SARS-CoV-2 Entry Inhibitors: Small Molecules and Peptides Targeting Virus or Host Cells



 ,
,
Abstract
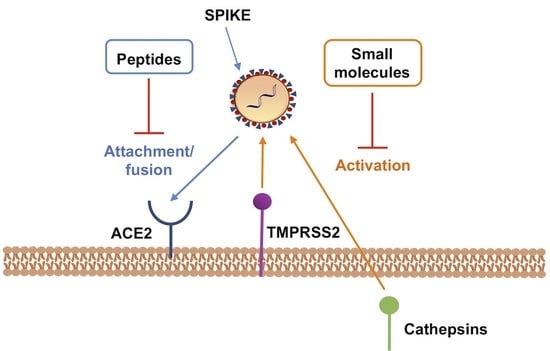
:
1. Introduction
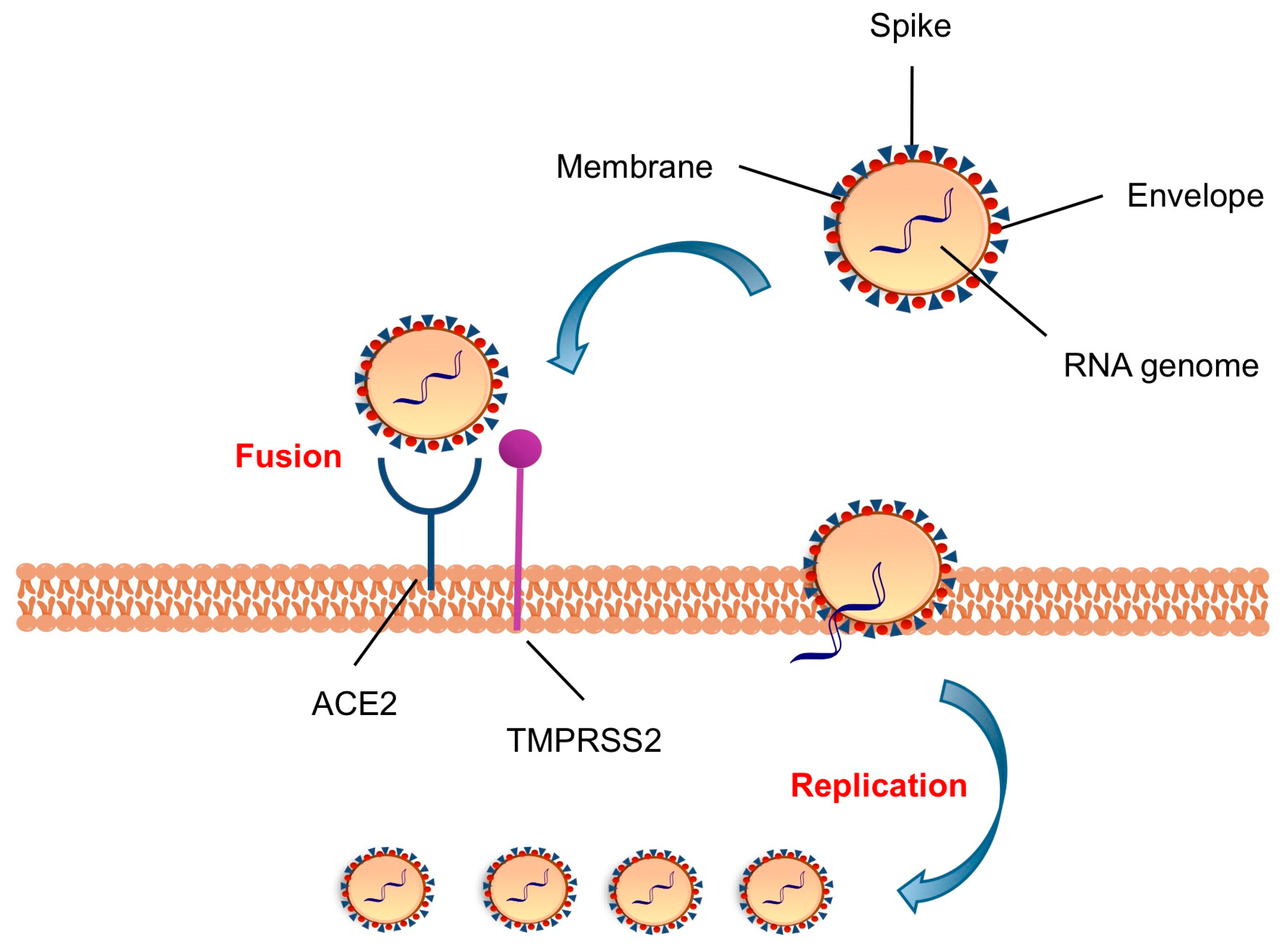
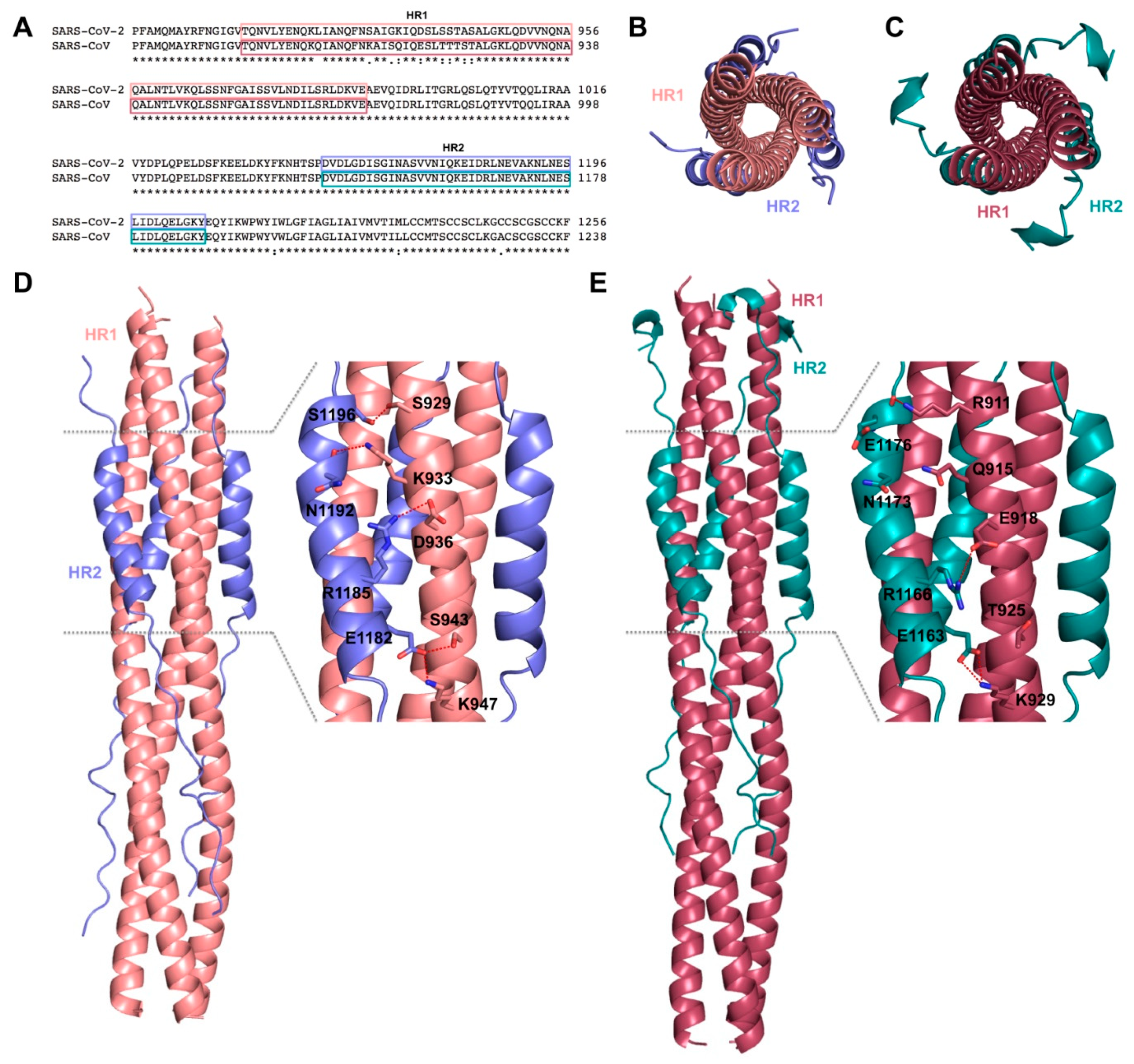
2. Targeting S Protein to Block Viral Entry
3. Targeting Host Protease to Block Viral Entry
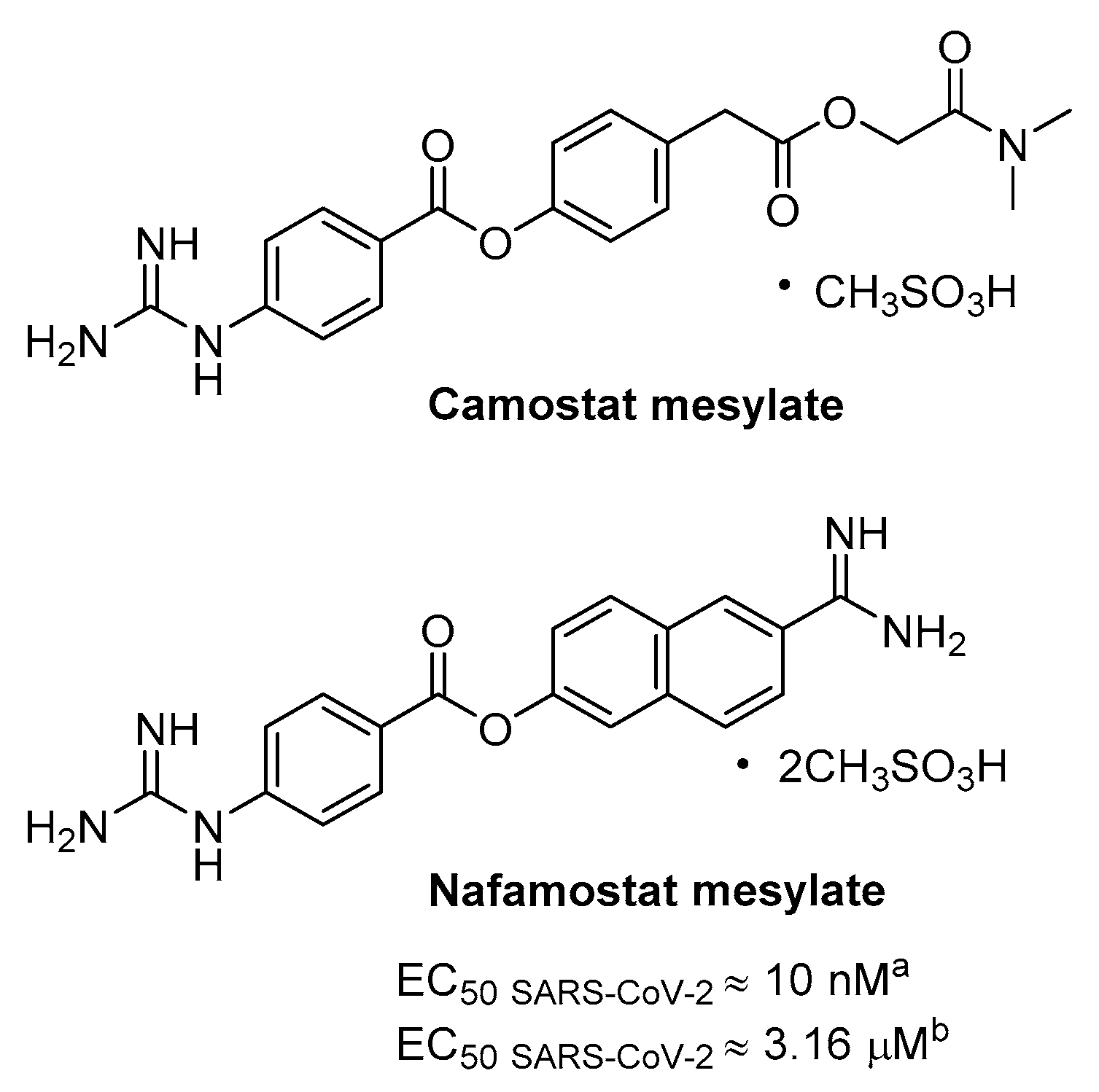
3.1. TMPRSS2 as Host Target and Its Inhibitors
3.2. CatB/L as Host Targets and Its Inhibitors
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| WHO | World Health Organization |
| CoV | Coronavirus |
| SARS-CoV-2 | Severe acute respiratory syndrome 2 |
| COVID-19 | Coronavirus disease 2019 |
| SARS-CoV | Severe acute respiratory syndrome virus |
| MERS-CoV | Middle East respiratory syndrome virus |
| ACE2 | Angiotensin-converting enzyme 2 |
| Cat | Cathepsin |
| TMPRSS2 | Transmembrane Protease Serine Type 2 Secretory |
| S | Spike protein |
| RBD | Receptor Binding Domain |
| HB | Helix Bundle |
| E | Envelope |
| M | Membrane |
| N | Nucleocapsid |
| PDB | Protein Data Bank |
References
- WHO. Ten Threats to Global Health in 2019; WHO: Geneva, Switzerland, 2019; Available online: https://www.who.int/emergencies/ten-threats-to-global-health-in-2019 (accessed on 8 June 2020).
- WHO. Prioritizing Diseases for Research and Development in Emergency Contexts; WHO: Geneva, Switzerland, 2020; Available online: Https://www.who.int/activities/prioritizing-diseases-for-research-anddevelopment-In-emergency-contexts (accessed on 8 June 2020).
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Rolling Updates on Coronavirus Disease (COVID-19). Available online: Https://www.who.int/emergencies/diseases/novel-coronavirus-2019/events-as-they-happen (accessed on 8 June 2020).
- Riddle, M.S.; Connor, B.A.; Beeching, N.J.; DuPont, H.L.; Hamer, D.H.; Kozarsky, P.; Libman, M.; Steffen, R.; Taylor, D.; Tribble, D.R.; et al. 2020 Hubei Lockdowns. J. Travel Med. 2020, 24, S63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saglietto, A.; D’Ascenzo, F.; Zoccai, G.B.; De Ferrari, G.M. COVID-19 in Europe: The Italian lesson. Lancet 2020, 395, 1110–1111. [Google Scholar] [CrossRef]
- Remuzzi, A.; Remuzzi, G. COVID-19 and Italy: What next? Lancet 2020, 395, 1225–1228. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2018, 18, 41–58. [Google Scholar] [CrossRef]
- Alpern, J.D.; Gertner, E. Off-Label Therapies for COVID-19-Are We All In This Together? Clin. Pharmacol. Ther. 2020, 108, 182–184. [Google Scholar] [CrossRef] [Green Version]
- Goldman, J.D.; Lye, D.C.B.; Hui, D.S.; Marks, K.M.; Bruno, R.; Montejano, R.; Spinner, C.D.; Galli, M.; Ahn, M.-Y.; Nahass, R.G.; et al. Remdesivir for 5 or 10 Days in Patients with Severe Covid-19. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of Covid-19—Preliminary Report. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Liu, C.; Zhou, Q.; Li, Y.; Garner, L.V.; Watkins, S.P.; Carter, L.J.; Smoot, J.; Gregg, A.C.; Daniels, A.D.; Jervey, S.; et al. Research and Development on Therapeutic Agents and Vaccines for COVID-19 and Related Human Coronavirus Diseases. ACS Cent. Sci. 2020, 6, 315–331. [Google Scholar] [CrossRef]
- Chan, J.F.W.; Kok, K.H.; Zhu, Z.; Chu, H.; To, K.K.W.; Yuan, S.; Yuen, K.Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; de Groot, R.J.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.L.; Lauber, C.; Leontovich, A.M.; Neuman, B.W.; et al. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shereen, M.A.; Khan, S.; Kazmi, A.; Bashir, N.; Siddique, R. COVID-19 infection: Origin, transmission, and characteristics of human coronaviruses. J. Adv. Res. 2020, 24, 91–98. [Google Scholar] [CrossRef]
- Sanche, S.; Lin, Y.T.; Xu, C.; Romero-Severson, E.; Hengartner, N.; Ke, R. High Contagiousness and Rapid Spread of Severe Acute Respiratory Syndrome Coronavirus 2. Emerg. Infect. Dis. 2020, 26, 1470–1477. [Google Scholar] [CrossRef]
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. In Coronaviruses: Methods and Protocols; Springer: New York, NY, USA, 2015; Volume 1282, pp. 1–23. ISBN 9781493924387. [Google Scholar]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Bosch, B.J.; van der Zee, R.; de Haan, C.A.M.; Rottier, P.J.M. The Coronavirus Spike Protein Is a Class I Virus Fusion Protein: Structural and Functional Characterization of the Fusion Core Complex. J. Virol. 2003, 77, 8801–8811. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, K.; Lokugamage, K.G.; Makino, S. Viral and Cellular mRNA Translation in Coronavirus-Infected Cells. In Advances in Virus Research; Academic Press Inc.: Cambridge, MA, USA, 2016; Volume 96, pp. 165–192. [Google Scholar]
- Zhao, L.; Jha, B.K.; Wu, A.; Elliott, R.; Ziebuhr, J.; Gorbalenya, A.E.; Silverman, R.H.; Weiss, S.R. Antagonism of the interferon-induced OAS-RNase L pathway by murine coronavirus ns2 protein is required for virus replication and liver pathology. Cell Host Microbe 2012, 11, 607–616. [Google Scholar] [CrossRef] [Green Version]
- MacArthur, R.D.; Novak, R.M. Reviews Of Anti-infective Agents: Maraviroc: The First of a New Class of Antiretroviral Agents. Clin. Infect. Dis. 2008, 47, 236–241. [Google Scholar] [CrossRef]
- Matthews, T.; Salgo, M.; Greenberg, M.; Chung, J.; DeMasi, R.; Bolognesi, D. Enfuvirtide: The first therapy to inhibit the entry of HIV-1 into host CD4 lymphocytes. Nat. Rev. Drug Discov. 2004, 3, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Meanwell, N.A.; Krystal, M.R.; Nowicka-Sans, B.; Langley, D.R.; Conlon, D.A.; Eastgate, M.D.; Grasela, D.M.; Timmins, P.; Wang, T.; Kadow, J.F. Inhibitors of HIV-1 Attachment: The Discovery and Development of Temsavir and its Prodrug Fostemsavir. J. Med. Chem. 2018, 61, 62–80. [Google Scholar] [PubMed]
- Teissier, E.; Penin, F.; Pécheur, E.I. Targeting cell entry of enveloped viruses as an antiviral strategy. Molecules 2011, 16, 221–250. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Xiong, X.; Park, Y.J.; Tortorici, M.A.; Snijder, J.; Quispe, J.; Cameroni, E.; Gopal, R.; Dai, M.; Lanzavecchia, A.; et al. Unexpected Receptor Functional Mimicry Elucidates Activation of Coronavirus Fusion. Cell 2019, 176, 1026–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walls, A.C.; Tortorici, M.A.; Bosch, B.J.; Frenz, B.; Rottier, P.J.M.; DiMaio, F.; Rey, F.A.; Veesler, D. Cryo-electron microscopy structure of a coronavirus spike glycoprotein trimer. Nature 2016, 531, 114–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Y.; Cao, D.; Zhang, Y.; Ma, J.; Qi, J.; Wang, Q.; Lu, G.; Wu, Y.; Yan, J.; Shi, Y.; et al. Cryo-EM structures of MERS-CoV and SARS-CoV spike glycoproteins reveal the dynamic receptor binding domains. Nat. Commun. 2017, 8, 1–9. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef]
- Walls, A.C.; Tortorici, M.A.; Snijder, J.; Xiong, X.; Bosch, B.J.; Rey, F.A.; Veesler, D. Tectonic conformational changes of a coronavirus spike glycoprotein promote membrane fusion. Proc. Natl. Acad. Sci. USA 2017, 114, 11157–11162. [Google Scholar] [CrossRef] [Green Version]
- Pallesen, J.; Wang, N.; Corbett, K.S.; Wrapp, D.; Kirchdoerfer, R.N.; Turner, H.L.; Cottrell, C.A.; Becker, M.M.; Wang, L.; Shi, W.; et al. Immunogenicity and structures of a rationally designed prefusion MERS-CoV spike antigen. Proc. Natl. Acad. Sci. USA 2017, 114, E7348–E7357. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Li, W.; Farzan, M.; Harrison, S.C. Structural biology: Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 2005, 309, 1864–1868. [Google Scholar] [CrossRef]
- Song, W.; Gui, M.; Wang, X.; Xiang, Y. Cryo-EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2. PLOS Pathog. 2018, 14, e1007236. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Chen, P.; Wang, J.; Feng, J.; Zhou, H.; Li, X.; Zhong, W.; Hao, P. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Sci. China Life Sci. 2020, 63, 457–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [Green Version]
- Jaimes, J.A.; André, N.M.; Chappie, J.S.; Millet, J.K.; Whittaker, G.R. Phylogenetic analysis and structural modeling of SARS-CoV-2 Spike protein reveals an evolutionary distinct and proteolytically sensitive activation loop. J. Mol. Biol. 2020, 432, 3309–3325. [Google Scholar] [CrossRef]
- Duquerroy, S.; Vigouroux, A.; Rottier, P.J.M.; Rey, F.A.; Jan Bosch, B. Central ions and lateral asparagine/glutamine zippers stabilize the post-fusion hairpin conformation of the SARS coronavirus spike glycoprotein. Virology 2005, 335, 276–285. [Google Scholar] [CrossRef] [Green Version]
- Xia, S.; Liu, M.; Wang, C.; Xu, W.; Lan, Q.; Feng, S.; Qi, F.; Bao, L.; Du, L.; Liu, S.; et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020, 30, 343–355. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Pomplun, S.; Loftis, A.R.; Loas, A.; Pentelute, B.L. The first-in-class peptide binder to the SARS-CoV-2 spike protein. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Xia, S.; Zhu, Y.; Liu, M.; Lan, Q.; Xu, W.; Wu, Y.; Ying, T.; Liu, S.; Shi, Z.; Jiang, S.; et al. Fusion mechanism of 2019-nCoV and fusion inhibitors targeting HR1 domain in spike protein. Cell. Mol. Immunol. 2020, 17, 765–767. [Google Scholar] [CrossRef]
- Bosch, B.J.; Martina, B.E.E.; Van Der Zee, R.; Lepault, J.; Haijema, B.J.; Versluis, C.; Heck, A.J.R.; De Groot, R.; Osterhaus, A.D.M.E.; Rottier, P.J.M. Severe acute respiratory syndrome coronavirus (SARS-CoV) infection inhibition using spike protein heptad repeat-derived peptides. Proc. Natl. Acad. Sci. USA 2004, 101, 8455–8460. [Google Scholar] [CrossRef] [Green Version]
- Xia, S.; Yan, L.; Xu, W.; Agrawal, A.S.; Algaissi, A.; Tseng, C.T.K.; Wang, Q.; Du, L.; Tan, W.; Wilson, I.A.; et al. A pan-coronavirus fusion inhibitor targeting the HR1 domain of human coronavirus spike. Sci. Adv. 2019, 5, eaav4580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, L.; Liu, Q.; Zhu, Y.; Chan, K.H.; Qin, L.; Li, Y.; Wang, Q.; Chan, J.F.W.; Du, L.; Yu, F.; et al. Structure-based discovery of Middle East respiratory syndrome coronavirus fusion inhibitor. Nat. Commun. 2014, 5, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Xiao, G.; Chen, Y.; He, Y.; Niu, J.; Escalante, C.R.; Xiong, H.; Farmar, J.; Debnath, A.K.; Tien, P.; et al. Interaction between heptad repeat 1 and 2 regions in spike protein of SARS-associated coronavirus: Implications for virus fusogenic mechanism and identification of fusion inhibitors. Lancet 2004, 363, 938–947. [Google Scholar] [CrossRef] [Green Version]
- Eckert, D.M.; Malashkevich, V.N.; Hong, L.H.; Carr, P.A.; Kim, P.S. Inhibiting HIV-1 entry: Discovery of D-peptide inhibitors that target the gp41 coiled-coil pocket. Cell 1999, 99, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Lambert, D.M.; Barney, S.; Lambert, A.L.; Guthrie, K.; Medinas, R.; Davis, D.E.; Bucy, T.; Erickson, J.; Merutka, G.; Petteway, S.R. Peptides from conserved regions of paramyxovirus fusion (F) proteins are potent inhibitors of viral fusion. Proc. Natl. Acad. Sci. USA 1996, 93, 2186–2191. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Li, W.; Jiang, S. Peptide fusion inhibitors targeting the HIV-1 gp41: A patent review (2009–2014). Expert Opin. Ther. Pat. 2015, 25, 159–173. [Google Scholar] [CrossRef]
- Pang, W.; Tam, S.-C.; Zheng, Y.-T. Current peptide HIV type-1 fusion inhibitors. Antivir. Chem. Chemother. 2009, 20, 1–18. [Google Scholar] [CrossRef]
- Chong, H.; Xue, J.; Xiong, S.; Cong, Z.; Ding, X.; Zhu, Y.; Liu, Z.; Chen, T.; Feng, Y.; He, L.; et al. A Lipopeptide HIV-1/2 Fusion Inhibitor with Highly Potent In Vitro, Ex Vivo, and In Vivo Antiviral Activity. J. Virol. 2017, 91, e00288-18. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Zhao, L.; Xia, S.; Zhang, T.; Cao, R.; Liang, G.; Li, Y.; Meng, G.; Wang, W.; Shi, W.; et al. De Novo Design of α-Helical Lipopeptides Targeting Viral Fusion Proteins: A Promising Strategy for Relatively Broad-Spectrum Antiviral Drug Discovery. J. Med. Chem. 2018, 61, 8734–8745. [Google Scholar]
- Zhu, Y.; Yu, D.; Yan, H.; Chong, H.; He, Y. Design of potent membrane fusion inhibitors against SARS-CoV-2, an emerging coronavirus with high fusogenic activity. J. Virol. 2020, 94, e00635-20. [Google Scholar] [CrossRef]
- Yamamoto, N.; Yang, R.; Yoshinaka, Y.; Amari, S.; Nakano, T.; Cinatl, J.; Rabenau, H.; Doerr, H.W.; Hunsmann, G.; Otaka, A.; et al. HIV protease inhibitor nelfinavir inhibits replication of SARS-associated coronavirus. Biochem. Biophys. Res. Commun. 2004, 318, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Musarrat, F.; Chouljenko, V.; Dahal, A.; Nabi, R.; Chouljenko, T.; Jois, S.D.; Kousoulas, K.G. The anti-HIV drug nelfinavir mesylate (Viracept) is a potent inhibitor of cell fusion caused by the SARSCoV-2 spike (S) glycoprotein warranting further evaluation as an antiviral against COVID-19 infections. J. Med. Virol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Kawase, M.; Shirato, K.; van der Hoek, L.; Taguchi, F.; Matsuyama, S. Simultaneous Treatment of Human Bronchial Epithelial Cells with Serine and Cysteine Protease Inhibitors Prevents Severe Acute Respiratory Syndrome Coronavirus Entry. J. Virol. 2012, 86, 6537–6545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwata-Yoshikawa, N.; Okamura, T.; Shimizu, Y.; Hasegawa, H.; Takeda, M.; Nagata, N. TMPRSS2 Contributes to Virus Spread and Immunopathology in the Airways of Murine Models after Coronavirus Infection. J. Virol. 2019, 93, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuyama, S.; Nagata, N.; Shirato, K.; Kawase, M.; Takeda, M.; Taguchi, F. Efficient Activation of the Severe Acute Respiratory Syndrome Coronavirus Spike Protein by the Transmembrane Protease TMPRSS2. J. Virol. 2010, 84, 12658–12664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glowacka, I.; Bertram, S.; Muller, M.A.; Allen, P.; Soilleux, E.; Pfefferle, S.; Steffen, I.; Tsegaye, T.S.; He, Y.; Gnirss, K.; et al. Evidence that TMPRSS2 Activates the Severe Acute Respiratory Syndrome Coronavirus Spike Protein for Membrane Fusion and Reduces Viral Control by the Humoral Immune Response. J. Virol. 2011, 85, 4122–4134. [Google Scholar] [CrossRef] [Green Version]
- Simmons, G.; Gosalia, D.N.; Rennekamp, A.J.; Reeves, J.D.; Diamond, S.L.; Bates, P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. USA 2005, 102, 11876–11881. [Google Scholar] [CrossRef] [Green Version]
- Szabo, R.; Bugge, T.H. Type II transmembrane serine proteases in development and disease. Int. J. Biochem. Cell Biol. 2008, 40, 1297–1316. [Google Scholar] [CrossRef]
- Fukushi, S.; Mizutani, T.; Saijo, M.; Matsuyama, S.; Miyajima, N.; Taguchi, F.; Itamura, S.; Kurane, I.; Morikawa, S. Vesicular stomatitis virus pseudotyped with severe acute respiratory syndrome coronavirus spike protein. J. Gen. Virol. 2005, 86, 2269–2274. [Google Scholar] [CrossRef]
- Choi, S.Y.; Bertram, S.; Glowacka, I.; Park, Y.W.; Pöhlmann, S. Type II transmembrane serine proteases in cancer and viral infections. Trends Mol. Med. 2009, 15, 303–312. [Google Scholar] [CrossRef]
- Wilson, S.; Greer, B.; Hooper, J.; Zijlstra, A.; Walker, B.; Quigley, J.; Hawthorne, S. The membrane-anchored serine protease, TMPRSS2, activates PAR-2 in prostate cancer cells. Biochem. J. 2005, 388, 967–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bottcher-Friebertshauser, E.; Freuer, C.; Sielaff, F.; Schmidt, S.; Eickmann, M.; Uhlendorff, J.; Steinmetzer, T.; Klenk, H.D.; Garten, W. Cleavage of Influenza Virus Hemagglutinin by Airway Proteases TMPRSS2 and HAT Differs in Subcellular Localization and Susceptibility to Protease Inhibitors. J. Virol. 2010, 84, 5605–5614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- TMPRSS2—An overview | ScienceDirect Topics. Available online: https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/tmprss2 (accessed on 8 June 2020).
- Shulla, A.; Heald-Sargent, T.; Subramanya, G.; Zhao, J.; Perlman, S.; Gallagher, T. A Transmembrane Serine Protease Is Linked to the Severe Acute Respiratory Syndrome Coronavirus Receptor and Activates Virus Entry. J. Virol. 2011, 85, 873–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Vedantham, P.; Lu, K.; Agudelo, J.; Carrion, R.; Nunneley, J.W.; Barnard, D.; Pöhlmann, S.; McKerrow, J.H.; Renslo, A.R.; et al. Protease inhibitors targeting coronavirus and filovirus entry. Antiviral Res. 2015, 116, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Matsuyama, S.; Li, X.; Takeda, M.; Kawaguchi, Y.; Inoue, J.I.; Matsuda, Z. Identification of nafamostat as a potent inhibitor of middle east respiratory syndrome Coronavirus s protein-mediated membrane fusion using the split-protein-based cell-cell fusion assay. Antimicrob. Agents Chemother. 2016, 60, 6532–6539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, M.; Kiso, M.; Sakai-Tagawa, Y.; Iwatsuki-Horimoto, K.; Imai, M.; Takeda, M.; Kinoshita, N.; Ohmagari, N.; Gohda, J.; Semba, K.; et al. The anticoagulant nafamostat potently inhibits SARS-CoV-2 S protein-mediated fusion in a cell fusion assay system and viral infection in vitro in a cell-type-dependent manner. Viruses 2020, 12, 629. [Google Scholar] [CrossRef] [PubMed]
- Muto, S.; Imai, M.; Asano, Y. Mechanisms of hyperkalemia caused by nafamostat mesilate. Gen. Pharmacol. 1995, 26, 1627–1632. [Google Scholar] [CrossRef]
- Lucas, J.M.; Heinlein, C.; Kim, T.; Hernandez, S.A.; Malik, M.S.; True, L.D.; Morrissey, C.; Corey, E.; Montgomery, B.; Mostaghel, E.; et al. The androgen-regulated protease TMPRSS2 activates a proteolytic cascade involving components of the tumor microenvironment and promotes prostate cancer metastasis. Cancer Discov. 2014, 4, 1310–1325. [Google Scholar] [CrossRef] [Green Version]
- Sisay, M.T.; Steinmetzer, T.; Stirnberg, M.; Maurer, E.; Hammami, M.; Bajorath, J.; Gütschow, M. Identification of the first low-molecular-weight inhibitors of matriptase-2. J. Med. Chem. 2010, 53, 5523–5535. [Google Scholar] [CrossRef]
- Stürzebecher, J.; Prasa, D.; Hauptmann, J.; Vieweg, H.; Wikström, P. Synthesis and structure-activity relationships of potent thrombin inhibitors: Piperazides of 3-amidinophenylalanine. J. Med. Chem. 1997, 40, 3091–3099. [Google Scholar] [CrossRef]
- Sielaff, F.; Böttcher-Friebertshäuser, E.; Meyer, D.; Saupe, S.M.; Volk, I.M.; Garten, W.; Steinmetzer, T. Development of substrate analogue inhibitors for the human airway trypsin-like protease HAT. Bioorganic Med. Chem. Lett. 2011, 21, 4860–4864. [Google Scholar] [CrossRef] [PubMed]
- Meyer, D.; Sielaff, F.; Hammami, M.; Böttcher-Friebertshäuser, E.; Garten, W.; Steinmetzer, T. Identification of the first synthetic inhibitors of the type II transmembrane serine protease TMPRSS2 suitable for inhibition of influenza virus activation. Biochem. J. 2013, 452, 331–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bestle, D.; Heindl, M.R.; Limburg, H.; Van Lam van, T.; Pilgram, O.; Moulton, H.; Stein, D.A.; Hardes, K.; Eickmann, M.; Dolnik, O.; et al. TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. Life Sci. Alliance 2020, 3, e202000786. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.-W.; Qian, M.-Q.; Yu, K.; Narva, S.; Yu, F.; Wu, Y.-L.; Zhang, W. Inhibition of Influenza A virus propagation by benzoselenoxanthenes stabilizing TMPRSS2 Gene G-quadruplex and hence down-regulating TMPRSS2 expression. Sci. Rep. 2020, 10, 7635. [Google Scholar] [CrossRef] [PubMed]
- Shirato, K.; Kawase, M.; Matsuyama, S. Wild-type human coronaviruses prefer cell-surface TMPRSS2 to endosomal cathepsins for cell entry. Virology 2018, 517, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Shirato, K.; Kanou, K.; Kawase, M.; Matsuyama, S. Clinical Isolates of Human Coronavirus 229E Bypass the Endosome for Cell Entry. J. Virol. 2017, 91, e01387-16. [Google Scholar] [CrossRef] [Green Version]
- Reiser, J.; Adair, B.; Reinheckel, T. Specialized roles for cysteine cathepsins in health and disease. J. Clin. Investig. 2010, 120, 3421–3431. [Google Scholar] [CrossRef] [Green Version]
- Kang, K.; Kim, W. Recent developments of cathepsin inhibitors and their selectivity. Expert Opin. Ther. Pat. 2002, 12, 419–432. [Google Scholar] [CrossRef]
- Lee-Dutra, A.; Wiener, D.K.; Sun, S. Cathepsin S inhibitors: 2004–2010. Expert Opin. Ther. Pat. 2011, 21, 311–337. [Google Scholar] [CrossRef]
- Hernandez, A.A.; Roush, W.R. Recent advances in the synthesis, design and selection of cysteine protease inhibitors. Curr. Opin. Chem. Biol. 2002, 6, 459–465. [Google Scholar] [CrossRef]
- Dana, D.; Pathak, S.K. A review of small molecule inhibitors and functional probes of human cathepsin L. Molecules 2020, 25, 698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simmons, G.; Zmora, P.; Gierer, S.; Heurich, A.; Pöhlmann, S. Proteolytic activation of the SARS-coronavirus spike protein: Cutting enzymes at the cutting edge of antiviral research. Antivir. Res. 2013, 100, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Pager, C.T.; Craft, W.W.; Patch, J.; Dutch, R.E. A mature and fusogenic form of the Nipah virus fusion protein requires proteolytic processing by cathepsin L. Virology 2006, 346, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Y.; Fang, J.; Ao, G.Z. Cathepsin B and L inhibitors: A patent review (2010 - present). Expert Opin. Ther. Pat. 2017, 27, 643–656. [Google Scholar] [CrossRef]
- Musil, D.; Zucic, D.; Turk, D.; Engh, R.A.; Mayr, I.; Huber, R.; Popovic, T.; Turk, V.; Towatari, T.; Katunuma, N. The refined 2.15 A X-ray crystal structure of human liver cathepsin B: The structural basis for its specificity. EMBO J. 1991, 10, 2321–2330. [Google Scholar] [CrossRef]
- Cotrin, S.S.; Puzer, L.; De Souza Judice, W.A.; Juliano, L.; Carmona, A.K.; Juliano, M.A. Positional-scanning combinatorial libraries of fluorescence resonance energy transfer peptides to define substrate specificity of carboxydipeptidases: Assays with human cathepsin B. Anal. Biochem. 2004, 335, 244–252. [Google Scholar] [CrossRef]
- Shenoy, R.T.; Sivaraman, J. Structural basis for reversible and irreversible inhibition of human cathepsin L by their respective dipeptidyl glyoxal and diazomethylketone inhibitors. J. Struct. Biol. 2011, 173, 14–19. [Google Scholar] [CrossRef]
- Varughese, K.I.; Ahmed, F.R.; Carey, P.R.; Hasnain, S.; Huber, C.P.; Storer, A.C. Crystal Structure of a Papain–E-64 Complex. Biochemistry 1989, 28, 1330–1332. [Google Scholar] [CrossRef]
- Barrett, A.J.; Kembhavi, A.A.; Brown, M.A.; Kirschke, H.; Knight, C.G.; Tamai, M.; Hanada, K. L-trans-Epoxysuccinyl-leucylamido(4-guanidino)butane (E-64) and its analogues as inhibitors of cysteine proteinases including cathepsins, B, H and L. Biochem. J. 1982, 201, 189–198. [Google Scholar] [CrossRef] [Green Version]
- Katunuma, N.; Murata, E.; Kakegawa, H.; Matsui, A.; Tsuzuki, H.; Tsuge, H.; Turk, D.; Turk, V.; Fukushima, M.; Tada, Y.; et al. Structure based development of novel specific inhibitors for cathepsin L and cathepsin S in vitro and in vivo. FEBS Lett. 1999, 458, 6–10. [Google Scholar] [CrossRef] [Green Version]
- Tsuge, H.; Nishimura, T.; Tada, Y.; Asao, T.; Turk, D.; Turk, V.; Katunuma, N. Inhibition Mechanism of Cathepsin L-Specific Inhibitors Based on the Crystal Structure of Papain–CLIK148 Complex. Biochem. Biophys. Res. Commun. 1999, 266, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Engel, J.C.; Doyle, P.S.; Hsieh, I.; McKerrow, J.H. Cysteine protease inhibitors cure an experimental Trypanosoma cruzi infection. J. Exp. Med. 1998, 188, 725–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choy, J.W.; Bryant, C.; Calvet, C.M.; Doyle, P.S.; Gunatilleke, S.S.; Leung, S.S.F.; Ang, K.K.H.; Chen, S.; Gut, J.; Oses-Prieto, J.A.; et al. Chemical-biological characterization of a cruzain inhibitor reveals a second target and a mammalian off-target. Beilstein J. Org. Chem. 2013, 9, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Barr, S.C.; Warner, K.L.; Kornreic, B.G.; Piscitelli, J.; Wolfe, A.; Benet, L.; McKerrow, J.H. A cysteine protease inhibitor protects dogs from cardiac damage during infection by Trypanosoma cruzi. Antimicrob. Agents Chemother. 2005, 49, 5160–5161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKerrow, J.H. Designing drugs for parasitic diseases of the developing world. PLoS Med. 2005, 2, e210. [Google Scholar] [CrossRef] [Green Version]
- Jacobsen, W.; Christians, U.; Benet, L.Z. In vitro evaluation of the disposition of a novel cysteine protease inhibitor. Drug Metab. Dispos. 2000, 28, 1343–1351. [Google Scholar]
- Abdulla, M.H.; Lim, K.C.; Sajid, M.; McKerrow, J.H.; Caffrey, C.R. Schistosomiasis mansoni: Novel chemotherapy using a cysteine protease inhibitor. PLoS Med. 2007, 4, e14. [Google Scholar] [CrossRef]
- Pauly, T.A.; Sulea, T.; Ammirati, M.; Sivaraman, J.; Danley, D.E.; Griffor, M.C.; Kamath, A.V.; Wang, I.-K.; Laird, E.R.; Seddon, A.P.; et al. Specificity Determinants of Human Cathepsin S Revealed by Crystal Structures of Complexes. Biochemistry 2003, 42, 3203–3213. [Google Scholar] [CrossRef]
- McGrath, M.E.; Klaus, J.L.; Barnes, M.G.; Bromme, D. Crystal structure of human cathepsin K complexed with a potent inhibitor. Nat. Struct. Biol. 1997, 4, 105–109. [Google Scholar] [CrossRef]
- Somoza, J.R.; Palmer, J.T.; Ho, J.D. The Crystal Structure of Human Cathepsin F and Its Implications for the Development of Novel Immunomodulators. J. Mol. Biol. 2002, 322, 559–568. [Google Scholar] [CrossRef]
- Mendieta, L.; Picó, A.; Tarragó, T.; Teixidó, M.; Castillo, M.; Rafecas, L.; Moyano, A.; Giralt, E. Novel peptidyl aryl vinyl sulfones as highly potent and selective inhibitors of cathepsins L and B. Chem. Med. Chem. 2010, 5, 1556–1567. [Google Scholar] [CrossRef] [PubMed]
- Barnard, D.L.; Hubbard, V.D.; Burton, J.; Smee, D.F.; Morrey, J.D.; Otto, M.J.; Sidwell, R.W. Inhibition of severe acute respiratory syndrome-associated coronavirus (SARSCoV) by calpain inhibitors and β-D-N4-hydroxycytidine. Antivir. Chem. Chemother. 2004, 15, 15–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.-A.; Howlett, W.; He, Q.P.; Miyashita, H.; Siddiqui, M.; Shuaib, A. Postischemic treatment with calpain inhibitor MDL 28170 ameliorates brain damage in a gerbil model of global ischemia. Neurosci. Lett. 1998, 247, 17–20. [Google Scholar] [CrossRef]
- Kharatmal, S.B.; Singh, J.N.; Sharma, S.S. Calpain inhibitor, MDL 28170 confer electrophysiological, nociceptive and biochemical improvement in diabetic neuropathy. Neuropharmacology 2015, 97, 113–121. [Google Scholar] [CrossRef]
- Myers, M.C.; Shah, P.P.; Beavers, M.P.; Napper, A.D.; Diamond, S.L.; Smith, A.B.; Huryn, D.M. Design, synthesis, and evaluation of inhibitors of cathepsin L: Exploiting a unique thiocarbazate chemotype. Bioorganic Med. Chem. Lett. 2008, 18, 3646–3651. [Google Scholar] [CrossRef] [Green Version]
- Beavers, M.P.; Myers, M.C.; Shah, P.P.; Purvis, J.E.; Diamond, S.L.; Cooperman, B.S.; Huryn, D.M.; Smith, A.B. Molecular Docking of Cathepsin L Inhibitors in the Binding Site of Papain. J. Chem. Inf. Model. 2010, 50, 2274. [Google Scholar] [CrossRef] [Green Version]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
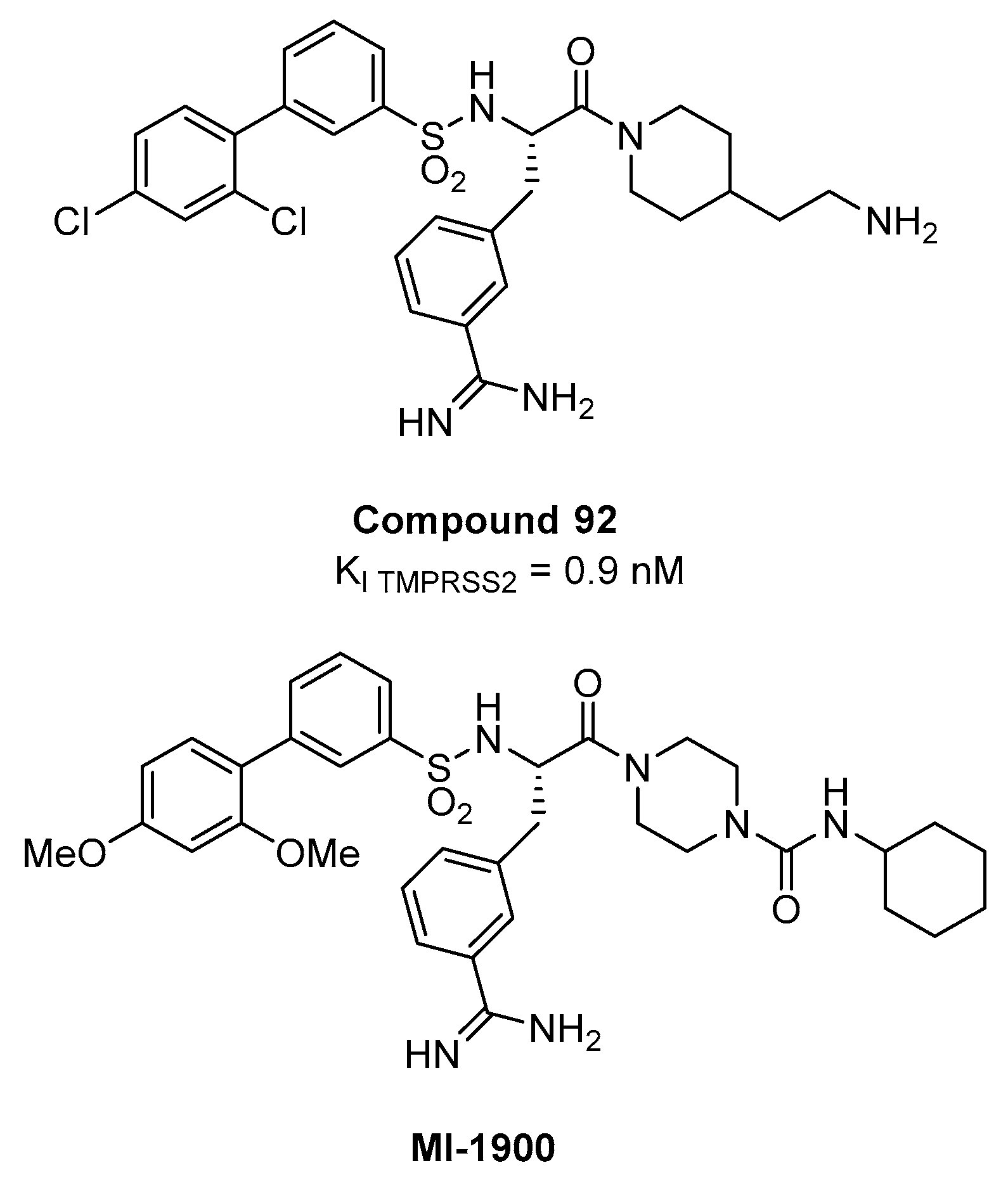{kind=link}
{kind=link}
{kind=link}
{kind=link}
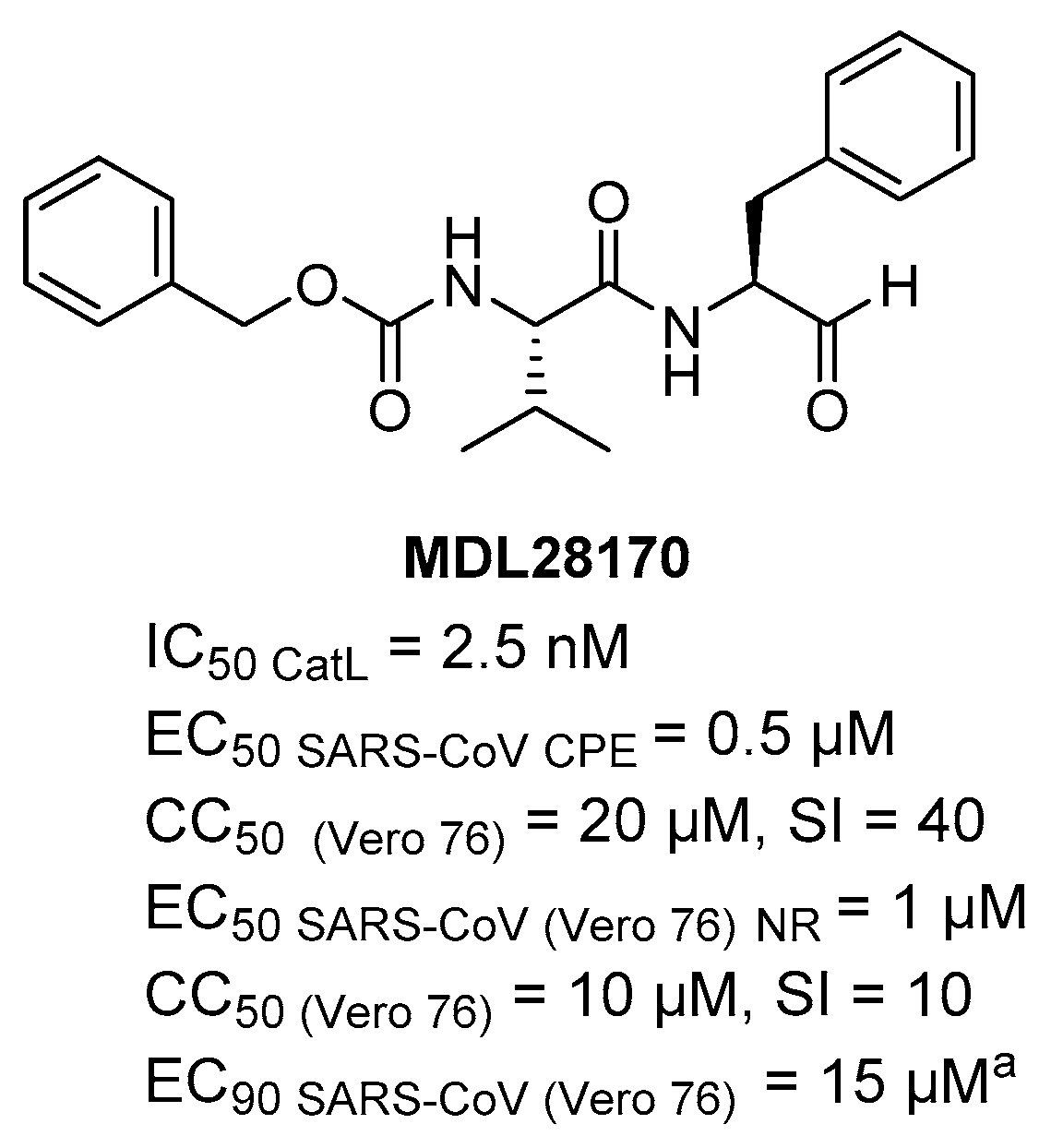{kind=link}
{kind=link}
| Compound | SARS-CoV | CPE Inhibition | NR Assay | Virus Yield Reduction | ||||
|---|---|---|---|---|---|---|---|---|
| EC50 (µM) | CC50 (µM) | SI | EC50 (µM) | CC50 (µM) | SI | EC90 (µM) | ||
| K11777 | Urbani | <0.05 | >106.6 | >2111 | <0.52 | >100 | >193 | 0.35 |
| Toronto-2 | <0.05 | 85.2 | >1704 | <0.35 | 52.7 | >151 | 1.04 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cannalire, R.; Stefanelli, I.; Cerchia, C.; Beccari, A.R.; Pelliccia, S.; Summa, V. SARS-CoV-2 Entry Inhibitors: Small Molecules and Peptides Targeting Virus or Host Cells. Int. J. Mol. Sci. 2020, 21, 5707. https://doi.org/10.3390/ijms21165707
Cannalire R, Stefanelli I, Cerchia C, Beccari AR, Pelliccia S, Summa V. SARS-CoV-2 Entry Inhibitors: Small Molecules and Peptides Targeting Virus or Host Cells. International Journal of Molecular Sciences. 2020; 21(16):5707. https://doi.org/10.3390/ijms21165707
Chicago/Turabian StyleCannalire, Rolando, Irina Stefanelli, Carmen Cerchia, Andrea R. Beccari, Sveva Pelliccia, and Vincenzo Summa. 2020. "SARS-CoV-2 Entry Inhibitors: Small Molecules and Peptides Targeting Virus or Host Cells" International Journal of Molecular Sciences 21, no. 16: 5707. https://doi.org/10.3390/ijms21165707
APA StyleCannalire, R., Stefanelli, I., Cerchia, C., Beccari, A. R., Pelliccia, S., & Summa, V. (2020). SARS-CoV-2 Entry Inhibitors: Small Molecules and Peptides Targeting Virus or Host Cells. International Journal of Molecular Sciences, 21(16), 5707. https://doi.org/10.3390/ijms21165707