Philadelphia Chromosome-Positive Leukemia in the Lymphoid Lineage—Similarities and Differences with the Myeloid Lineage and Specific Vulnerabilities


Abstract
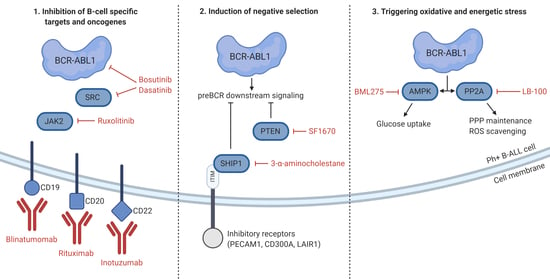
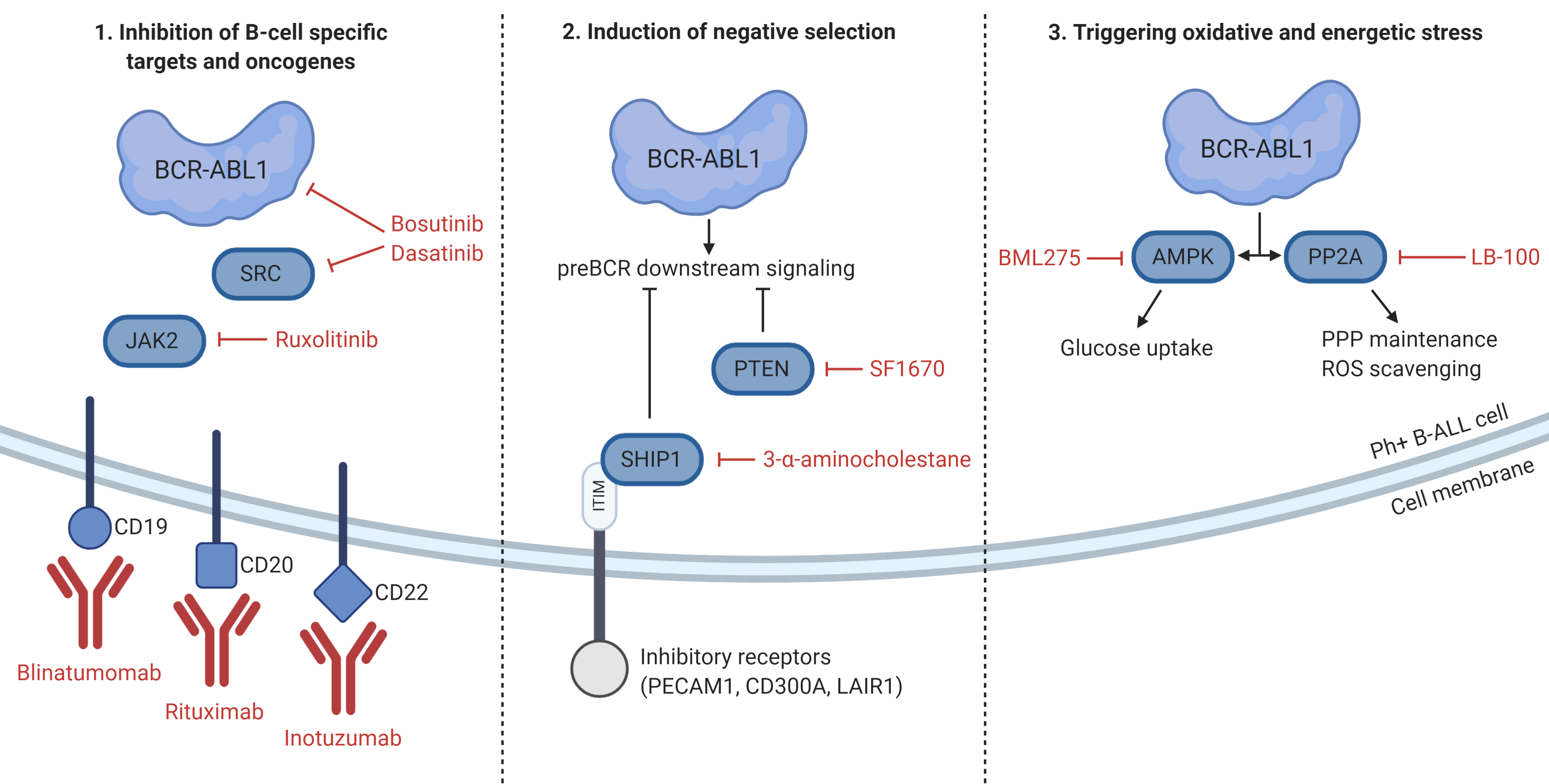
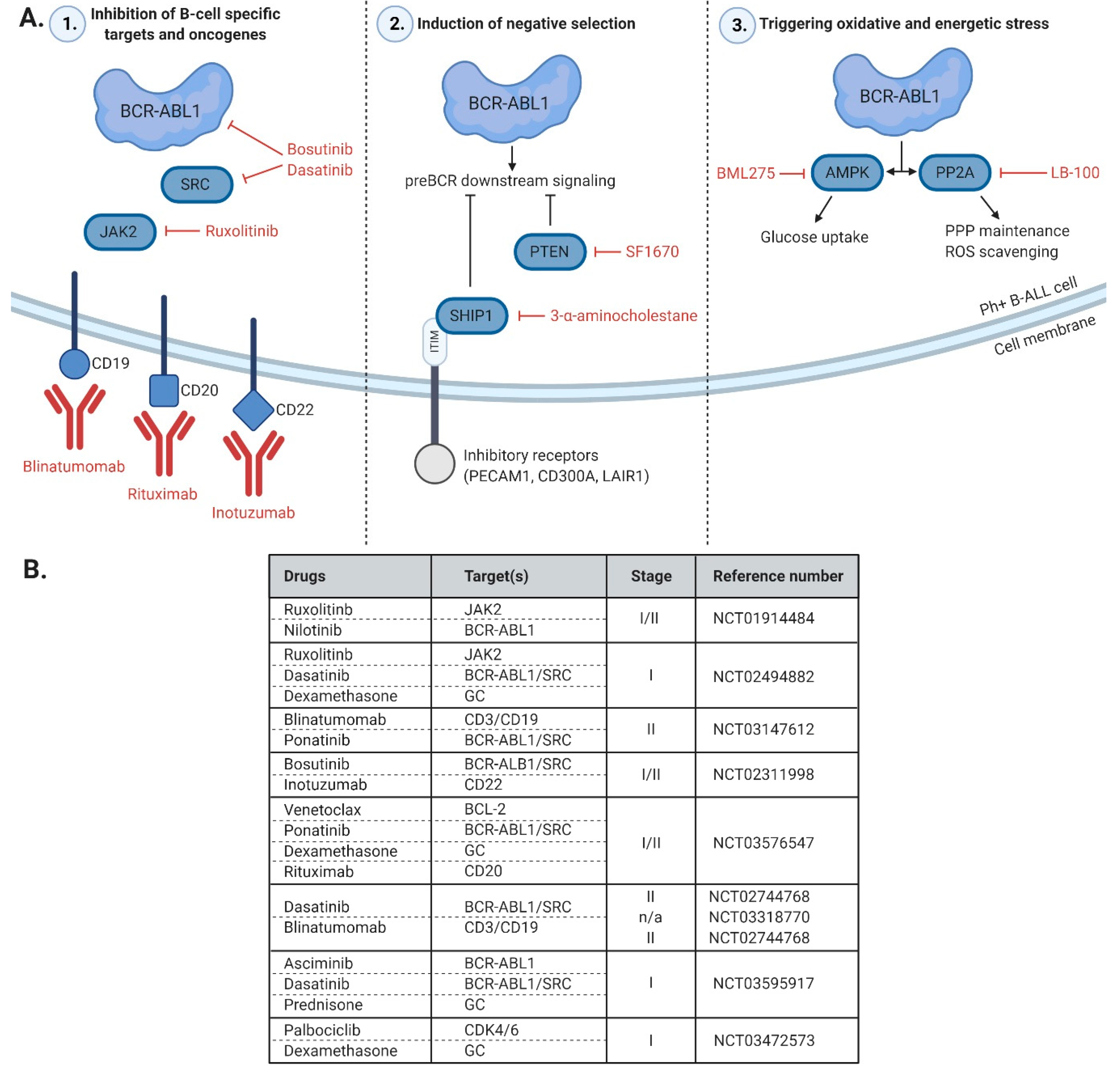
:
1. Introduction
2. Genetic Alterations and Characteristics of Different Types of Leukemia with BCR-ABL1 Translocation
2.1. Chronic Myeloid Leukemia
2.1.1. Chronic Phase CML
2.1.2. Advanced Phases of CML
2.2. Philadelphia Positive B Cell Acute Lymphoblastic Leukemia
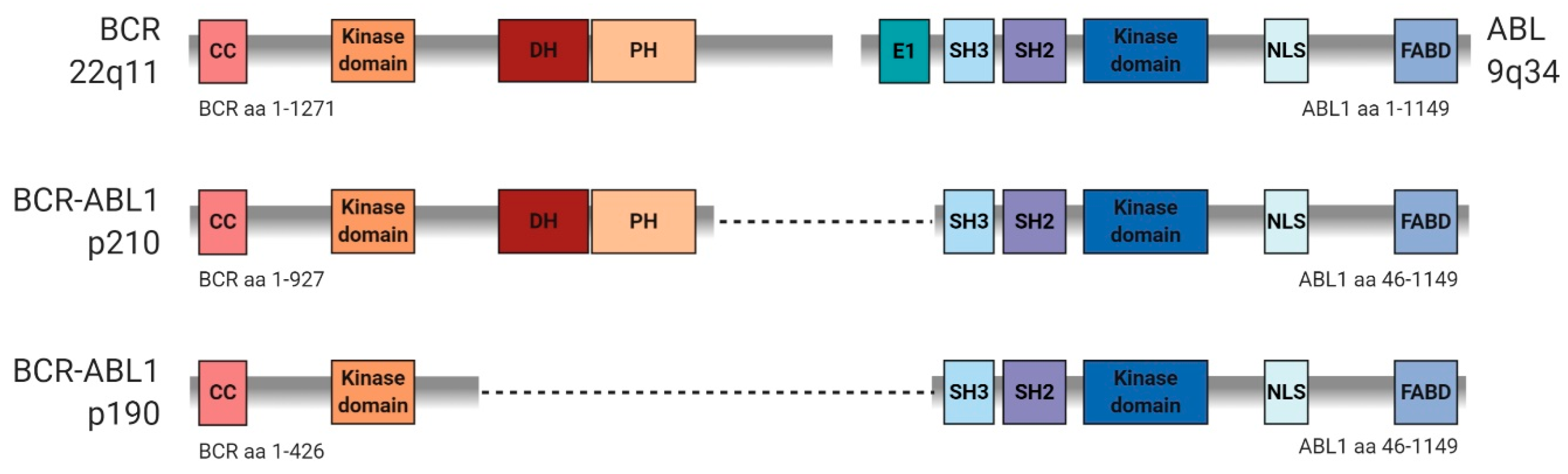
3. Similarities and Differences in Signaling Mediated by p190 and p210 BCR-ABL Variants
4. Tyrosine Kinase Inhibitors as a Core Treatment of Different Types of Ph+ Leukemia
4.1. TKIs for the Treatment of CML
4.1.1. Treatment of CML in Chronic Phase
4.1.2. Treatment of CML in Advanced Phases
4.2. TKIs in the Treatment of Ph+ ALL
4.3. Resistance to TKIs in CML and Ph+ B-ALL
4.4. Novel Tyrosine Kinase Inhibitors Tested in Clinical Trials
5. Unique Features and Lineage-Specific Drug Sensitivities of Ph+ Lymphoid Leukemia
6. Concluding Remarks and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABL1 | ABL proto-oncogene 1 |
| AKT | protein kinase B |
| ALL | acute lymphoblastic leukemia |
| allo-HSCT | allogeneic hematopoietic stem cell transplantation |
| AMPK | 5′AMP-activated protein kinase |
| AP | accelerated phase |
| aPKC | atypical protein kinase C |
| ASXL1 | additional sex combs like 1 |
| B-ALL | B cell acute lymphoblastic leukemia |
| BCL-2 | B-cell lymphoma 2 |
| BCR | breakpoint cluster region |
| BCOR | BCL6 corepressor |
| BCORL1 | BCL6 corepressor like 1 |
| BCR-ABL1 | breakpoint cluster region - ABL proto-oncogene 1 |
| BET | bromodomain and extra terminal protein |
| BM | bone marrow |
| BML275 | small molecule AMPK inhibitor |
| BP | blast phase |
| BTG1 | B-cell translocation gene 1 |
| CaMK2G | calcium/calmodulin-dependent protein kinase type II gamma chain |
| CaMKIIγ | calcium-calmodulin-dependent kinase IIγ |
| CAR | chimeric antigen receptor |
| CAT | catalase |
| CBL | Cbl proto-oncogene |
| CBLB | Cbl proto-oncogene B |
| CC | coiled-coil |
| CCyR | complete cytogenic response |
| CDKN2A/B | cyclin dependent kinase inhibitor 2A/B |
| CDKs | cyclin dependent kinases |
| CHR | complete hematologic response |
| c-KIT | proto-oncogene c-KIT |
| CML | chronic myeloid leukemia |
| CMR | complete molecular response |
| CNR2 | cannabinoid receptor type 2 |
| CNS | central nervous system |
| CP | chronic phase |
| CRK | CRK proto-oncogene |
| DH | Dbl-homology |
| DMR | deep molecular response |
| DNMT3A | DNA methyltransferase 3A |
| DOK1 | docking protein 1 |
| EBF1 | early B-cell factor 1 |
| EGR1 | early growth response protein 1 |
| ELN | European LeukamiaNet |
| EMA | European Medicines Agency |
| EPH | erythropoietin-producing human hepatocellular receptor |
| ERK | extracellular signal-regulated kinases |
| ETV6 | translocation-Ets-leukemia virus |
| EUTOS | European Treatment and Outcome Study |
| EZH2 | enhancer of zeste homolog 2 |
| FABD | F-actin binding domain |
| FAK | focal adhesion kinase |
| FDA | Food and Drug Administration |
| FGFR1 | fibroblast growth factor receptor |
| FLAG-Ida | fludarabine, arabinoside cytosine, G-CSF, idarubicin regimen |
| FLT3 | fms like tyrosine kinase 3 |
| G6PD | glucose-6-phosphate dehydrogenase |
| GC | glucocorticoid receptor |
| GLUT1/3/6 | glucose transporter 1/3/6 |
| GRB2 | growth factor receptor bound protein 2 |
| GvHL | graft versus host leukemia |
| HCLS1 | hematopoietic cell specific LYN substrate 1 |
| HCVAD | hyperfractionated cyclophosphamide, vincristine, adriamycin, dexamethasone |
| HK2/3 | hexokinase 2/3 |
| HR | hematologic response |
| HSCs | hematopoietic stem cells |
| IKZF1 | IKAROS family zinc finger 1 |
| IRIS | International Randomized Study of Interferon and STI571 |
| ITAM | immunoreceptor tyrosine-based activation motifs |
| ITIM | immunoreceptor tyrosine-based inhibition motif |
| JAK2 | Janus kinase 2 |
| KDM1A | (K)-specific demethylase 1A |
| KIT | proto-oncogene c-KIT |
| LAIR1 | leukocyte-associated immunoglobulin-like receptor 1 |
| LB-100 | small molecule PP2A inhibitor |
| LBP | lymphoid blast phase |
| LCK | lymphocyte-specific protein tyrosine kinase |
| LSCs | leukemia stem cells |
| mAbs | monoclonal antibodies |
| MAPK | mitogen-activated protein kinases |
| MBP | myeloid blast phase |
| MCyR | major cytogenetic response |
| MEK | mitogen-activated protein kinase kinase |
| MMR | major molecular response |
| MSCs | mesenchymal stem cells |
| MSH6 | mutS homolog 6 |
| mTOR | mammalian target of rapamycin |
| mTORC1 | mammalian target of rapamycin complex 1 |
| MYC | MYC proto-oncogene |
| NCK1/2 | NCK adaptor protein 1/2 |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NGS | next generation sequencing |
| NLS | nuclear localization signal |
| NR4A2 | nuclear receptor subfamily 4 group A member 2 |
| NRC3C1 | nuclear receptor subfamily 3 group C member 1 |
| OS | overall survival |
| PAX5 | paired box 5 |
| PDGFR | platelet-derived growth factor receptor |
| PECAM1 | platelet endothelial cell adhesion molecule 1 |
| PFS | progression free survival |
| Ph | Philadelphia chromosome |
| Ph- | Ph negative |
| Ph+ B-ALL | Ph-positive B cell acute lymphoblastic leukemia |
| PI3K | phosphoinositide 3-kinases |
| PIK3R2 | PI3K regulatory subunit 2 |
| PP2A | serine-threonine phosphatase 2A |
| PPP | pentose phosphate pathway |
| PRC1/2 | polycomb repressive complex 1/2 |
| PRDX2/4 | peroxiredoxin 2/4 |
| preBCR | preB-cell receptor |
| PTEN | phosphatase and tensin homolog deleted on chromosome ten |
| R/R | relapsed/refractory |
| RAG | recombinase-activating gene |
| RB1 | retinoblastoma protein 1 |
| RBP2 | retinoblastoma binding protein 2 |
| RET | RET proto-oncogene |
| ROS | reactive oxygen species |
| RUNX1 | runt-related transcription factor 1 |
| SET | SET nuclear proto-oncogene |
| SF1670 | small molecule PTEN inhibitor |
| SHC1 | SHC adaptor protein 1 |
| SHIP1/2 | SH2 domain-containing inositol 5′-phosphatase 1/2 |
| SOD2 | superoxide dismutase 2 |
| SOS1/2 | son of sevenless homolog 1/2 |
| SRC | proto-oncogene tyrosine-protein kinase Src |
| STAT | signal transducer and activator of transcription |
| TEC | tyrosine-protein kinase Tec |
| TET2/3 | Tet methylcytosine dioxygenase 2/3 |
| TKIs | tyrosine kinase inhibitors |
| TNFα/β | transforming growth factor α/β |
| TP53 | tumor protein P53 |
| TXNIP | thioredoxin-interacting protein |
| UBASH3b | ubiquitin associated and SH3 domain containing B |
| VEGFR | vascular endothelial growth factor |
References
- Ren, R. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat. Rev. Cancer 2005, 5, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Nowell, P.; Hungerford, D. A minute chromosome in human chronic granulocytic leukemia. Science 1960, 132, 1497. [Google Scholar]
- Chereda, B.; Melo, J.V. The biology and pathogenesis of chronic myeloid leukemia. In Chronic Myeloid Leukemia, 1st ed.; Hehlmann, R., Ed.; Springer: Cham, Switzerland, 2016; pp. 17–39. [Google Scholar] [CrossRef]
- Mattarucchi, E.; Guerini, V.; Rambaldi, A.; Campiotti, L.; Venco, A.; Pasquali, F.; Lo Curto, F.; Porta, G. Microhomologies and interspersed repeat elements at genomic breakpoints in chronic myeloid leukemia. Genes Chromosomes Cancer 2008, 47, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Score, J.; Calasanz, M.J.; Ottman, O.; Pane, F.; Yeh, R.F.; Sobrinho-Simoes, M.A.; Kreil, S.; Ward, D.; Hidalgo-Curtis, C.; Melo, J.V.; et al. Analysis of genomic breakpoints in p190 and p210 BCR-ABL indicate distinct mechanisms of formation. Leukemia 2010, 24, 1742–1750. [Google Scholar] [CrossRef] [Green Version]
- Reckel, S.; Gehin, C.; Tardivon, D.; Georgeon, S.; Kukenshoner, T.; Lohr, F.; Koide, A.; Buchner, L.; Panjkovich, A.; Reynaud, A.; et al. Structural and functional dissection of the DH and PH domains of oncogenic Bcr-Abl tyrosine kinase. Nat. Commun. 2017, 8, 2101. [Google Scholar] [CrossRef]
- Bernt, K.M.; Hunger, S.P. Current concepts in pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia. Front. Oncol. 2014, 4, 54. [Google Scholar] [CrossRef]
- Soverini, S.; Bassan, R.; Lion, T. Treatment and monitoring of Philadelphia chromosome-positive leukemia patients: Recent advances and remaining challenges. J. Hematol. Oncol. 2019, 12, 39. [Google Scholar] [CrossRef]
- Hochhaus, A.; Baccarani, M.; Silver, R.T.; Schiffer, C.; Apperley, J.F.; Cervantes, F.; Clark, R.E.; Cortes, J.E.; Deininger, M.W.; Guilhot, F.; et al. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia 2020, 34, 966–984. [Google Scholar] [CrossRef] [Green Version]
- Abou Dalle, I.; Kantarjian, H.M.; Short, N.J.; Konopleva, M.; Jain, N.; Garcia-Manero, G.; Garris, R.; Qiao, W.; Cortes, J.E.; O’Brien, S.; et al. Philadelphia chromosome-positive acute lymphoblastic leukemia at first relapse in the era of tyrosine kinase inhibitors. Am. J. Hematol 2019, 94, 1388–1395. [Google Scholar] [CrossRef]
- Lichty, B.D.; Keating, A.; Callum, J.; Yee, K.; Croxford, R.; Corpus, G.; Nwachukwu, B.; Kim, P.; Guo, J.; Kamel-Reid, S. Expression of p210 and p190 BCR-ABL due to alternative splicing in chronic myelogenous leukaemia. Br. J. Haematol. 1998, 103, 711–715. [Google Scholar] [CrossRef]
- Cutler, J.A.; Tahir, R.; Sreenivasamurthy, S.K.; Mitchell, C.; Renuse, S.; Nirujogi, R.S.; Patil, A.H.; Heydarian, M.; Wong, X.; Wu, X.; et al. Differential signaling through p190 and p210 BCR-ABL fusion proteins revealed by interactome and phosphoproteome analysis. Leukemia 2017, 31, 1513–1524. [Google Scholar] [CrossRef] [PubMed]
- Chan, L.N.; Chen, Z.; Braas, D.; Lee, J.W.; Xiao, G.; Geng, H.; Cosgun, K.N.; Hurtz, C.; Shojaee, S.; Cazzaniga, V.; et al. Metabolic gatekeeper function of B-lymphoid transcription factors. Nature 2017, 542, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Chan, L.N.; Muschen, M. B-cell identity as a metabolic barrier against malignant transformation. Exp. Hematol. 2017, 53, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Muschen, M. Metabolic gatekeepers to safeguard against autoimmunity and oncogenic B cell transformation. Nat. Rev. Immunol. 2019, 19, 337–348. [Google Scholar] [CrossRef]
- Shojaee, S.; Chan, L.N.; Buchner, M.; Cazzaniga, V.; Cosgun, K.N.; Geng, H.; Qiu, Y.H.; von Minden, M.D.; Ernst, T.; Hochhaus, A.; et al. PTEN opposes negative selection and enables oncogenic transformation of pre-B cells. Nat. Med. 2016, 22, 379–387. [Google Scholar] [CrossRef]
- Xiao, G.; Chan, L.N.; Klemm, L.; Braas, D.; Chen, Z.; Geng, H.; Zhang, Q.C.; Aghajanirefah, A.; Cosgun, K.N.; Sadras, T.; et al. B-cell-specific diversion of glucose carbon utilization reveals a unique vulnerability in B cell malignancies. Cell 2018, 173, 470–484.e418. [Google Scholar] [CrossRef]
- Li, S.; Ilaria, R.L., Jr.; Million, R.P.; Daley, G.Q.; Van Etten, R.A. The P190, P210, and P230 forms of the BCR/ABL oncogene induce a similar chronic myeloid leukemia-like syndrome in mice but have different lymphoid leukemogenic activity. J. Exp. Med. 1999, 189, 1399–1412. [Google Scholar] [CrossRef] [Green Version]
- Howlader, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. SEER Cancer Statistics Review (CSR) 1975–2017, 04.2020 ed.; National Cancer Institute: Bethesda, MD, USA, 2020. [Google Scholar]
- Foulon, S.; Cony-Makhoul, P.; Guerci-Bresler, A.; Delord, M.; Solary, E.; Monnereau, A.; Bonastre, J.; Tubert-Bitter, P. Using healthcare claims data to analyze the prevalence of BCR-ABL-positive chronic myeloid leukemia in France: A nationwide population-based study. Cancer Med. 2019, 8, 3296–3304. [Google Scholar] [CrossRef]
- Chereda, B.; Melo, J.V. Natural course and biology of CML. Ann. Hematol. 2015, 94 (Suppl. 2), S107–S121. [Google Scholar] [CrossRef]
- Soverini, S.; Mancini, M.; Bavaro, L.; Cavo, M.; Martinelli, G. Chronic myeloid leukemia: The paradigm of targeting oncogenic tyrosine kinase signaling and counteracting resistance for successful cancer therapy. Mol. Cancer 2018, 17, 49. [Google Scholar] [CrossRef]
- Clarkson, B.; Strife, A.; Wisniewski, D.; Lambek, C.L.; Liu, C. Chronic myelogenous leukemia as a paradigm of early cancer and possible curative strategies. Leukemia 2003, 17, 1211–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieborowska-Skorska, M.; Sullivan, K.; Dasgupta, Y.; Podszywalow-Bartnicka, P.; Hoser, G.; Maifrede, S.; Martinez, E.; Di Marcantonio, D.; Bolton-Gillespie, E.; Cramer-Morales, K.; et al. Gene expression and mutation-guided synthetic lethality eradicates proliferating and quiescent leukemia cells. J. Clin. Investig. 2017, 127, 2392–2406. [Google Scholar] [CrossRef] [PubMed]
- Houshmand, M.; Simonetti, G.; Circosta, P.; Gaidano, V.; Cignetti, A.; Martinelli, G.; Saglio, G.; Gale, R.P. Chronic myeloid leukemia stem cells. Leukemia 2019, 33, 1543–1556. [Google Scholar] [CrossRef] [Green Version]
- Warfvinge, R.; Geironson, L.; Sommarin, M.N.E.; Lang, S.; Karlsson, C.; Roschupkina, T.; Stenke, L.; Stentoft, J.; Olsson-Stromberg, U.; Hjorth-Hansen, H.; et al. Single-cell molecular analysis defines therapy response and immunophenotype of stem cell subpopulations in CML. Blood 2017, 129, 2384–2394. [Google Scholar] [CrossRef]
- Giustacchini, A.; Thongjuea, S.; Barkas, N.; Woll, P.S.; Povinelli, B.J.; Booth, C.A.G.; Sopp, P.; Norfo, R.; Rodriguez-Meira, A.; Ashley, N.; et al. Single-cell transcriptomics uncovers distinct molecular signatures of stem cells in chronic myeloid leukemia. Nat. Med. 2017, 23, 692–702. [Google Scholar] [CrossRef]
- Chen, Z.; Cortes, J.E.; Jorgensen, J.L.; Wang, W.; Yin, C.C.; You, M.J.; Jabbour, E.; Kantarjian, H.M.; Medeiros, L.J.; Hu, S. Differential impact of additional chromosomal abnormalities in myeloid vs lymphoid blast phase of chronic myelogenous leukemia in the era of tyrosine kinase inhibitor therapy. Leukemia 2016, 30, 1606–1609. [Google Scholar] [CrossRef] [Green Version]
- Togasaki, E.; Takeda, J.; Yoshida, K.; Shiozawa, Y.; Takeuchi, M.; Oshima, M.; Saraya, A.; Iwama, A.; Yokote, K.; Sakaida, E.; et al. Frequent somatic mutations in epigenetic regulators in newly diagnosed chronic myeloid leukemia. Blood Cancer J. 2017, 7, e559. [Google Scholar] [CrossRef]
- Schmidt, M.; Rinke, J.; Schafer, V.; Schnittger, S.; Kohlmann, A.; Obstfelder, E.; Kunert, C.; Ziermann, J.; Winkelmann, N.; Eigendorff, E.; et al. Molecular-defined clonal evolution in patients with chronic myeloid leukemia independent of the BCR-ABL status. Leukemia 2014, 28, 2292–2299. [Google Scholar] [CrossRef]
- Reid, A.G.; De Melo, V.A.; Elderfield, K.; Clark, I.; Marin, D.; Apperley, J.; Naresh, K.N. Phenotype of blasts in chronic myeloid leukemia in blastic phase-Analysis of bone marrow trephine biopsies and correlation with cytogenetics. Leuk Res. 2009, 33, 418–425. [Google Scholar] [CrossRef]
- Perrotti, D.; Jamieson, C.; Goldman, J.; Skorski, T. Chronic myeloid leukemia: Mechanisms of blastic transformation. J. Clin. Investig. 2010, 120, 2254–2264. [Google Scholar] [CrossRef] [Green Version]
- Branford, S.; Wang, P.; Yeung, D.T.; Thomson, D.; Purins, A.; Wadham, C.; Shahrin, N.H.; Marum, J.E.; Nataren, N.; Parker, W.T.; et al. Integrative genomic analysis reveals cancer-associated mutations at diagnosis of CML in patients with high-risk disease. Blood 2018, 132, 948–961. [Google Scholar] [CrossRef] [PubMed]
- Adnan Awad, S.; Kankainen, M.; Ojala, T.; Koskenvesa, P.; Eldfors, S.; Ghimire, B.; Kumar, A.; Kytola, S.; Kamel, M.M.; Heckman, C.A.; et al. Mutation accumulation in cancer genes relates to nonoptimal outcome in chronic myeloid leukemia. Blood Adv. 2020, 4, 546–559. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Li, Y.L.; Zhou, X.; Cao, R.; Li, H.; Lu, Q.S.; Li, L.; Lu, Z.Y.; Huang, J.X.; Sun, J.; et al. CDKN2 Gene deletion as poor prognosis predictor involved in the progression of adult B-lineage acute lymphoblastic leukemia patients. J. Cancer 2015, 6, 1114–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marke, R.; van Leeuwen, F.N.; Scheijen, B. The many faces of IKZF1 in B-cell precursor acute lymphoblastic leukemia. Haematologica 2018, 103, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Thomson, D.W.; Shahrin, N.H.; Wang, P.P.S.; Wadham, C.; Shanmuganathan, N.; Scott, H.S.; Dinger, M.E.; Hughes, T.P.; Schreiber, A.W.; Branford, S. Aberrant RAG-mediated recombination contributes to multiple structural rearrangements in lymphoid blast crisis of chronic myeloid leukemia. Leukemia 2020, 34, 2051–2063. [Google Scholar] [CrossRef]
- Pippa, R.; Odero, M.D. The role of MYC and PP2A in the initiation and progression of myeloid leukemias. Cells 2020, 9, 544. [Google Scholar] [CrossRef] [Green Version]
- Neviani, P.; Santhanam, R.; Trotta, R.; Notari, M.; Blaser, B.W.; Liu, S.; Mao, H.; Chang, J.S.; Galietta, A.; Uttam, A.; et al. The tumor suppressor PP2A is functionally inactivated in blast crisis CML through the inhibitory activity of the BCR/ABL-regulated SET protein. Cancer Cell 2005, 8, 355–368. [Google Scholar] [CrossRef] [Green Version]
- Sharma, N.; Magistroni, V.; Piazza, R.; Citterio, S.; Mezzatesta, C.; Khandelwal, P.; Pirola, A.; Gambacorti-Passerini, C. BCR/ABL1 and BCR are under the transcriptional control of the MYC oncogene. Mol. Cancer 2015, 14, 132. [Google Scholar] [CrossRef] [Green Version]
- Giotopoulos, G.; van der Weyden, L.; Osaki, H.; Rust, A.G.; Gallipoli, P.; Meduri, E.; Horton, S.J.; Chan, W.I.; Foster, D.; Prinjha, R.K.; et al. A novel mouse model identifies cooperating mutations and therapeutic targets critical for chronic myeloid leukemia progression. J. Exp. Med. 2015, 212, 1551–1569. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.; Zheng, W.; Zhang, J.; Gan, X.; Ma, X.; Meng, Z.; Chen, T.; Lu, X.; Wu, Z.; Huang, W.; et al. Aberrant activation of CaMKIIgamma accelerates chronic myeloid leukemia blast crisis. Leukemia 2016, 30, 1282–1289. [Google Scholar] [CrossRef]
- Amabile, G.; Di Ruscio, A.; Muller, F.; Welner, R.S.; Yang, H.; Ebralidze, A.K.; Zhang, H.; Levantini, E.; Qi, L.; Martinelli, G.; et al. Dissecting the role of aberrant DNA methylation in human leukaemia. Nat. Commun. 2015, 6, 7091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heller, G.; Topakian, T.; Altenberger, C.; Cerny-Reiterer, S.; Herndlhofer, S.; Ziegler, B.; Datlinger, P.; Byrgazov, K.; Bock, C.; Mannhalter, C.; et al. Next-generation sequencing identifies major DNA methylation changes during progression of Ph+ chronic myeloid leukemia. Leukemia 2016, 30, 1861–1868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, X.; Zhou, M.; Fu, Y.; Yang, L.; Xu, M.; Sun, T.; Wang, X.; Huang, T.; Chen, C. Histone demethylase RBP2 mediates the blast crisis of chronic myeloid leukemia through an RBP2/PTEN/BCR-ABL cascade. Cell Signal. 2019, 63, 109360. [Google Scholar] [CrossRef] [PubMed]
- Ko, T.K.; Javed, A.; Lee, K.L.; Pathiraja, T.N.; Liu, X.; Malik, S.; Soh, S.X.; Heng, X.T.; Takahashi, N.; Tan, J.H.J.; et al. An integrative model of pathway convergence in genetically heterogeneous blast crisis chronic myeloid leukemia. Blood 2020, 135, 2337–2353. [Google Scholar] [CrossRef]
- Hoelzer, D.; Bassan, R.; Dombret, H.; Fielding, A.; Ribera, J.M.; Buske, C.; Committee, E.G. Acute lymphoblastic leukaemia in adult patients: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2016, 27, v69–v82. [Google Scholar] [CrossRef]
- Rehe, K.; Wilson, K.; Bomken, S.; Williamson, D.; Irving, J.; den Boer, M.L.; Stanulla, M.; Schrappe, M.; Hall, A.G.; Heidenreich, O.; et al. Acute B lymphoblastic leukaemia-propagating cells are present at high frequency in diverse lymphoblast populations. EMBO Mol. Med. 2013, 5, 38–51. [Google Scholar] [CrossRef]
- Hunger, S.P.; Mullighan, C.G. Redefining ALL classification: Toward detecting high-risk ALL and implementing precision medicine. Blood 2015, 125, 3977–3987. [Google Scholar] [CrossRef] [Green Version]
- Primo, D.; Tabernero, M.D.; Perez, J.J.; Rasillo, A.; Sayagues, J.M.; Espinosa, A.B.; Lopez-Berges, M.C.; Garcia-Sanz, R.; Gutierrez, N.C.; Hernandez, J.M.; et al. Genetic heterogeneity of BCR/ABL+ adult B-cell precursor acute lymphoblastic leukemia: Impact on the clinical, biological and immunophenotypical disease characteristics. Leukemia 2005, 19, 713–720. [Google Scholar] [CrossRef] [Green Version]
- Dupuis, A.; Gaub, M.P.; Legrain, M.; Drenou, B.; Mauvieux, L.; Lutz, P.; Herbrecht, R.; Chan, S.; Kastner, P. Biclonal and biallelic deletions occur in 20% of B-ALL cases with IKZF1 mutations. Leukemia 2013, 27, 503–507. [Google Scholar] [CrossRef] [Green Version]
- Churchman, M.L.; Low, J.; Qu, C.; Paietta, E.M.; Kasper, L.H.; Chang, Y.; Payne-Turner, D.; Althoff, M.J.; Song, G.; Chen, S.C.; et al. Efficacy of retinoids in IKZF1-mutated BCR-ABL1 acute lymphoblastic leukemia. Cancer Cell 2015, 28, 343–356. [Google Scholar] [CrossRef]
- Mullighan, C.G.; Miller, C.B.; Radtke, I.; Phillips, L.A.; Dalton, J.; Ma, J.; White, D.; Hughes, T.P.; Le Beau, M.M.; Pui, C.H.; et al. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature 2008, 453, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Cen, J.; Chen, S.; Shen, H.; Chen, Y.; He, J.; Chen, Z. IK6 isoform with associated cytogenetic and molecular abnormalities in Chinese patients with Philadelphia chromosome-positive adult acute lymphoblastic leukemia. Leuk. Lymphoma 2013, 54, 1626–1632. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Liu, F.; Wu, C.; Li, S.; Zhao, X.; Zhang, P.; Jiao, J.; Yu, X.; Ji, Y.; Zhang, M. Illegitimate RAG-mediated recombination events are involved in IKZF1 Delta3-6 deletion in BCR-ABL1 lymphoblastic leukaemia. Clin. Exp. Immunol. 2016, 185, 320–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeifer, H.; Raum, K.; Markovic, S.; Nowak, V.; Fey, S.; Oblander, J.; Pressler, J.; Bohm, V.; Bruggemann, M.; Wunderle, L.; et al. Genomic CDKN2A/2B deletions in adult Ph(+) ALL are adverse despite allogeneic stem cell transplantation. Blood 2018, 131, 1464–1475. [Google Scholar] [CrossRef]
- Xu, N.; Li, Y.L.; Li, X.; Zhou, X.; Cao, R.; Li, H.; Li, L.; Lu, Z.Y.; Huang, J.X.; Fan, Z.P.; et al. Correlation between deletion of the CDKN2 gene and tyrosine kinase inhibitor resistance in adult Philadelphia chromosome-positive acute lymphoblastic leukemia. J. Hematol. Oncol. 2016, 9, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Familiades, J.; Bousquet, M.; Lafage-Pochitaloff, M.; Bene, M.C.; Beldjord, K.; De Vos, J.; Dastugue, N.; Coyaud, E.; Struski, S.; Quelen, C.; et al. PAX5 mutations occur frequently in adult B-cell progenitor acute lymphoblastic leukemia and PAX5 haploinsufficiency is associated with BCR-ABL1 and TCF3-PBX1 fusion genes: A GRAALL study. Leukemia 2009, 23, 1989–1998. [Google Scholar] [CrossRef]
- Martin-Lorenzo, A.; Auer, F.; Chan, L.N.; Garcia-Ramirez, I.; Gonzalez-Herrero, I.; Rodriguez-Hernandez, G.; Bartenhagen, C.; Dugas, M.; Gombert, M.; Ginzel, S.; et al. Loss of Pax5 Exploits Sca1-BCR-ABL(p190) Susceptibility to Confer the Metabolic Shift Essential for pB-ALL. Cancer Res. 2018, 78, 2669–2679. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Choi, J.E.; She, C.J.; Hwang, S.M.; Shin, H.Y.; Ahn, H.S.; Yoon, S.S.; Kim, B.K.; Park, M.H.; Lee, D.S. PAX5 deletion is common and concurrently occurs with CDKN2A deletion in B-lineage acute lymphoblastic leukemia. Blood Cells Mol. Dis. 2011, 47, 62–66. [Google Scholar] [CrossRef]
- Hock, H.; Shimamura, A. ETV6 in hematopoiesis and leukemia predisposition. Semin. Hematol. 2017, 54, 98–104. [Google Scholar] [CrossRef] [Green Version]
- Ramirez-Komo, J.A.; Delaney, M.A.; Straign, D.; Lukin, K.; Tsang, M.; Iritani, B.M.; Hagman, J. Spontaneous loss of B lineage transcription factors leads to pre-B leukemia in Ebf1(+/-)Bcl-xL(Tg) mice. Oncogenesis 2017, 6, e355. [Google Scholar] [CrossRef] [Green Version]
- Yuniati, L.; Scheijen, B.; van der Meer, L.T.; van Leeuwen, F.N. Tumor suppressors BTG1 and BTG2: Beyond growth control. J. Cell Physiol. 2019, 234, 5379–5389. [Google Scholar] [CrossRef] [Green Version]
- Knudsen, E.S.; Pruitt, S.C.; Hershberger, P.A.; Witkiewicz, A.K.; Goodrich, D.W. Cell Cycle and Beyond: Exploiting New RB1 Controlled Mechanisms for Cancer Therapy. Trends Cancer 2019, 5, 308–324. [Google Scholar] [CrossRef]
- Greuber, E.K.; Smith-Pearson, P.; Wang, J.; Pendergast, A.M. Role of ABL family kinases in cancer: From leukaemia to solid tumours. Nat. Rev. Cancer 2013, 13, 559–571. [Google Scholar] [CrossRef] [Green Version]
- Dasgupta, Y.; Koptyra, M.; Hoser, G.; Kantekure, K.; Roy, D.; Gornicka, B.; Nieborowska-Skorska, M.; Bolton-Gillespie, E.; Cerny-Reiterer, S.; Muschen, M.; et al. Normal ABL1 is a tumor suppressor and therapeutic target in human and mouse leukemias expressing oncogenic ABL1 kinases. Blood 2016, 127, 2131–2143. [Google Scholar] [CrossRef] [Green Version]
- Aloisi, A.; Di Gregorio, S.; Stagno, F.; Guglielmo, P.; Mannino, F.; Sormani, M.P.; Bruzzi, P.; Gambacorti-Passerini, C.; Saglio, G.; Venuta, S.; et al. BCR-ABL nuclear entrapment kills human CML cells: Ex vivo study on 35 patients with the combination of imatinib mesylate and leptomycin B. Blood 2006, 107, 1591–1598. [Google Scholar] [CrossRef] [Green Version]
- Cilloni, D.; Saglio, G. Molecular pathways: BCR-ABL. Clin. Cancer Res. 2012, 18, 930–937. [Google Scholar] [CrossRef] [Green Version]
- Jeong, S.Y.; Kim, J.A.; Oh, I.H. The adaptive remodeling of stem cell niche in stimulated bone marrow counteracts the leukemic niche. Stem Cells 2018, 36, 1617–1629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reckel, S.; Hamelin, R.; Georgeon, S.; Armand, F.; Jolliet, Q.; Chiappe, D.; Moniatte, M.; Hantschel, O. Differential signaling networks of Bcr-Abl p210 and p190 kinases in leukemia cells defined by functional proteomics. Leukemia 2017, 31, 1502–1512. [Google Scholar] [CrossRef] [PubMed]
- Hantschel, O.; Warsch, W.; Eckelhart, E.; Kaupe, I.; Grebien, F.; Wagner, K.U.; Superti-Furga, G.; Sexl, V. BCR-ABL uncouples canonical JAK2-STAT5 signaling in chronic myeloid leukemia. Nat. Chem. Biol. 2012, 8, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Smithgall, T.E.; Glazer, R.I. K562 leukemia cells transfected with the human c-fes gene acquire the ability to undergo myeloid differentiation. J. Biol. Chem. 1989, 264, 10276–10281. [Google Scholar] [PubMed]
- Mian, A.A.; Baumann, I.; Liebermann, M.; Grebien, F.; Superti-Furga, G.; Ruthardt, M.; Ottmann, O.G.; Hantschel, O. The phosphatase UBASH3B/Sts-1 is a negative regulator of Bcr-Abl kinase activity and leukemogenesis. Leukemia 2019, 33, 2319–2323. [Google Scholar] [CrossRef] [PubMed]
- Cutler, J.A.; Udainiya, S.; Madugundu, A.K.; Renuse, S.; Xu, Y.; Jung, J.; Kim, K.P.; Wu, X.; Pandey, A. Integrative phosphoproteome and interactome analysis of the role of Ubash3b in BCR-ABL signaling. Leukemia 2020, 34, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Arora, A.; Scholar, E.M. Role of tyrosine kinase inhibitors in cancer therapy. J. Pharmacol. Exp. Ther. 2005, 315, 971–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pophali, P.A.; Patnaik, M.M. The role of new tyrosine kinase inhibitors in chronic myeloid leukemia. Cancer J. 2016, 22, 40–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jabbour, E.; Kantarjian, H. Chronic myeloid leukemia: 2018 update on diagnosis, therapy and monitoring. Am. J. Hematol. 2018, 93, 442–459. [Google Scholar] [CrossRef] [Green Version]
- Fausel, C. Targeted chronic myeloid leukemia therapy: Seeking a cure. Am. J. Health Syst. Pharm. 2007, 64, S9–S15. [Google Scholar] [CrossRef] [Green Version]
- Buchdunger, E.; Cioffi, C.L.; Law, N.; Stover, D.; Ohno-Jones, S.; Druker, B.J.; Lydon, N.B. Abl protein-tyrosine kinase inhibitor STI571 inhibits in vitro signal transduction mediated by c-kit and platelet-derived growth factor receptors. J. Pharmacol. Exp. Ther. 2000, 295, 139–145. [Google Scholar]
- Olivieri, A.; Manzione, L. Dasatinib: A new step in molecular target therapy. Ann. Oncol. 2007, 18 (Suppl. 6), vi42–vi46. [Google Scholar] [CrossRef]
- Steinberg, M. Dasatinib: A tyrosine kinase inhibitor for the treatment of chronic myelogenous leukemia and philadelphia chromosome-positive acute lymphoblastic leukemia. Clin. Ther. 2007, 29, 2289–2308. [Google Scholar] [CrossRef]
- Brattas, M.K.; Reikvam, H.; Tvedt, T.H.A.; Bruserud, O. Dasatinib as an investigational drug for the treatment of Philadelphia chromosome-positive acute lymphoblastic leukemia in adults. Expert Opin. Investig. Drugs 2019, 28, 411–420. [Google Scholar] [CrossRef]
- Weisberg, E.; Manley, P.; Mestan, J.; Cowan-Jacob, S.; Ray, A.; Griffin, J.D. AMN107 (nilotinib): A novel and selective inhibitor of BCR-ABL. Br. J. Cancer 2006, 94, 1765–1769. [Google Scholar] [CrossRef] [PubMed]
- Puttini, M.; Coluccia, A.M.; Boschelli, F.; Cleris, L.; Marchesi, E.; Donella-Deana, A.; Ahmed, S.; Redaelli, S.; Piazza, R.; Magistroni, V.; et al. In vitro and in vivo activity of SKI-606, a novel Src-Abl inhibitor, against imatinib-resistant Bcr-Abl+ neoplastic cells. Cancer Res. 2006, 66, 11314–11322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redaelli, S.; Piazza, R.; Rostagno, R.; Magistroni, V.; Perini, P.; Marega, M.; Gambacorti-Passerini, C.; Boschelli, F. Activity of bosutinib, dasatinib, and nilotinib against 18 imatinib-resistant BCR/ABL mutants. J. Clin. Oncol. 2009, 27, 469–471. [Google Scholar] [CrossRef] [PubMed]
- Eskazan, A.E.; Keskin, D. Radotinib and its clinical potential in chronic-phase chronic myeloid leukemia patients: An update. Ther. Adv. Hematol. 2017, 8, 237–243. [Google Scholar] [CrossRef] [Green Version]
- Tan, F.H.; Putoczki, T.L.; Stylli, S.S.; Luwor, R.B. Ponatinib: A novel multi-tyrosine kinase inhibitor against human malignancies. Onco Targets Ther. 2019, 12, 635–645. [Google Scholar] [CrossRef] [Green Version]
- Hochhaus, A.; Larson, R.A.; Guilhot, F.; Radich, J.P.; Branford, S.; Hughes, T.P.; Baccarani, M.; Deininger, M.W.; Cervantes, F.; Fujihara, S.; et al. Long-Term outcomes of imatinib treatment for chronic myeloid leukemia. N. Engl. J. Med. 2017, 376, 917–927. [Google Scholar] [CrossRef]
- Hehlmann, R.; Lauseker, M.; Saussele, S.; Pfirrmann, M.; Krause, S.; Kolb, H.J.; Neubauer, A.; Hossfeld, D.K.; Nerl, C.; Gratwohl, A.; et al. Assessment of imatinib as first-line treatment of chronic myeloid leukemia: 10-year survival results of the randomized CML study IV and impact of non-CML determinants. Leukemia 2017, 31, 2398–2406. [Google Scholar] [CrossRef]
- Sacha, T.; Gora-Tybor, J.; Szarejko, M.; Bober, G.; Grzybowska-Izydorczyk, O.; Niesiobedzka-Krezel, J.; Dudzinski, M.; Wasilewska, E.; Mysliwiec, K.; Gil, J.; et al. A multicenter prospective study on efficacy and safety of imatinib generics: A report from Polish Adult Leukemia Group imatinib generics registry. Am. J. Hematol. 2017, 92, E125–E128. [Google Scholar] [CrossRef] [Green Version]
- Madhav, D. Generic imatinib in chronic myeloid leukemia: Survival of the cheapest. Blood 2016, 128, 630. [Google Scholar] [CrossRef]
- Cortes, J.E.; Saglio, G.; Kantarjian, H.M.; Baccarani, M.; Mayer, J.; Boque, C.; Shah, N.P.; Chuah, C.; Casanova, L.; Bradley-Garelik, B.; et al. Final 5-year study results of DASISION: The dasatinib versus imatinib study in treatment-naive chronic myeloid leukemia patients trial. J. Clin. Oncol. 2016, 34, 2333–2340. [Google Scholar] [CrossRef]
- Hochhaus, A.; Saglio, G.; Hughes, T.P.; Larson, R.A.; Kim, D.W.; Issaragrisil, S.; le Coutre, P.D.; Etienne, G.; Dorlhiac-Llacer, P.E.; Clark, R.E.; et al. Long-term benefits and risks of frontline nilotinib vs imatinib for chronic myeloid leukemia in chronic phase: 5-year update of the randomized ENESTnd trial. Leukemia 2016, 30, 1044–1054. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.E.; Gambacorti-Passerini, C.; Deininger, M.W.; Mauro, M.J.; Chuah, C.; Kim, D.W.; Dyagil, I.; Glushko, N.; Milojkovic, D.; le Coutre, P.; et al. Bosutinib Versus Imatinib for Newly Diagnosed Chronic Myeloid Leukemia: Results From the Randomized BFORE Trial. J. Clin. Oncol. 2018, 36, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Efficace, F.; Stagno, F.; Iurlo, A.; Breccia, M.; Cottone, F.; Bonifacio, M.; Abruzzese, E.; Castagnetti, F.; Caocci, G.; Crugnola, M.; et al. Health-related quality of life of newly diagnosed chronic myeloid leukemia patients treated with first-line dasatinib versus imatinib therapy. Leukemia 2020, 34, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Claudiani, S.; Apperley, J.F. The argument for using imatinib in CML. Hematol. Am. Soc. Hematol. Educ. Program 2018, 2018, 161–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, R.E.; Polydoros, F.; Apperley, J.F.; Milojkovic, D.; Pocock, C.; Smith, G.; Byrne, J.L.; de Lavallade, H.; O’Brien, S.G.; Coffey, T.; et al. De-escalation of tyrosine kinase inhibitor dose in patients with chronic myeloid leukaemia with stable major molecular response (DESTINY): An interim analysis of a non-randomised, phase 2 trial. Lancet Haematol. 2017, 4, e310–e316. [Google Scholar] [CrossRef] [Green Version]
- Saussele, S.; Richter, J.; Guilhot, J.; Gruber, F.X.; Hjorth-Hansen, H.; Almeida, A.; Janssen, J.; Mayer, J.; Koskenvesa, P.; Panayiotidis, P.; et al. Discontinuation of tyrosine kinase inhibitor therapy in chronic myeloid leukaemia (EURO-SKI): A prespecified interim analysis of a prospective, multicentre, non-randomised, trial. Lancet Oncol. 2018, 19, 747–757. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, V.S.; Baccarani, M.; Hasford, J.; Lindoerfer, D.; Burgstaller, S.; Sertic, D.; Costeas, P.; Mayer, J.; Indrak, K.; Everaus, H.; et al. The EUTOS population-based registry: Incidence and clinical characteristics of 2904 CML patients in 20 European Countries. Leukemia 2015, 29, 1336–1343. [Google Scholar] [CrossRef]
- Hehlmann, R.; Saussele, S.; Voskanyan, A.; Silver, R.T. Management of CML-blast crisis. Best Pract. Res. Clin. Haematol. 2016, 29, 295–307. [Google Scholar] [CrossRef]
- Iacoboni, S.J.; Plunkett, W.; Kantarjian, H.M.; Estey, E.; Keating, M.J.; McCredie, K.B.; Freireich, E.J. High-dose cytosine arabinoside: Treatment and cellular pharmacology of chronic myelogenous leukemia blast crisis. J. Clin. Oncol. 1986, 4, 1079–1088. [Google Scholar] [CrossRef]
- Bonifacio, M.; Stagno, F.; Scaffidi, L.; Krampera, M.; Di Raimondo, F. Management of Chronic Myeloid Leukemia in Advanced Phase. Front. Oncol. 2019, 9, 1132. [Google Scholar] [CrossRef] [Green Version]
- Baccarani, M.; Cortes, J.; Pane, F.; Niederwieser, D.; Saglio, G.; Apperley, J.; Cervantes, F.; Deininger, M.; Gratwohl, A.; Guilhot, F.; et al. Chronic myeloid leukemia: An update of concepts and management recommendations of European LeukemiaNet. J. Clin. Oncol. 2009, 27, 6041–6051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Q.; Xu, L.P.; Liu, D.H.; Liu, K.Y.; Chen, S.S.; Jiang, B.; Jiang, H.; Chen, H.; Chen, Y.H.; Han, W.; et al. Imatinib mesylate versus allogeneic hematopoietic stem cell transplantation for patients with chronic myelogenous leukemia in the accelerated phase. Blood 2011, 117, 3032–3040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palandri, F.; Castagnetti, F.; Testoni, N.; Luatti, S.; Marzocchi, G.; Bassi, S.; Breccia, M.; Alimena, G.; Pungolino, E.; Rege-Cambrin, G.; et al. Chronic myeloid leukemia in blast crisis treated with imatinib 600 mg: Outcome of the patients alive after a 6-year follow-up. Haematologica 2008, 93, 1792–1796. [Google Scholar] [CrossRef] [Green Version]
- Balsat, M.; Alcazer, V.; Etienne, G.; Fossard, G.; Huguet, F.; Berger, M.G.; Cayssials, E.; Charbonnier, A.; Escoffre-Barbe, M.; Johnson-Ansah, H.; et al. First-line second generation tyrosine kinase inhibitors in newly diagnosed accelerated phase chronic myeloid leukemia patients. Blood 2018, 132, 48. [Google Scholar] [CrossRef]
- Kantarjian, H.; Cortes, J.; Kim, D.W.; Dorlhiac-Llacer, P.; Pasquini, R.; DiPersio, J.; Muller, M.C.; Radich, J.P.; Khoury, H.J.; Khoroshko, N.; et al. Phase 3 study of dasatinib 140 mg once daily versus 70 mg twice daily in patients with chronic myeloid leukemia in accelerated phase resistant or intolerant to imatinib: 15-month median follow-up. Blood 2009, 113, 6322–6329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gambacorti-Passerini, C.; Kantarjian, H.M.; Kim, D.W.; Khoury, H.J.; Turkina, A.G.; Brummendorf, T.H.; Matczak, E.; Bardy-Bouxin, N.; Shapiro, M.; Turnbull, K.; et al. Long-term efficacy and safety of bosutinib in patients with advanced leukemia following resistance/intolerance to imatinib and other tyrosine kinase inhibitors. Am. J. Hematol. 2015, 90, 755–768. [Google Scholar] [CrossRef] [Green Version]
- Cortes, J.E.; Kim, D.W.; Pinilla-Ibarz, J.; le Coutre, P.; Paquette, R.; Chuah, C.; Nicolini, F.E.; Apperley, J.F.; Khoury, H.J.; Talpaz, M.; et al. A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias. N. Engl. J. Med. 2013, 369, 1783–1796. [Google Scholar] [CrossRef] [Green Version]
- Saglio, G.; Hochhaus, A.; Goh, Y.T.; Masszi, T.; Pasquini, R.; Maloisel, F.; Erben, P.; Cortes, J.; Paquette, R.; Bradley-Garelik, M.B.; et al. Dasatinib in imatinib-resistant or imatinib-intolerant chronic myeloid leukemia in blast phase after 2 years of follow-up in a phase 3 study: Efficacy and tolerability of 140 milligrams once daily and 70 milligrams twice daily. Cancer 2010, 116, 3852–3861. [Google Scholar] [CrossRef] [Green Version]
- Giles, F.J.; Kantarjian, H.M.; le Coutre, P.D.; Baccarani, M.; Mahon, F.X.; Blakesley, R.E.; Gallagher, N.J.; Gillis, K.; Goldberg, S.L.; Larson, R.A.; et al. Nilotinib is effective in imatinib-resistant or-intolerant patients with chronic myeloid leukemia in blastic phase. Leukemia 2012, 26, 959–962. [Google Scholar] [CrossRef] [Green Version]
- Strati, P.; Kantarjian, H.; Thomas, D.; O’Brien, S.; Konoplev, S.; Jorgensen, J.L.; Luthra, R.; Abruzzo, L.; Jabbour, E.; Quintas-Cardama, A.; et al. HCVAD plus imatinib or dasatinib in lymphoid blastic phase chronic myeloid leukemia. Cancer 2014, 120, 373–380. [Google Scholar] [CrossRef]
- Jain, P.; Kantarjian, H.M.; Ghorab, A.; Sasaki, K.; Jabbour, E.J.; Nogueras Gonzalez, G.; Kanagal-Shamanna, R.; Issa, G.C.; Garcia-Manero, G.; Kc, D.; et al. Prognostic factors and survival outcomes in patients with chronic myeloid leukemia in blast phase in the tyrosine kinase inhibitor era: Cohort study of 477 patients. Cancer 2017, 123, 4391–4402. [Google Scholar] [CrossRef] [PubMed]
- Copland, M.; Slade, D.; Byrne, J. FLAG-IDA and ponatinib in patients with blast phase chronic myeloid leukaemia: Results from the phase I/II UK trials acceleration programme matchpoint trial. Blood 2019, 134 (Suppl. 1), 497. [Google Scholar] [CrossRef]
- Fielding, A.K. Curing Ph+ ALL: Assessing the relative contributions of chemotherapy, TKIs, and allogeneic stem cell transplant. Hematol. Am. Soc. Hematol. Educ. Program 2019, 2019, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Boer, J.M.; den Boer, M.L. BCR-ABL1-like acute lymphoblastic leukaemia: From bench to bedside. Eur. J. Cancer 2017, 82, 203–218. [Google Scholar] [CrossRef]
- Short, N.J.; Kantarjian, H.; Pui, C.H.; Goldstone, A.; Jabbour, E. SOHO state of the art update and next questions: Philadelphia chromosome-positive acute lymphoblastic leukemia. Clin. Lymphoma Myeloma Leuk. 2018, 18, 439–446. [Google Scholar] [CrossRef]
- Ravandi, F. Current management of Philadelphia chromosome positive ALL and the role of stem cell transplantation. Hematol. Am. Soc. Hematol. Educ Program 2017, 2017, 22–27. [Google Scholar] [CrossRef] [Green Version]
- Fielding, A.K.; Rowe, J.M.; Buck, G.; Foroni, L.; Gerrard, G.; Litzow, M.R.; Lazarus, H.; Luger, S.M.; Marks, D.I.; McMillan, A.K.; et al. UKALLXII/ECOG2993: Addition of imatinib to a standard treatment regimen enhances long-term outcomes in Philadelphia positive acute lymphoblastic leukemia. Blood 2014, 123, 843–850. [Google Scholar] [CrossRef]
- Chalandon, Y.; Thomas, X.; Hayette, S.; Cayuela, J.M.; Abbal, C.; Huguet, F.; Raffoux, E.; Leguay, T.; Rousselot, P.; Lepretre, S.; et al. Randomized study of reduced-intensity chemotherapy combined with imatinib in adults with Ph-positive acute lymphoblastic leukemia. Blood 2015, 125, 3711–3719. [Google Scholar] [CrossRef] [Green Version]
- Foa, R.; Vitale, A.; Vignetti, M.; Meloni, G.; Guarini, A.; De Propris, M.S.; Elia, L.; Paoloni, F.; Fazi, P.; Cimino, G.; et al. Dasatinib as first-line treatment for adult patients with Philadelphia chromosome-positive acute lymphoblastic leukemia. Blood 2011, 118, 6521–6528. [Google Scholar] [CrossRef] [Green Version]
- Ravandi, F.; O’Brien, S.M.; Cortes, J.E.; Thomas, D.M.; Garris, R.; Faderl, S.; Burger, J.A.; Rytting, M.E.; Ferrajoli, A.; Wierda, W.G.; et al. Long-term follow-up of a phase 2 study of chemotherapy plus dasatinib for the initial treatment of patients with Philadelphia chromosome-positive acute lymphoblastic leukemia. Cancer 2015, 121, 4158–4164. [Google Scholar] [CrossRef]
- Rousselot, P.; Coude, M.M.; Gokbuget, N.; Gambacorti Passerini, C.; Hayette, S.; Cayuela, J.M.; Huguet, F.; Leguay, T.; Chevallier, P.; Salanoubat, C.; et al. Dasatinib and low-intensity chemotherapy in elderly patients with Philadelphia chromosome-positive ALL. Blood 2016, 128, 774–782. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Joo, Y.D.; Lim, S.N.; Kim, S.D.; Lee, J.H.; Lee, J.H.; Kim, D.H.; Kim, K.; Jung, C.W.; Kim, I.; et al. Nilotinib combined with multiagent chemotherapy for newly diagnosed Philadelphia-positive acute lymphoblastic leukemia. Blood 2015, 126, 746–756. [Google Scholar] [CrossRef] [Green Version]
- Ottmann, O.; Pfeifer, H.; Cayuela, J.M.; Spiekermann, K.; Beck, J.; Jung, W.E.; Viardot, A.; Schäfer-Eckart, K.; Albrecht, R.; Maury, S.; et al. Nilotinib (Tasigna®) and chemotherapy for first-line treatment in elderly patients with De Novo Philadelphia chromosome/BCR-ABL1 positive acute lymphoblastic leukemia (ALL): A trial of the European Working Group for Adult ALL (EWALL-PH-02). Blood 2014, 124, 798. [Google Scholar] [CrossRef]
- Gambacorti Passerini, C.; Kantarjian, H.; Bruemmendorf, T.; Martinelli, G.; Baccarani, M.; Fischer, T.; Pogliani, E.M.; Hewes, B.; Volkert, A.D.G.; Bardy-Bouxin, N.; et al. Bosutinib (SKI-606) demonstrates clinical activity and is well tolerated among patients with AP and BP CML and Ph+ ALL. Blood 2007, 110, 473. [Google Scholar] [CrossRef]
- Jabbour, E.; DerSarkissian, M.; Duh, M.S.; McCormick, N.; Cheng, W.Y.; McGarry, L.J.; Souroutzidis, A.; Huang, H.; O’Brien, S.; Ravandi, F.; et al. Efficacy of ponatinib versus earlier generation tyrosine kinase inhibitors for front-line treatment of newly diagnosed Philadelphia-positive acute lymphoblastic leukemia. Clin. Lymphoma Myeloma Leuk. 2018, 18, 257–265. [Google Scholar] [CrossRef]
- Martinelli, G.; Piciocchi, A.; Papayannidis, C.; Paolini, S.; Robustelli, V.; Soverini, S.; Terragna, C.; Lemoli, R.M.; Guolo, F.; Bartolomeo, P.; et al. First report of the gimema LAL1811 phase II prospective study of the combination of steroids with ponatinib as frontline therapy of elderly or unfit patients with Philadelphia chromosome-positive acute lymphoblastic leukemia. Blood 2017, 130, 99. [Google Scholar]
- Shen, S.; Chen, X.; Cai, J.; Yu, J.; Gao, J.; Hu, S.; Zhai, X.; Liang, C.; Ju, X.; Jiang, H.; et al. Effect of dasatinib vs imatinib in the treatment of pediatric philadelphia chromosome-positive acute lymphoblastic leukemia: A randomized clinical trial. JAMA Oncol. 2020, 6, 358–366. [Google Scholar] [CrossRef]
- Sasaki, K.; Jabbour, E.J.; Ravandi, F.; Short, N.J.; Thomas, D.A.; Garcia-Manero, G.; Daver, N.G.; Kadia, T.M.; Konopleva, M.Y.; Jain, N.; et al. Hyper-CVAD plus ponatinib versus hyper-CVAD plus dasatinib as frontline therapy for patients with Philadelphia chromosome-positive acute lymphoblastic leukemia: A propensity score analysis. Cancer 2016, 122, 3650–3656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Chen, F.; Yin, C.; Liu, Q.; Sun, J.; Xuan, L.; Fan, Z.; Wang, Q.; Liu, X.; Jiang, Q.; et al. Upfront treatment with the first and second-generation tyrosine kinase inhibitors in Ph-positive acute lymphoblastic leukemia. Oncotarget 2017, 8, 107022–107032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vignetti, M.; Fazi, P.; Cimino, G.; Martinelli, G.; Di Raimondo, F.; Ferrara, F.; Meloni, G.; Ambrosetti, A.; Quarta, G.; Pagano, L.; et al. Imatinib plus steroids induces complete remissions and prolonged survival in elderly Philadelphia chromosome-positive patients with acute lymphoblastic leukemia without additional chemotherapy: Results of the Gruppo Italiano Malattie Ematologiche dell’Adulto (GIMEMA) LAL0201-B protocol. Blood 2007, 109, 3676–3678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinelli, G.; Boissel, N.; Chevallier, P.; Ottmann, O.; Gokbuget, N.; Topp, M.S.; Fielding, A.K.; Rambaldi, A.; Ritchie, E.K.; Papayannidis, C.; et al. Complete hematologic and molecular response in adult patients with relapsed/refractory Philadelphia chromosome-positive B-precursor acute lymphoblastic leukemia following treatment with blinatumomab: Results from a phase II, single-arm, multicenter study. J. Clin. Oncol. 2017, 35, 1795–1802. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Yeshurun, M.; Weisdorf, D.; Rowe, J.M.; Tallman, M.S.; Zhang, M.J.; Wang, H.L.; Saber, W.; de Lima, M.; Sandmaier, B.M.; Uy, G.; et al. The impact of the graft-versus-leukemia effect on survival in acute lymphoblastic leukemia. Blood Adv. 2019, 3, 670–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speziali, C.; Paulson, K.; Seftel, M. Hematopoietic cell transplantation for acute lymphoblastic leukemia in adults. Curr. Hematol. Malig. Rep. 2016, 11, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Litzow, M.R. Should anyone with Philadelphia chromosome-positive ALL who is negative for minimal residual disease receive a hematopoietic stem cell transplant in first remission? Best Pract. Res. Clin. Haematol. 2016, 29, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Short, N.J.; Ravandi, F.; Daver, N.G.; Pemmaraju, N.; Thomas, D.A.; Yilmaz, M.; Kadia, T.M.; Sasaki, K.; Garris, R.; Garcia-Manero, G.; et al. Frontline hyper-CVAD plus ponatinib for patients with Philadelphia chromosome-positive acute lymphoblastic leukemia: Updated results of a phase II study. J. Clin. Oncol. 2017, 35, 7013. [Google Scholar] [CrossRef]
- O’Hare, T.; Zabriskie, M.S.; Eiring, A.M.; Deininger, M.W. Pushing the limits of targeted therapy in chronic myeloid leukaemia. Nat. Rev. Cancer 2012, 12, 513–526. [Google Scholar] [CrossRef]
- Soverini, S.; De Benedittis, C.; Mancini, M.; Martinelli, G. Best practices in chronic myeloid leukemia monitoring and management. Oncologist 2016, 21, 626–633. [Google Scholar] [CrossRef] [Green Version]
- Soverini, S.; Martinelli, G.; Rosti, G.; Iacobucci, I.; Baccarani, M. Advances in treatment of chronic myeloid leukemia with tyrosine kinase inhibitors: The evolving role of Bcr-Abl mutations and mutational analysis. Pharmacogenomics 2012, 13, 1271–1284. [Google Scholar] [CrossRef]
- Chandrasekhar, C.; Kumar, P.S.; Sarma, P. Novel mutations in the kinase domain of BCR-ABL gene causing imatinib resistance in chronic myeloid leukemia patients. Sci Rep. 2019, 9, 2412. [Google Scholar] [CrossRef]
- Soverini, S.; Bavaro, L.; De Benedittis, C.; Martelli, M.; Iurlo, A.; Orofino, N.; Sica, S.; Sora, F.; Lunghi, F.; Ciceri, F.; et al. Prospective assessment of NGS-detectable mutations in CML patients with nonoptimal response: The NEXT-in-CML study. Blood 2020, 135, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Baccarani, M.; Deininger, M.W.; Rosti, G.; Hochhaus, A.; Soverini, S.; Apperley, J.F.; Cervantes, F.; Clark, R.E.; Cortes, J.E.; Guilhot, F.; et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood 2013, 122, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Liu, H.; Wang, F.; Zhang, Y.; Chen, X.; Nie, D.; Li, Y.; Tan, Y.; Xu, Y.; Ma, X. Dynamic evolution of ponatinib resistant BCR-ABL1 T315 and compound mutations. Blood 2019, 134, 3796. [Google Scholar] [CrossRef]
- Miller, G.D.; Bruno, B.J.; Lim, C.S. Resistant mutations in CML and Ph(+)ALL—Role of ponatinib. Biologics 2014, 8, 243–254. [Google Scholar] [CrossRef] [Green Version]
- Zabriskie, M.S.; Eide, C.A.; Tantravahi, S.K.; Vellore, N.A.; Estrada, J.; Nicolini, F.E.; Khoury, H.J.; Larson, R.A.; Konopleva, M.; Cortes, J.E.; et al. BCR-ABL1 compound mutations combining key kinase domain positions confer clinical resistance to ponatinib in Ph chromosome-positive leukemia. Cancer Cell 2014, 26, 428–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bavaro, L.; Martelli, M.; Cavo, M.; Soverini, S. Mechanisms of disease progression and resistance to tyrosine kinase inhibitor therapy in chronic myeloid leukemia: An update. Int. J. Mol. Sci. 2019, 20, 6141. [Google Scholar] [CrossRef] [Green Version]
- Braun, T.P.; Eide, C.A.; Druker, B.J. Response and resistance to BCR-ABL1-targeted therapies. Cancer Cell 2020, 37, 530–542. [Google Scholar] [CrossRef]
- Burchert, A.; Wang, Y.; Cai, D.; von Bubnoff, N.; Paschka, P.; Muller-Brusselbach, S.; Ottmann, O.G.; Duyster, J.; Hochhaus, A.; Neubauer, A. Compensatory PI3-kinase/Akt/mTor activation regulates imatinib resistance development. Leukemia 2005, 19, 1774–1782. [Google Scholar] [CrossRef] [Green Version]
- Eiring, A.M.; Page, B.D.G.; Kraft, I.L.; Mason, C.C.; Vellore, N.A.; Resetca, D.; Zabriskie, M.S.; Zhang, T.Y.; Khorashad, J.S.; Engar, A.J.; et al. Combined STAT3 and BCR-ABL1 inhibition induces synthetic lethality in therapy-resistant chronic myeloid leukemia. Leukemia 2015, 29, 586–597. [Google Scholar] [CrossRef] [Green Version]
- Wagle, M.; Eiring, A.M.; Wongchenko, M.; Lu, S.; Guan, Y.; Wang, Y.; Lackner, M.; Amler, L.; Hampton, G.; Deininger, M.W.; et al. A role for FOXO1 in BCR-ABL1-independent tyrosine kinase inhibitor resistance in chronic myeloid leukemia. Leukemia 2016, 30, 1493–1501. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, M.; Abe, A.; Imagama, S.; Nomura, Y.; Tanizaki, R.; Minami, Y.; Hayakawa, F.; Ito, Y.; Katsumi, A.; Yamamoto, K.; et al. BCR-ABL-independent and RAS/MAPK pathway-dependent form of imatinib resistance in Ph-positive acute lymphoblastic leukemia cell line with activation of EphB4. Eur. J. Haematol. 2010, 84, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Mian, A.A.; Zafar, U.; Ottmann, O.; Ruthardt, M.; Lalani, E.M.A. Activation of AKT/mTOR pathway in Ph+ acute lymphoblastic leukemia (ALL) leads to non-mutational resistance. Blood 2019, 134, 2570. [Google Scholar] [CrossRef]
- Mallampati, S.; Leng, X.; Ma, H.; Zeng, J.; Li, J.; Wang, H.; Lin, K.; Lu, Y.; Yang, Y.; Sun, B.; et al. Tyrosine kinase inhibitors induce mesenchymal stem cell-mediated resistance in BCR-ABL+ acute lymphoblastic leukemia. Blood 2015, 125, 2968–2973. [Google Scholar] [CrossRef] [PubMed]
- Wylie, A.A.; Schoepfer, J.; Jahnke, W.; Cowan-Jacob, S.W.; Loo, A.; Furet, P.; Marzinzik, A.L.; Pelle, X.; Donovan, J.; Zhu, W.; et al. The allosteric inhibitor ABL001 enables dual targeting of BCR-ABL1. Nature 2017, 543, 733–737. [Google Scholar] [CrossRef]
- Hughes, T.P.; Mauro, M.J.; Cortes, J.E.; Minami, H.; Rea, D.; DeAngelo, D.J.; Breccia, M.; Goh, Y.T.; Talpaz, M.; Hochhaus, A.; et al. Asciminib in chronic myeloid leukemia after ABL kinase inhibitor failure. N. Engl. J. Med. 2019, 381, 2315–2326. [Google Scholar] [CrossRef]
- Eide, C.A.; Zabriskie, M.S.; Savage Stevens, S.L.; Antelope, O.; Vellore, N.A.; Than, H.; Schultz, A.R.; Clair, P.; Bowler, A.D.; Pomicter, A.D.; et al. Combining the allosteric inhibitor asciminib with ponatinib suppresses emergence of and restores efficacy against highly resistant BCR-ABL1 mutants. Cancer Cell 2019, 36, 431–443.e435. [Google Scholar] [CrossRef]
- LeBien, T.W. Fates of human B-cell precursors. Blood 2000, 96, 9–23. [Google Scholar] [CrossRef]
- Chen, Z.; Shojaee, S.; Buchner, M.; Geng, H.; Lee, J.W.; Klemm, L.; Titz, B.; Graeber, T.G.; Park, E.; Tan, Y.X.; et al. Signalling thresholds and negative B-cell selection in acute lymphoblastic leukaemia. Nature 2015, 521, 357–361. [Google Scholar] [CrossRef]
- Hay, N. Reprogramming glucose metabolism in cancer: Can it be exploited for cancer therapy? Nat. Rev. Cancer 2016, 16, 635–649. [Google Scholar] [CrossRef] [Green Version]
- Nayak, R.C.; Hegde, S.; Althoff, M.J.; Wellendorf, A.M.; Mohmoud, F.; Perentesis, J.; Reina-Campos, M.; Reynaud, D.; Zheng, Y.; Diaz-Meco, M.T.; et al. The signaling axis atypical protein kinase C lambda/iota-Satb2 mediates leukemic transformation of B-cell progenitors. Nat. Commun. 2019, 10, 46. [Google Scholar] [CrossRef]
- Luong-Gardiol, N.; Siddiqui, I.; Pizzitola, I.; Jeevan-Raj, B.; Charmoy, M.; Huang, Y.; Irmisch, A.; Curtet, S.; Angelov, G.S.; Danilo, M.; et al. Gamma-catenin-dependent signals maintain BCR-ABL1(+) B cell acute lymphoblastic leukemia. Cancer Cell 2019, 35, 649–663.e610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Chen, Y.; Douglas, L.; Li, S. beta-Catenin is essential for survival of leukemic stem cells insensitive to kinase inhibition in mice with BCR-ABL-induced chronic myeloid leukemia. Leukemia 2009, 23, 109–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, N.; Cortes, J.E.; Ravandi, F.; Konopleva, M.; Alvarado, Y.; Kadia, T.; Wierda, W.G.; Borthakur, G.; Naqvi, K.; Pemmaraju, N.; et al. Inotuzumab ozogamicin in combination with bosutinib for patients with relapsed or refractory Ph+ ALL or CML in lymphoid blast phase. Blood 2017, 130, 143. [Google Scholar]
- Ganzel, C.; Kharit, M.; Duksin, C.; Rowe, J.M. Daratumumab for relapsed/refractory Philadelphia-positive acute lymphoblastic leukemia. Haematologica 2018, 103, e489–e490. [Google Scholar] [CrossRef] [PubMed]
- Assi, R.; Kantarjian, H.; Short, N.J.; Daver, N.; Takahashi, K.; Garcia-Manero, G.; DiNardo, C.; Burger, J.; Cortes, J.; Jain, N.; et al. Safety and efficacy of blinatumomab in combination with a tyrosine kinase inhibitor for the treatment of relapsed Philadelphia chromosome-positive leukemia. Clin. Lymphoma Myeloma Leuk. 2017, 17, 897–901. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Disease | Gene Name | Frequency of Mutations | Gene Function | References |
|---|---|---|---|---|
| CML CP | ASXL1 | 10% | Epigenetic regulator; a member of the Polycomb group of proteins | [29,30,33,34] |
| TET2/3 | 8% | Epigenetic regulator; catalyzes the conversion of methylcytosine to 5-hydroxymethylcytosine | [29,30] | |
| DNMT3A | 8% | Epigenetic regulator; methylates CpG sites | ||
| KDM1A | 3% | Epigenetic regulator; demethylates H3K4me2 | ||
| MSH6 | 3% | Component of DNA mismatch repair mechanism | ||
| CML MBP | ASXL1 | 40% | Epigenetic regulator; a member of the Polycomb group of proteins | [33,34] |
| RUNX1 | 20–40% | Transcription factor; regulates HSCs differentiation | [32,33,34] | |
| TP53 | 20% | Regulation of cell cycle, DNA repair, apoptosis | [32] | |
| BCoRL1 | 10% | Apoptosis regulator; interacts with histone deacetylases | [33,34] | |
| CML LBP | IKZF1 | 55% | Transcriptional regulator; regulates B cell development | [32,33,34] |
| CDKN2A/B | 50% | Regulation of cell cycle, apoptosis; inhibits cyclin dependent kinases, stabilizes p53 | [32] | |
| RUNX1 | 25–35% | Transcription factor; regulates HSCs differentiation | [33,34] | |
| BCoR | 15–25% | Apoptosis regulator; interacts with histone deacetylases | ||
| Ph+ B-ALL | IKZF1 | 70% | Transcriptional regulator; regulates B cell development | [51,52] |
| CDKN2A/B | 45% | Regulation of cell cycle, apoptosis; inhibits cyclin dependent kinases, stabilizes p53 | [56] | |
| PAX5 | 30–40% | Transcription factor; regulates B cell development | [56,58] | |
| BTG1 | 18% | Negative regulation of cell proliferation | [56] | |
| RB1 | 14% | Regulation of cell cycle progression | ||
| EBF1 | 13% | Transcription factor; regulates B cell development | ||
| ETV6 | 5% | Transcription factor; regulates development of hematopoietic cells |
| TKI Generation | Name | Major Targets | Indications | Mechanism of Action Unique Properties | Side Effects |
|---|---|---|---|---|---|
| First generation | Imatinib mesylate [78,79] | BCR-ABL1, PDGFR, c-KIT, EPH | Newly diagnosed adult and pediatric patients with CML in chronic phase, blast crisis, adult patients with newly diagnosed, relapsed or refractory Ph+ B-ALL | ATP-competitive TKI; binds to the inactive conformation of ABL1 | Gastrointestinal symptoms, joints pain, skin rash, fatigue (frequent); cardiovascular events (5%) |
| Second generation | Dasatinib [80,81,82] | BCR-ABL1, PDGFR, c-KIT, EPH, FGFR1, APKK, CDK2, AKT, p38, FAK, SRC, LCK, c-KIT | Imatinib-resistant or intolerant CML and Ph+ B-ALL, newly diagnosed chronic phase CML | ATP-competitive TKI; higher inhibitory potential against BCR-ABL1 than imatinib; binds to the active conformation of ABL1; penetrates to the central nervous system | Pleural effusions (37%); pulmonary arterial hypertension (rare) |
| Nilotinib [83] | BCR-ABL1, PDGFR, c-KIT, EPH | Imatinib-resistant or intolerant chronic and accelerated phase CML, newly diagnosed CML | ATP-competitive TKI; better topographical fit for the ABL1 than imatinib; binds to the inactive conformation of ABL1 | Cardiovascular events (20%); pancreatitis (5%) | |
| Bosutinib [84,85] | BCR-ABL1, SRC, LCK, TEC, CaMK2G, PDGFR, c-KIT | Imatinib- or dasatinib- or nilotinib-resistant CML patients | ATP-competitive TKI; binds to the active and inactive conformation of ABL1 | Transient diarrhea, nausea, gastrointestinal symptoms (30%) | |
| Radotinib [86] | BCR-ABL1, PDGFR, c-KIT, SRC | Approved in South Korea for CML chronic phase in patients newly diagnosed or with insufficient response to other TKIs | ATP-competitive TKI; structurally similar to imatinib and to nilotinib | Fatigue, nausea, asthenia (rare) | |
| Third generation | Ponatinib [87] | BCR-ABL1 including BCR-ABL1T315I, RET, FLT3, KIT, FGFR, VEGFR1, VEGFR2, PDGFR, SRC, EPH, Auora kinases | CML and Ph+ B-ALL patients with the BCR/ABL1T315I mutation or resistant to two or more TKIs | ATP-competitive TKI; binds to the inactive conformation of ABL1 | Gastrointestinal symptoms, joints pain, skin rash, fatigue (frequent); cardiovascular events (30%) |
| Drug(s) | Number of Patients | Hematologic Response | Cytogenetic/Molecular Response | Survival |
|---|---|---|---|---|
| First generation TKI in AP at CML diagnosis | ||||
| Imatinib [104] | 87 | CHR 85% | CCyR 47% MMR 34% | 6-years PFS 48% |
| First generation TKI in BP at CML diagnosis | ||||
| Imatinib [105] | 92 | MBP: CHR 24% LBP: CHR 35% | MCyR 12% CCyR 10% | Median survival 7 months |
| Second-generation TKI in AP at CML diagnosis | ||||
| Nilotinib [106] Dasatinib [106] | 66 | CHR 97% | CCyR 84% MMR70% | 7-years OS 87% |
| Second- and third-generation TKI in AP CML after imatinib and/or other TKI | ||||
| Dasatinib [107] | 317 | CHR 47–52% | CCyR 32–33% | 2-years OS 63–72% |
| Bosutinib [108] | 79 | HR 57% | MCyR 40% | 4-years OS 59% |
| Ponatinib [109] | 83 | HR 55% | CCyR 24% | 1-year OS 84% |
| Second- and third-generation TKI in BP CML after imatinib and/or other TKI | ||||
| Dasatinib [110] | 149 MBP 61 LBP | HR 28% MBP HR 38% LBP | MCyR 27% MBP MCyR 46% LBP | 2-years OS 24–28%MPB 16–21% LBP |
| Ponatinib [109] | 52 MBP 10 LBP | HR 29% MBP HR 40% LBP | MCyR 19% MBP MCyR 40% LBP | - |
| Nilotinib [111] | 105 MBP 31 LBP | HR 60% MBP HR 59% LBP | MCyR 38% MBP MCyR 52% LBP | 2-years OS 32%MPB 10% LBP |
| Third-generation TKI in BP CML after dasatinib or nilotinib failure or intolerance | ||||
| Ponatinib [109] | 38 MBP 24 LBP | HR 32% MBP HR 29% LBP | MCyR 18% MBP MCyR 29% LBP | 2-years OS 29% MPB 29% LBP |
| TKIs in combination with chemotherapy | ||||
| HCVAD+imatinib/dasatinib [112] | 42 LBP | CHR 90% | CCyR 58% | Median survival 17 months |
| Different TKIs+chemotherapy [113] | 195 LBP/MBP | HR 64% | CCyR 29% | Median survival 12 months |
| FLAG-Ida+ponatinib [114] | 17 BP | CHR 17% | MCyR 52% | 1-year OS 45.8% |
| Regimen | N | Age Median (Range) | CMR Rate | Allo-HSCT Rate | OS Rate |
|---|---|---|---|---|---|
| Imatinib | |||||
| Imatinib + intensive chemotherapy [119] | 169 | 42 (16–64) | - | 72% | 38% (4 years) |
| Imatinib + intensive chemotherapy [120] | 133 | 45 (21–9) | 23% (two cycle) | 65% | 46% (5 years) |
| Imatinib + non-intensive chemotherapy [120] | 135 | 49 (18–59) | 29% (two cycles) | 62% | 46% (5 years) |
| Dasatinib | |||||
| Dasatinib + corticosteroids [121] | 53 | 54 (24–77) | 15% (day 85) | 42% | 31% (20 months) |
| Dasatinib + intensive chemotherapy [122] | 72 | 55 (21–0) | 65% (overall) | 17% | 46% (5 years) |
| Dasatinib + non-intensive chemotherapy [123] | 71 | 69 (55–83) | 24% (consolidation) | 10% | 36% (5 years) |
| Nilotinib | |||||
| Nilotinib + intensive chemotherapy [124] | 90 | 47 (17–71) | 77% (3 months) | 63% | 72% (2 years) |
| Nilotinib + chemotherapy [125] | 47 | 66 (55–85) | 30% (induction) | - | 47% (5 years) |
| Bosutinib | |||||
| Bosutinib monotherapy [126] | 24 | 59 (24–84) | - | - | Median OS 3.6 months |
| Ponatinib | |||||
| Ponatinib + intensive chemotherapy [127] | 37 | 51 (27–75) | 78% (overall) | 24% | 80% (2 years) |
| Ponatinib + corticosteroids [128] | 44 | > 60 years | 45% (8 weeks) | - | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Komorowski, L.; Fidyt, K.; Patkowska, E.; Firczuk, M. Philadelphia Chromosome-Positive Leukemia in the Lymphoid Lineage—Similarities and Differences with the Myeloid Lineage and Specific Vulnerabilities. Int. J. Mol. Sci. 2020, 21, 5776. https://doi.org/10.3390/ijms21165776
Komorowski L, Fidyt K, Patkowska E, Firczuk M. Philadelphia Chromosome-Positive Leukemia in the Lymphoid Lineage—Similarities and Differences with the Myeloid Lineage and Specific Vulnerabilities. International Journal of Molecular Sciences. 2020; 21(16):5776. https://doi.org/10.3390/ijms21165776
Chicago/Turabian StyleKomorowski, Lukasz, Klaudyna Fidyt, Elżbieta Patkowska, and Malgorzata Firczuk. 2020. "Philadelphia Chromosome-Positive Leukemia in the Lymphoid Lineage—Similarities and Differences with the Myeloid Lineage and Specific Vulnerabilities" International Journal of Molecular Sciences 21, no. 16: 5776. https://doi.org/10.3390/ijms21165776
APA StyleKomorowski, L., Fidyt, K., Patkowska, E., & Firczuk, M. (2020). Philadelphia Chromosome-Positive Leukemia in the Lymphoid Lineage—Similarities and Differences with the Myeloid Lineage and Specific Vulnerabilities. International Journal of Molecular Sciences, 21(16), 5776. https://doi.org/10.3390/ijms21165776