi, M.; Capsoni, S.; Cattaneo, A.; Fang, F.; Stern, D.M.; Chen, J.X.; Schmidt, A.M.; Arancio, O.; Yan, S.D.; et al. Receptor for advanced glycation end product-dependent activation of p38 mitogen-activated protein kinase contributes to amyloid-beta-mediated cortical synaptic dysfunction. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 3521–3530. [Google Scholar] [CrossRef] [Green Version]
- Origlia, N.; Criscuolo, C.; Arancio, O.; Yan, S.S.; Domenici, L. RAGE inhibition in microglia prevents ischemia-dependent synaptic dysfunction in an amyloid-enriched environment. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 8749–8760. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.; Du, Y.; Zhang, Y.; Huang, Z.; Fu, M.; Li, J.; Pang, Y.; Lei, P.; Wang, Y.T.; Song, W.; et al. MKP-1 reduces Abeta generation and alleviates cognitive impairments in Alzheimer’s disease models. Signal Transduct. Target. Ther. 2019, 4, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, R.N.; Jana, M.; Pahan, K. MAPK p38 regulates transcriptional activity of NF-kappaB in primary human astrocytes via acetylation of p65. J. Immunol. 2007, 179, 7101–7109. [Google Scholar] [CrossRef] [Green Version]
- Buxade, M.; Parra-Palau, J.L.; Proud, C.G. The Mnks: MAP kinase-interacting kinases (MAP kinase signal-integrating kinases). Front. Biosci. J. Virtual Libr. 2008, 13, 5359–5373. [Google Scholar] [CrossRef]
- Mody, N.; Leitch, J.; Armstrong, C.; Dixon, J.; Cohen, P. Effects of MAP kinase cascade inhibitors on the MKK5/ERK5 pathway. FEBS Lett. 2001, 502, 21–24. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; He, D.; Bai, Y. Microglia-Mediated Inflammation and Neurodegenerative Disease. Mol. Neurobiol. 2016, 53, 6709–6715. [Google Scholar] [CrossRef] [PubMed]
- Kelly, A.; Vereker, E.; Nolan, Y.; Brady, M.; Barry, C.; Loscher, C.E.; Mills, K.H.; Lynch, M.A. Activation of p38 plays a pivotal role in the inhibitory effect of lipopolysaccharide and interleukin-1 beta on long term potentiation in rat dentate gyrus. J. Biol. Chem. 2003, 278, 19453–19462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laye, S.; Parnet, P.; Goujon, E.; Dantzer, R. Peripheral administration of lipopolysaccharide induces the expression of cytokine transcripts in the brain and pituitary of mice. Brain Res. Mol. Brain Res. 1994, 27, 157–162. [Google Scholar] [CrossRef]
- Fan, L.; Young, P.R.; Barone, F.C.; Feuerstein, G.Z.; Smith, D.H.; McIntosh, T.K. Experimental brain injury induces expression of interleukin-1 beta mRNA in the rat brain. Brain Res. Mol. Brain Res. 1995, 30, 125–130. [Google Scholar] [CrossRef]
- Sonti, G.; Ilyin, S.E.; Plata-Salaman, C.R. Anorexia induced by cytokine interactions at pathophysiological concentrations. Am. J. Physiol. 1996, 270, R1394–R1402. [Google Scholar] [CrossRef] [PubMed]
- Maruta, E.; Yabuuchi, K.; Nishiyori, A.; Takami, S.; Minami, M.; Satoh, M. Beta2-adrenoceptors on the glial cells mediate the induction of interleukin-1beta mRNA in the rat brain. Brain Res. Mol. Brain Res. 1997, 49, 291–294. [Google Scholar] [CrossRef]
- Hoshino, K.; Hasegawa, K.; Kamiya, H.; Morimoto, Y. Synapse-specific effects of IL-1beta on long-term potentiation in the mouse hippocampus. Biomed. Res. 2017, 38, 183–188. [Google Scholar] [CrossRef] [Green Version]
- Hein, A.M.; Stasko, M.R.; Matousek, S.B.; Scott-McKean, J.J.; Maier, S.F.; Olschowka, J.A.; Costa, A.C.; O’Banion, M.K. Sustained hippocampal IL-1beta overexpression impairs contextual and spatial memory in transgenic mice. BrainBehav. Immun. 2010, 24, 243–253. [Google Scholar] [CrossRef] [Green Version]
- Tancredi, V.; D’Arcangelo, G.; Grassi, F.; Tarroni, P.; Palmieri, G.; Santoni, A.; Eusebi, F. Tumor necrosis factor alters synaptic transmission in rat hippocampal slices. Neurosci. Lett. 1992, 146, 176–178. [Google Scholar] [CrossRef]
- Butler, M.P.; O’Connor, J.J.; Moynagh, P.N. Dissection of tumor-necrosis factor-alpha inhibition of long-term potentiation (LTP) reveals a p38 mitogen-activated protein kinase-dependent mechanism which maps to early-but not late-phase LTP. Neuroscience 2004, 124, 319–326. [Google Scholar] [CrossRef]
- Fang, F.; Yu, Q.; Arancio, O.; Chen, D.; Gore, S.S.; Yan, S.S.; Yan, S.F. RAGE mediates Abeta accumulation in a mouse model of Alzheimer’s disease via modulation of beta- and gamma-secretase activity. Hum. Mol. Genet. 2018, 27, 1002–1014. [Google Scholar] [CrossRef] [PubMed]
- Kheiri, G.; Dolatshahi, M.; Rahmani, F.; Rezaei, N. Role of p38/MAPKs in Alzheimer’s disease: Implications for amyloid beta toxicity targeted therapy. Rev. Neurosci. 2018, 30, 9–30. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Walsh, D.M.; Rowan, M.J.; Selkoe, D.J.; Anwyl, R. Block of long-term potentiation by naturally secreted and synthetic amyloid beta-peptide in hippocampal slices is mediated via activation of the kinases c-Jun N-terminal kinase, cyclin-dependent kinase 5, and p38 mitogen-activated protein kinase as well as metabotropic glutamate receptor type 5. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 3370–3378. [Google Scholar] [CrossRef] [Green Version]
- Snyder, E.M.; Nong, Y.; Almeida, C.G.; Paul, S.; Moran, T.; Choi, E.Y.; Nairn, A.C.; Salter, M.W.; Lombroso, P.J.; Gouras, G.K.; et al. Regulation of NMDA receptor trafficking by amyloid-beta. Nat. Neurosci. 2005, 8, 1051–1058. [Google Scholar] [CrossRef]
- Zhu, X.; Rottkamp, C.A.; Hartzler, A.; Sun, Z.; Takeda, A.; Boux, H.; Shimohama, S.; Perry, G.; Smith, M.A. Activation of MKK6, an upstream activator of p38, in Alzheimer’s disease. J. Neurochem. 2001, 79, 311–318. [Google Scholar] [CrossRef]
- Ferrer, I. Stress kinases involved in tau phosphorylation in Alzheimer’s disease, tauopathies and APP transgenic mice. Neurotox. Res. 2004, 6, 469–475. [Google Scholar] [CrossRef]
- Maphis, N.; Jiang, S.; Xu, G.; Kokiko-Cochran, O.N.; Roy, S.M.; Van Eldik, L.J.; Watterson, D.M.; Lamb, B.T.; Bhaskar, K. Selective suppression of the alpha isoform of p38 MAPK rescues late-stage tau pathology. Alzheimer’s Res. Ther. 2016, 8, 54. [Google Scholar] [CrossRef] [Green Version]
- Yu, Q.; Wang, Y.; Du, F.; Yan, S.; Hu, G.; Origlia, N.; Rutigliano, G.; Sun, Q.; Yu, H.; Ainge, J.; et al. Overexpression of endophilin A1 exacerbates synaptic alterations in a mouse model of Alzheimer’s disease. Nat. Commun. 2018, 9, 2968. [Google Scholar] [CrossRef]
- Chen, K.; Lu, Y.; Liu, C.; Zhang, L.; Fang, Z.; Yu, G. Morroniside prevents H2O2 or Abeta1-42-induced apoptosis via attenuating JNK and p38 MAPK phosphorylation. Eur. J. Pharmacol. 2018, 834, 295–304. [Google Scholar] [CrossRef]
- Schnoder, L.; Hao, W.; Qin, Y.; Liu, S.; Tomic, I.; Liu, X.; Fassbender, K.; Liu, Y. Deficiency of Neuronal p38alpha MAPK Attenuates Amyloid Pathology in Alzheimer Disease Mouse and Cell Models through Facilitating Lysosomal Degradation of BACE1. J. Biol. Chem. 2016, 291, 2067–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefanoska, K.; Bertz, J.; Volkerling, A.M.; van der Hoven, J.; Ittner, L.M.; Ittner, A. Neuronal MAP kinase p38alpha inhibits c-Jun N-terminal kinase to modulate anxiety-related behaviour. Sci. Rep. 2018, 8, 14296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, S.M.; Minasov, G.; Arancio, O.; Chico, L.W.; Van Eldik, L.J.; Anderson, W.F.; Pelletier, J.C.; Watterson, D.M. A Selective and Brain Penetrant p38alphaMAPK Inhibitor Candidate for Neurologic and Neuropsychiatric Disorders That Attenuates Neuroinflammation and Cognitive Dysfunction. J. Med. Chem. 2019, 62, 5298–5311. [Google Scholar] [CrossRef]
- Ittner, A.; Chua, S.W.; Bertz, J.; Volkerling, A.; van der Hoven, J.; Gladbach, A.; Przybyla, M.; Bi, M.; van Hummel, A.; Stevens, C.H.; et al. Site-specific phosphorylation of tau inhibits amyloid-beta toxicity in Alzheimer’s mice. Science 2016, 354, 904–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ittner, A.A.; Gladbach, A.; Bertz, J.; Suh, L.S.; Ittner, L.M. p38 MAP kinase-mediated NMDA receptor-dependent suppression of hippocampal hypersynchronicity in a mouse model of Alzheimer’s disease. Acta Neuropathol Commun. 2014, 2, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bain, J.; Plater, L.; Elliott, M.; Shpiro, N.; Hastie, C.J.; McLauchlan, H.; Klevernic, I.; Arthur, J.S.; Alessi, D.R.; Cohen, P. The selectivity of protein kinase inhibitors: A further update. Biochem. J. 2007, 408, 297–315. [Google Scholar] [CrossRef] [Green Version]
- Kuma, Y.; Sabio, G.; Bain, J.; Shpiro, N.; Marquez, R.; Cuenda, A. BIRB796 inhibits all p38 MAPK isoforms in vitro and in vivo. J. Biol. Chem. 2005, 280, 19472–19479. [Google Scholar] [CrossRef] [Green Version]
- Chico, L.K.; Van Eldik, L.J.; Watterson, D.M. Targeting protein kinases in central nervous system disorders. Nat. Rev. Drug Discov. 2009, 8, 892–909. [Google Scholar] [CrossRef] [Green Version]
- Watterson, D.M.; Grum-Tokars, V.L.; Roy, S.M.; Schavocky, J.P.; Bradaric, B.D.; Bachstetter, A.D.; Xing, B.; Dimayuga, E.; Saeed, F.; Zhang, H.; et al. Development of Novel In Vivo Chemical Probes to Address CNS Protein Kinase Involvement in Synaptic Dysfunction. PLoS ONE 2013, 8, e66226. [Google Scholar] [CrossRef]
- Fabian, M.A.; Biggs, W.H.; Treiber, D.K.; Atteridge, C.E.; Azimioara, M.D.; Benedetti, M.G.; Carter, T.A.; Ciceri, P.; Edeen, P.T.; Floyd, M. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 2005, 23, 329–336. [Google Scholar] [CrossRef]
- Karaman, M.W.; Herrgard, S.; Treiber, D.K.; Gallant, P.; Atteridge, C.E.; Campbell, B.T.; Chan, K.W.; Ciceri, P.; Davis, M.I.; Edeen, P.T. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2008, 26, 127–132. [Google Scholar] [CrossRef]
- Zhou, Z.; Bachstetter, A.D.; Spani, C.B.; Roy, S.M.; Watterson, D.M.; Van Eldik, L.J. Retention of normal glia function by an isoform-selective protein kinase inhibitor drug candidate that modulates cytokine production and cognitive outcomes. J. Neuroinflammation 2017, 14, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancino, G.I.; Toledo, E.M.; Leal, N.R.; Hernandez, D.E.; Yevenes, L.F.; Inestrosa, N.C.; Alvarez, A.R. STI571 prevents apoptosis, tau phosphorylation and behavioural impairments induced by Alzheimer’s beta-amyloid deposits. Brain: A J. Neurol. 2008, 131, 2425–2442. [Google Scholar] [CrossRef] [PubMed]
- Lonskaya, I.; Hebron, M.L.; Desforges, N.M.; Franjie, A.; Moussa, C.E. Tyrosine kinase inhibition increases functional parkin-Beclin-1 interaction and enhances amyloid clearance and cognitive performance. EMBO Mol. Med. 2013, 5, 1247–1262. [Google Scholar] [CrossRef]
- Lonskaya, I.; Hebron, M.L.; Selby, S.T.; Turner, R.S.; Moussa, C.E. Nilotinib and bosutinib modulate pre-plaque alterations of blood immune markers and neuro-inflammation in Alzheimer’s disease models. Neuroscience 2015, 304, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Brennan, K.C.; Bates, E.A.; Shapiro, R.E.; Zyuzin, J.; Hallows, W.C.; Huang, Y.; Lee, H.Y.; Jones, C.R.; Fu, Y.H.; Charles, A.C. Casein kinase idelta mutations in familial migraine and advanced sleep phase. Sci. Transl. Med. 2013, 5, 183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutigliano, G.; Stazi, M.; Arancio, O.; Watterson, D.M.; Origlia, N. An isoform-selective p38alpha mitogen-activated protein kinase inhibitor rescues early entorhinal cortex dysfunctions in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2018, 70, 86–91. [Google Scholar] [CrossRef]
- Robson, M.J.; Quinlan, M.A.; Margolis, K.G.; Gajewski-Kurdziel, P.A.; Veenstra-VanderWeele, J.; Gershon, M.D.; Watterson, D.M.; Blakely, R.D. p38alpha MAPK signaling drives pharmacologically reversible brain and gastrointestinal phenotypes in the SERT Ala56 mouse. Proc. Natl. Acad. Sci. USA 2018, 115, E10245–E10254. [Google Scholar] [CrossRef] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Falcicchia, C.; Tozzi, F.; Arancio, O.; Watterson, D.M.; Origlia, N. Involvement of p38 MAPK in Synaptic Function and Dysfunction. Int. J. Mol. Sci. 2020, 21, 5624. https://doi.org/10.3390/ijms21165624
Falcicchia C, Tozzi F, Arancio O, Watterson DM, Origlia N. Involvement of p38 MAPK in Synaptic Function and Dysfunction. International Journal of Molecular Sciences. 2020; 21(16):5624. https://doi.org/10.3390/ijms21165624
Chicago/Turabian StyleFalcicchia, Chiara, Francesca Tozzi, Ottavio Arancio, Daniel Martin Watterson, and Nicola Origlia. 2020. "Involvement of p38 MAPK in Synaptic Function and Dysfunction" International Journal of Molecular Sciences 21, no. 16: 5624. https://doi.org/10.3390/ijms21165624
APA StyleFalcicchia, C., Tozzi, F., Arancio, O., Watterson, D. M., & Origlia, N. (2020). Involvement of p38 MAPK in Synaptic Function and Dysfunction. International Journal of Molecular Sciences, 21(16), 5624. https://doi.org/10.3390/ijms21165624