EphrinA1-Fc Attenuates Ventricular Remodeling and Dysfunction in Chronically Nonreperfused WT but not EphA2-R-M mice


Abstract
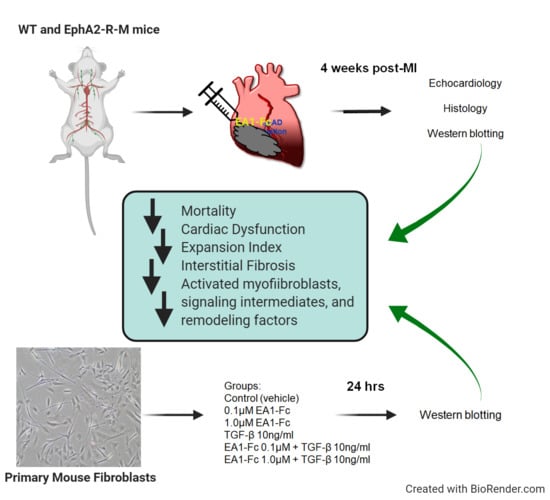
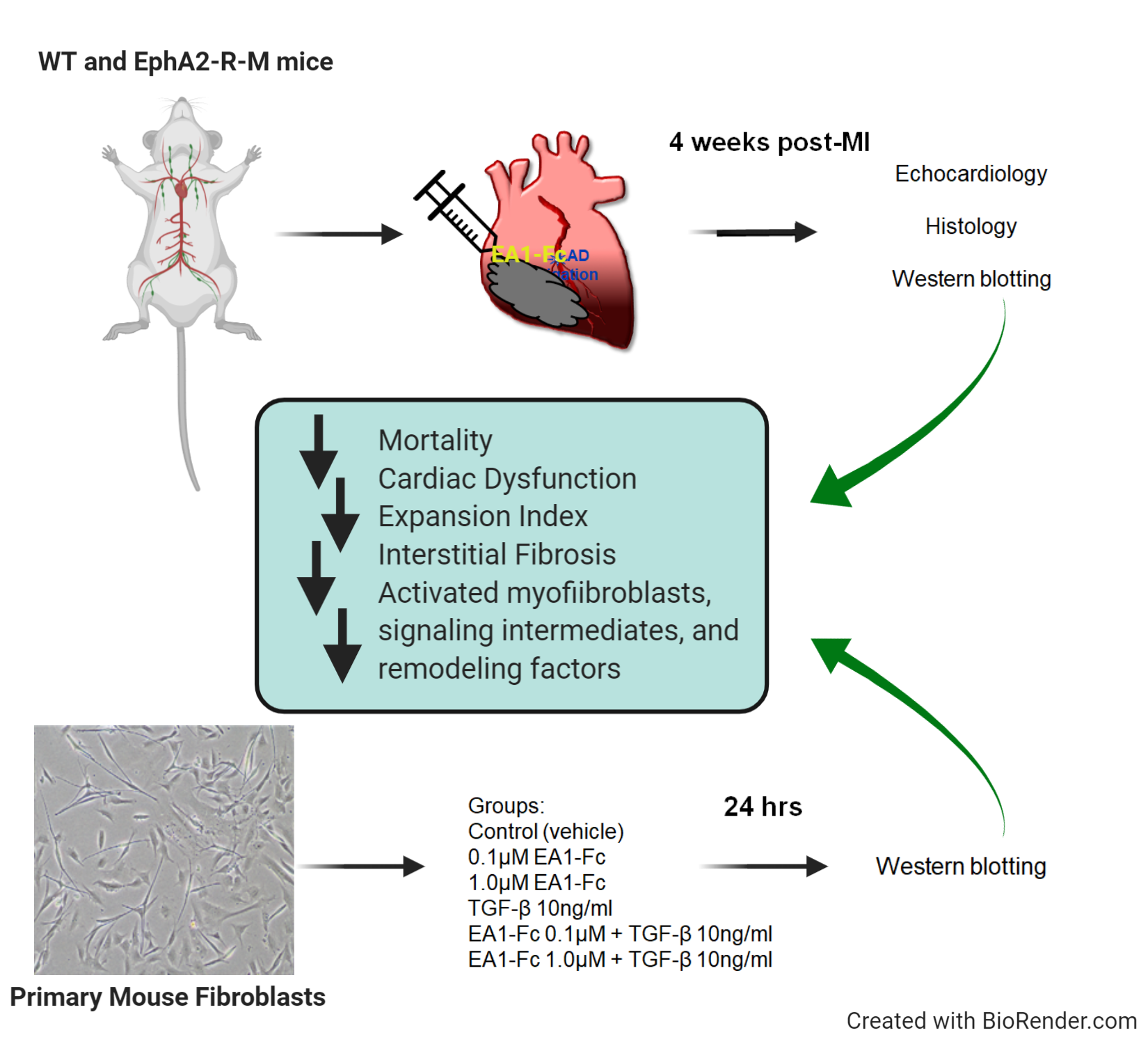
:
1. Introduction
2. Results
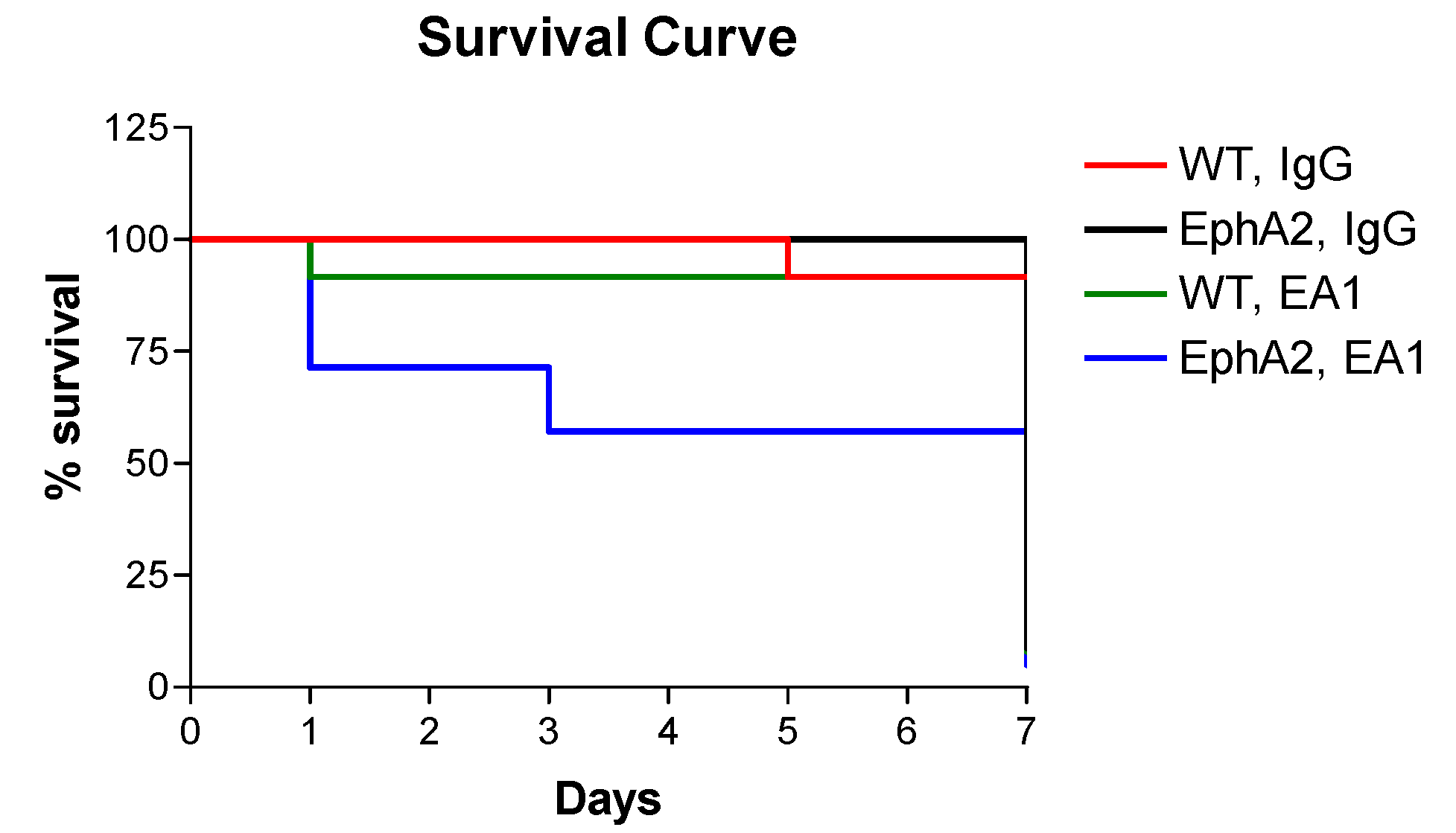
2.1. Survival Data
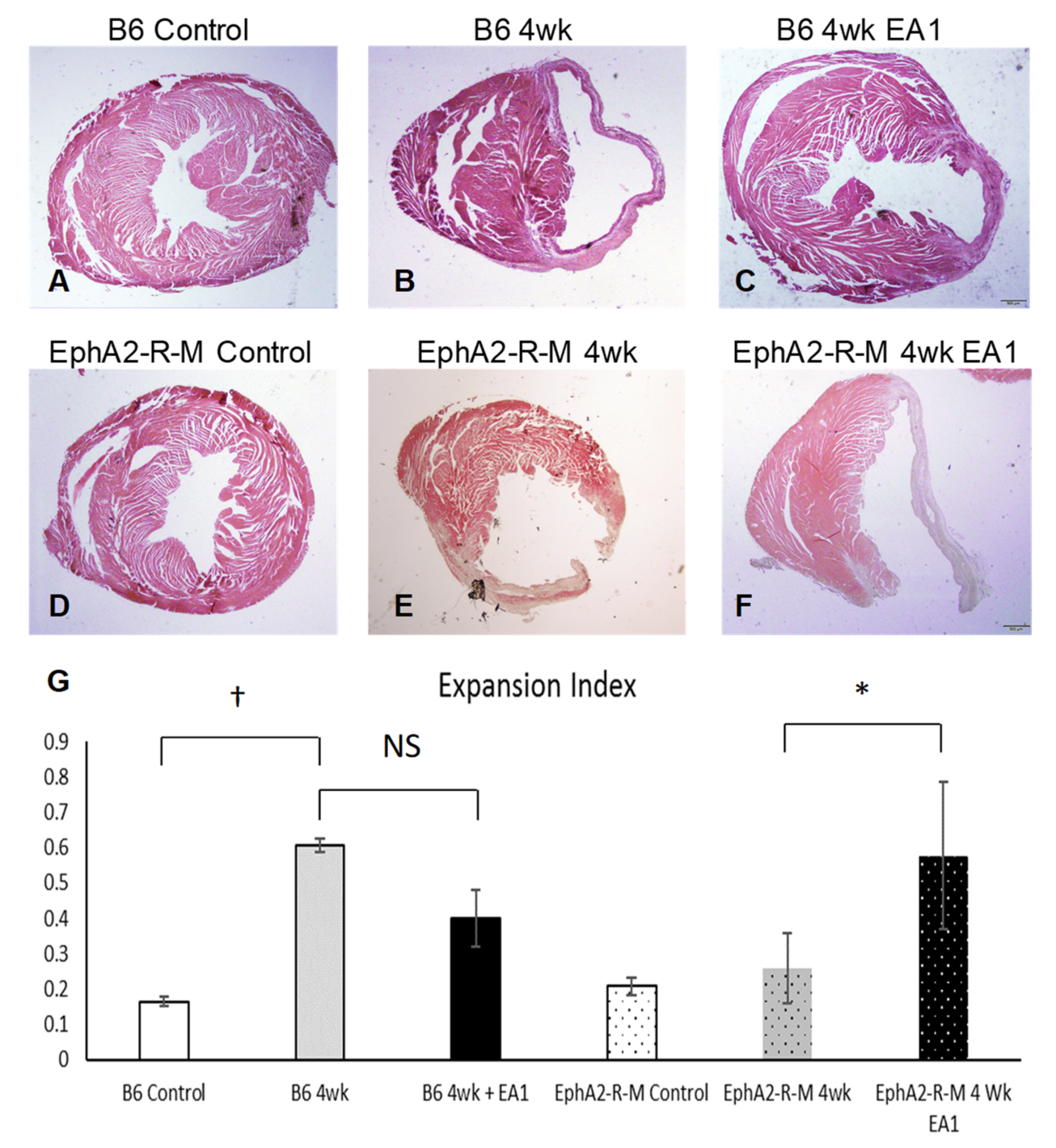
2.2. Morphometry
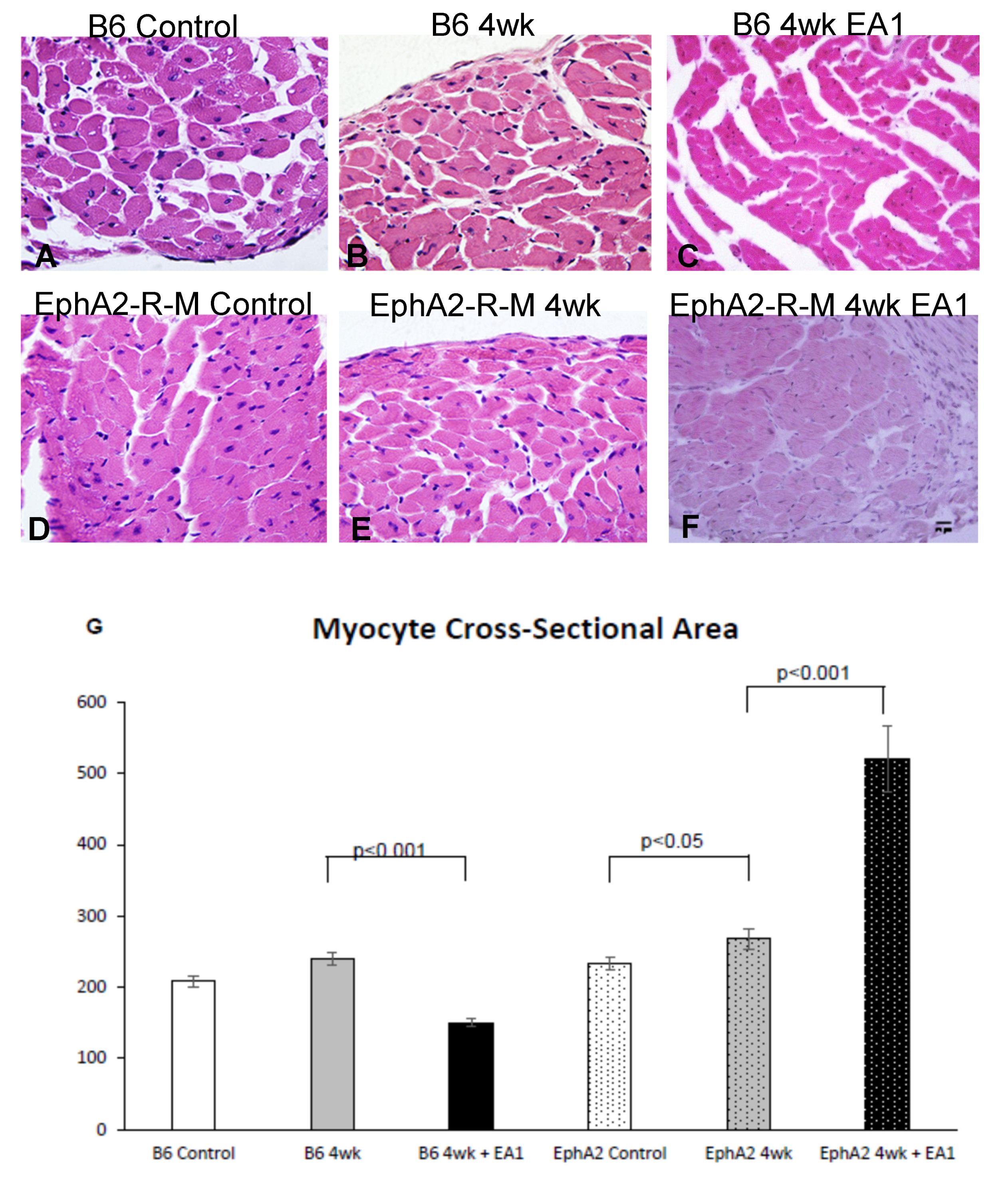
2.3. Myocyte Cross-Sectional Area
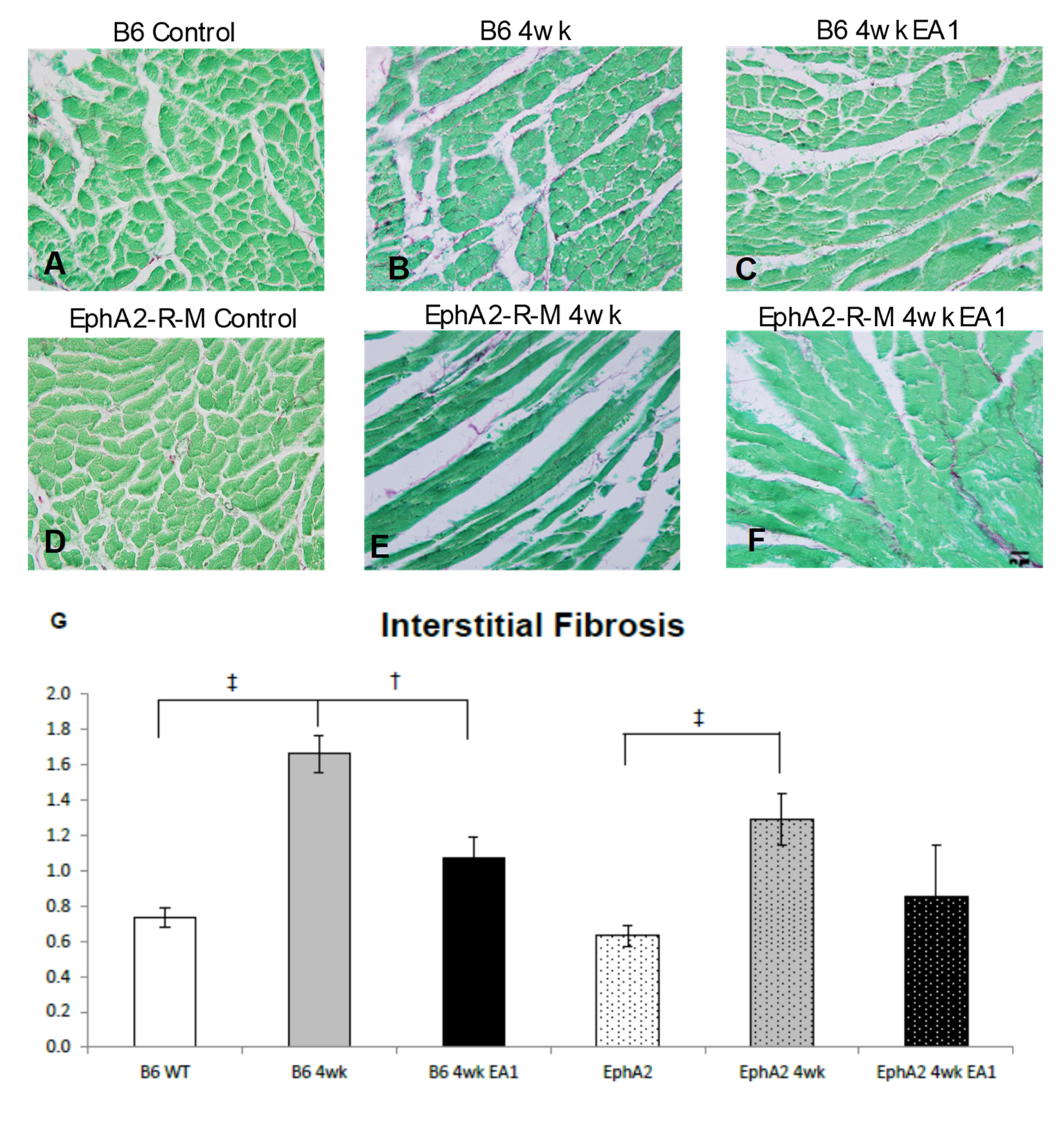
2.4. Fibrosis
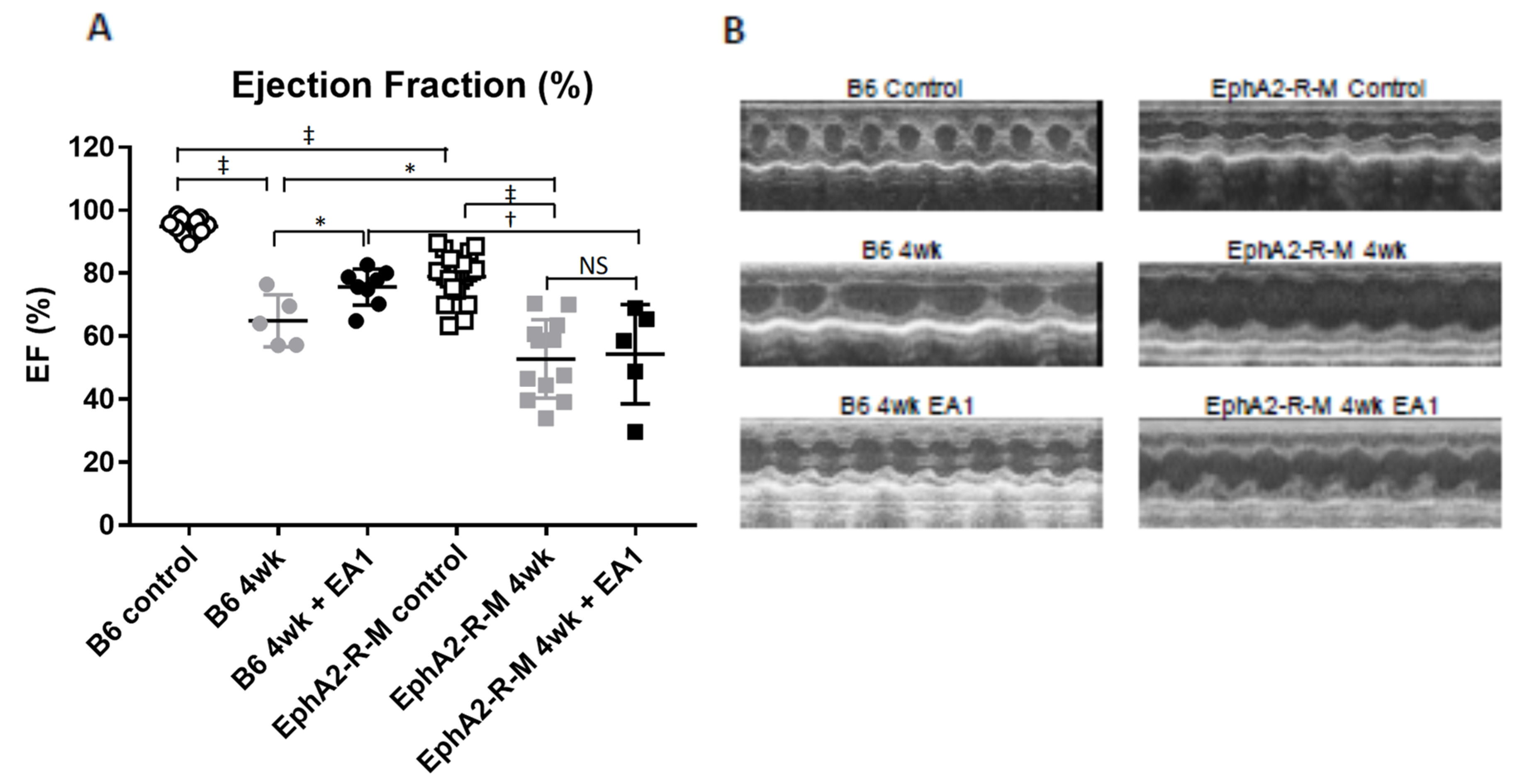
2.5. Cardiac Function
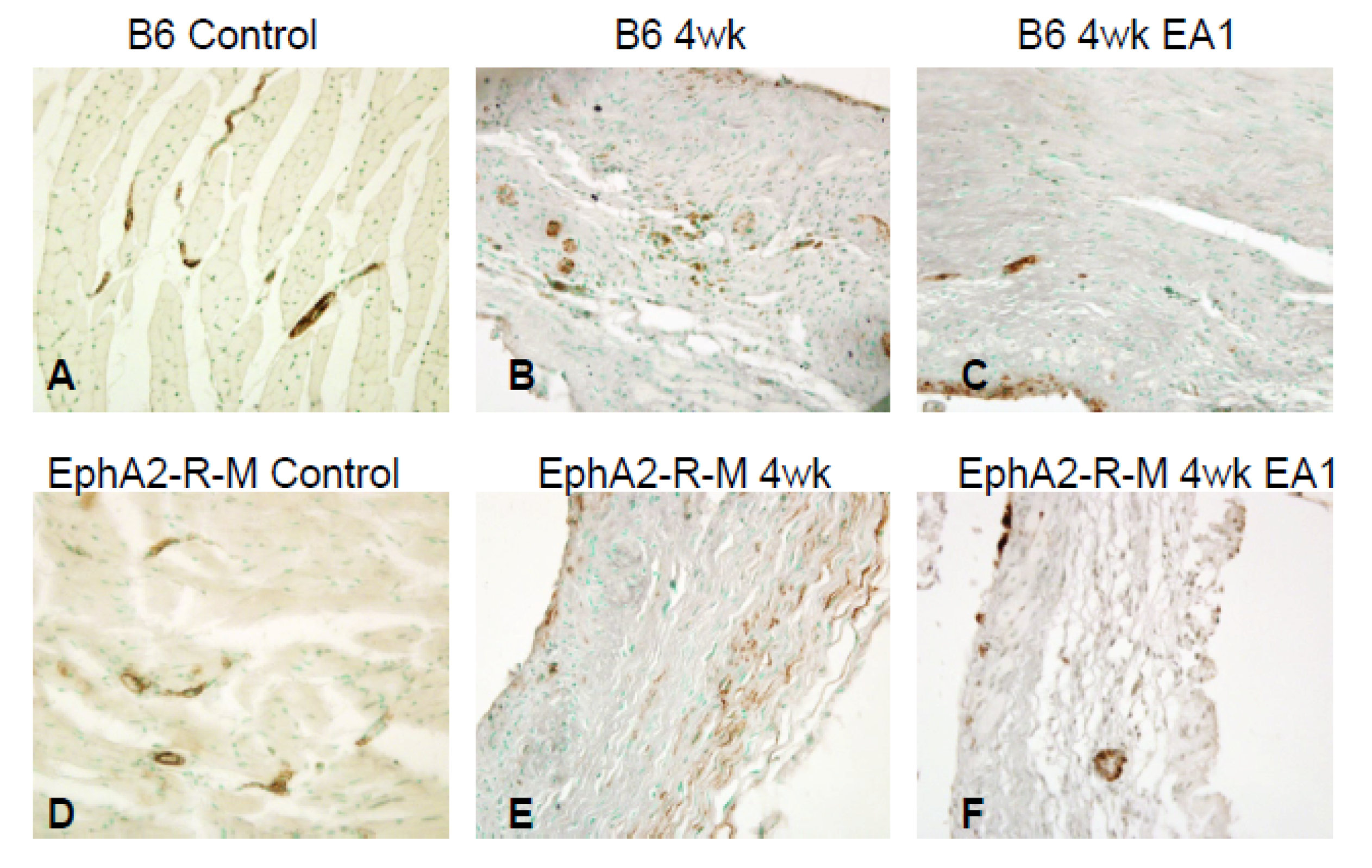
2.6. Immunohistochemical Detection of α-SMA+ Myofibroblasts in Infarcted Mouse Heart
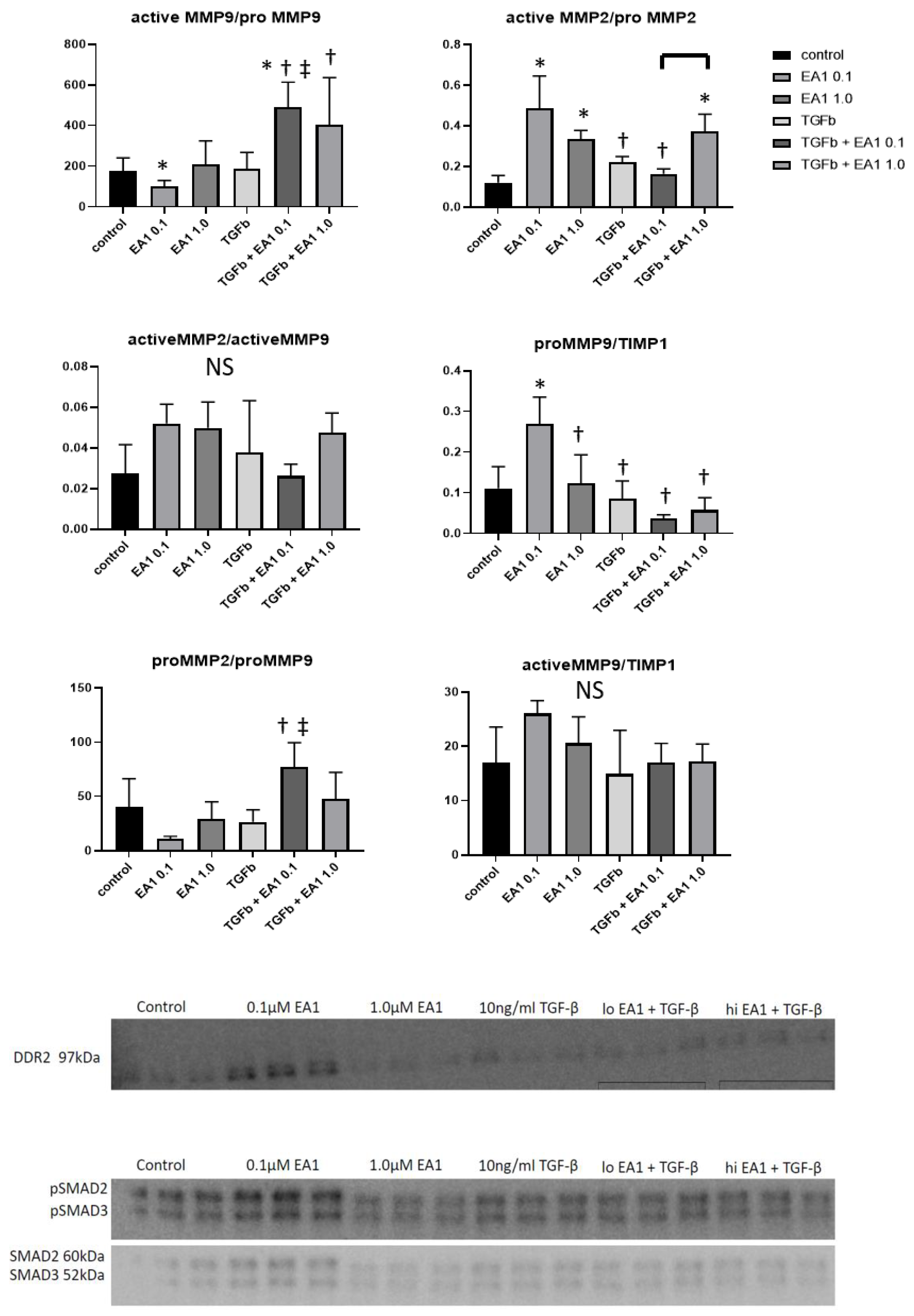
2.7. Western Blotting Analysis of Remodeling Indices in Isolated Primary Cardiac Fibroblasts
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Surgical Procedure and Tissue Collection
4.3. Echocardiography and Blood Pressure
4.4. Morphometry and Histology
4.5. Immunostaining
Isolation and Culture of Primary Mouse Cardiac Fibroblasts
4.6. Protein Extraction and Western Blotting
4.7. Statistics
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pasquale, E.B. Eph-ephrin bidirectional signaling in physiology and disease. Cell 2008, 133, 38–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kullander, K.; Klein, R. Mechanisms and functions of Eph and ephrin signalling. Nat. Rev. Mol. Cell Biol. 2002, 3, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Brantley, D.M.; Cheng, N.; Thompson, E.J.; Lin, Q.; Brekken, R.A.; Thorpe, P.E.; Muraoka, R.S.; Cerretti, D.P.; Pozzi, A.; Jackson, D.; et al. Soluble Eph A receptors inhibit tumor angiogenesis and progression in vivo. Oncogene 2002, 21, 7011–7026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarron, J.K.; Stringer, B.W.; Day, B.W.; Boyd, A.W. Ephrin expression and function in cancer. Future Oncol. 2010, 6, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Lisabeth, E.M.; Falivelli, G.; Pasquale, E.B. Eph receptor signaling and ephrins. Cold Spring Harb. Perspect. Biol. 2013, 5, a009159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, A.; Shao, H.; Marks, R.M.; Polverini, P.J.; Dixit, V.M. Role of B61, the ligand for the Eck receptor tyrosine kinase, in TNF-alpha-induced angiogenesis. Science 1995, 268, 567–569. [Google Scholar] [CrossRef]
- Cheng, N.; Brantley, D.M.; Chen, J. The ephrins and Eph receptors in angiogenesis. Cytokine Growth Factor Rev. 2002, 13, 75–85. [Google Scholar] [CrossRef]
- Brantley-Sieders, D.; Schmidt, S.; Parker, M.; Chen, J. Eph receptor tyrosine kinases in tumor and tumor microenvironment. Curr. Pharm. Des. 2004, 10, 3431–3442. [Google Scholar] [CrossRef]
- Brantley-Sieders, D.M.; Fang, W.B.; Hwang, Y.; Hicks, D.; Chen, J. Ephrin-A1 facilitates mammary tumor metastasis through an angiogenesis-dependent mechanism mediated by EphA receptor and vascular endothelial growth factor in mice. Cancer Res. 2006, 66, 10315–10324. [Google Scholar] [CrossRef] [Green Version]
- Beauchamp, A.; Debinski, W. Ephs and ephrins in cancer: Ephrin-A1 signalling. Semin. Cell Dev. Biol. 2011, 23, 109–115. [Google Scholar] [CrossRef] [Green Version]
- Okazaki, T.; Ni, A.; Baluk, P.; Ayeni, O.A.; Kearley, J.; Coyle, A.J.; Humbles, A.; McDonald, D.M. Capillary defects and exaggerated inflammatory response in the airways of EphA2-deficient mice. Am. J. Pathol. 2009, 174, 2388–2399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funk, S.D.; Orr, A.W. Ephs and ephrins resurface in inflammation, immunity, and atherosclerosis. Pharmacol. Res. 2013, 67, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Coulthard, M.G.; Morgan, M.; Woodruff, T.M.; Arumugam, T.V.; Taylor, S.M.; Carpenter, T.C.; Lackmann, M.; Boyd, A.W. Eph/Ephrin signaling in injury and inflammation. Am. J. Pathol. 2012, 181, 1493–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanov, A.I.; Romanovsky, A.A. Putative dual role of ephrin-Eph receptor interactions in inflammation. IUBMB Life 2006, 58, 389–394. [Google Scholar] [CrossRef]
- Jehle, J.; Staudacher, I.; Wiedmann, F.; Schweizer, P.; Becker, R.; Katus, H.; Thomas, D. Regulation of apoptosis in HL-1 cardiomyocytes by phosphorylation of the receptor tyrosine kinase EphA2 and protection by lithocholic acid. Br. J. Pharmacol. 2012, 167, 1563–1572. [Google Scholar] [CrossRef] [Green Version]
- DuSablon, A.; Parks, J.; Whitehurst, K.; Estes, H.; Chase, R.; Vlahos, E.; Sharma, U.; Wert, D.; Virag, J. EphrinA1-Fc attenuates myocardial ischemia/reperfusion injury in mice. PLoS ONE 2017, 12, e0189307. [Google Scholar] [CrossRef]
- Dries, J.L.; Kent, S.D.; Virag, J.A. Intramyocardial administration of chimeric ephrinA1-Fc promotes tissue salvage following myocardial infarction in mice. J. Physiol. 2011, 589 Pt 7, 1725–1740. [Google Scholar] [CrossRef]
- Torres, M.J.; McLaughlin, K.L.; Renegar, R.H.; Valsaraj, S.; Whitehurst, K.S.; Sharaf, O.M.; Sharma, U.M.; Horton, J.L.; Sarathy, B.; Parks, J.C.; et al. Intracardiac administration of ephrinA1-Fc preserves mitochondrial bioenergetics during acute ischemia/reperfusion injury. Life Sci. 2019, 239, 117053. [Google Scholar] [CrossRef]
- O’Neal, W.T.; Griffin, W.F.; Kent, S.D.; Faiz, F.; Hodges, J.; Vuncannon, J.; Virag, J.A. Deletion of the EphA2 receptor exacerbates myocardial injury and the progression of ischemic cardiomyopathy. Front. Physiol. 2014, 5, 132. [Google Scholar] [CrossRef] [Green Version]
- O’Neal, W.T.; Griffin, W.F.; Dries-Devlin, J.L.; Kent, S.D.; Chen, J.; Willis, M.S.; Virag, J.A. Ephrin-Eph signaling as a potential therapeutic target for the treatment of myocardial infarction. Med. Hypotheses 2013, 80, 738–744. [Google Scholar] [CrossRef] [Green Version]
- Whitehurst, K.S.; Estes, H.; Chase, R.; Sharma, U.; Virag, J.A. Abstract 590: EphrinA1-Fc Attenuates Dysfunction and Fibrosis in Nonreperfused Myocardium at 4 weeks Post-MI in WT B6 but Not EphA2-R Mutant Mice. Circ. Res. 2019, 125, A590. [Google Scholar] [CrossRef]
- McAlindon, E.; Bucciarelli-Ducci, C.; Suleiman, M.S.; Baumbach, A. Infarct size reduction in acute myocardial infarction. Heart 2015, 101, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Thundyil, J.; Manzanero, S.; Pavlovski, D.; Cully, T.R.; Lok, K.Z.; Widiapradja, A.; Chunduri, P.; Jo, D.G.; Naruse, C.; Asano, M.; et al. Evidence that the EphA2 receptor exacerbates ischemic brain injury. PLoS ONE 2013, 8, e53528. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, C.; Chen, Z.W.; Bedirian, A.; Yokota, N.; Nasr, S.H.; Rabb, H.; Lemay, S. Upregulation of EphA2 during in vivo and in vitro renal ischemia-reperfusion injury: Role of Src kinases. Am. J. Physiol. Ren. Physiol. 2006, 291, F960–F971. [Google Scholar] [CrossRef] [PubMed]
- de Boer, R.A.; De Keulenaer, G.; Bauersachs, J.; Brutsaert, D.; Cleland, J.G.; Diez, J.; Du, X.J.; Ford, P.; Heinzel, F.R.; Lipson, K.E.; et al. Towards better definition, quantification and treatment of fibrosis in heart failure. A scientific roadmap by the Committee of Translational Research of the Heart Failure Association (HFA) of the European Society of Cardiology. Eur. J. Heart Fail. 2019, 21, 272–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Squires, C.E.; Escobar, G.P.; Payne, J.F.; Leonardi, R.A.; Goshorn, D.K.; Sheats, N.J.; Mains, I.M.; Mingoia, J.T.; Flack, E.C.; Lindsey, M.L. Altered fibroblast function following myocardial infarction. J. Mol. Cell. Cardiol. 2005, 39, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Arpino, V.; Brock, M.; Gill, S.E. The role of TIMPs in regulation of extracellular matrix proteolysis. Matrix Biol. 2015, 44–46, 247–254. [Google Scholar] [CrossRef]
- Li, Y.Y.; McTiernan, C.F.; Feldman, A.M. Interplay of matrix metalloproteinases, tissue inhibitors of metalloproteinases and their regulators in cardiac matrix remodeling. Cardiovasc. Res. 2000, 46, 214–224. [Google Scholar] [CrossRef] [Green Version]
- Frangogiannis, N.G. The mechanistic basis of infarct healing. Antioxid. Redox Signal. 2006, 8, 1907–1939. [Google Scholar] [CrossRef]
- Beauchamp, A.; Lively, M.O.; Mintz, A.; Gibo, D.; Wykosky, J.; Debinski, W. EphrinA1 is released in three forms from cancer cells by matrix metalloproteases. Mol. Cell. Biol. 2012, 32, 3253–3264. [Google Scholar] [CrossRef] [Green Version]
- Souders, C.A.; Bowers, S.L.; Baudino, T.A. Cardiac fibroblast: The renaissance cell. Circ. Res. 2009, 105, 1164–1176. [Google Scholar] [CrossRef] [PubMed]
- Shyu, K.G.; Chao, Y.M.; Wang, B.W.; Kuan, P. Regulation of discoidin domain receptor 2 by cyclic mechanical stretch in cultured rat vascular smooth muscle cells. Hypertension 2005, 46, 614–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olaso, E.; Labrador, J.P.; Wang, L.; Ikeda, K.; Eng, F.J.; Klein, R.; Lovett, D.H.; Lin, H.C.; Friedman, S.L. Discoidin domain receptor 2 regulates fibroblast proliferation and migration through the extracellular matrix in association with transcriptional activation of matrix metalloproteinase-2. J. Biol. Chem. 2002, 277, 3606–3613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalil, H.; Kanisicak, O.; Prasad, V.; Correll, R.N.; Fu, X.; Schips, T.; Vagnozzi, R.J.; Liu, R.; Huynh, T.; Lee, S.J.; et al. Fibroblast-specific TGF-beta-Smad2/3 signaling underlies cardiac fibrosis. J. Clin. Investig. 2017, 127, 3770–3783. [Google Scholar] [CrossRef]
- Kamato, D.; Burch, M.L.; Piva, T.J.; Rezaei, H.B.; Rostam, M.A.; Xu, S.; Zheng, W.; Little, P.J.; Osman, N. Transforming growth factor-beta signalling: Role and consequences of Smad linker region phosphorylation. Cell. Signal. 2013, 25, 2017–2024. [Google Scholar] [CrossRef]
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell. Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef] [Green Version]
- Pokharel, S.; van Geel, P.P.; Sharma, U.C.; Cleutjens, J.P.; Bohnemeier, H.; Tian, X.L.; Schunkert, H.; Crijns, H.J.; Paul, M.; Pinto, Y.M. Increased myocardial collagen content in transgenic rats overexpressing cardiac angiotensin-converting enzyme is related to enhanced breakdown of N-acetyl-Ser-Asp-Lys-Pro and increased phosphorylation of Smad2/3. Circulation 2004, 110, 3129–3135. [Google Scholar] [CrossRef] [Green Version]
- Sakata, Y.; Chancey, A.L.; Divakaran, V.G.; Sekiguchi, K.; Sivasubramanian, N.; Mann, D.L. Transforming growth factor-beta receptor antagonism attenuates myocardial fibrosis in mice with cardiac-restricted overexpression of tumor necrosis factor. Basic Res. Cardiol. 2008, 103, 60–68. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Gupte, M.; Umbarkar, P.; Singh, A.P.; Sui, J.Y.; Force, T.; Lal, H. Entanglement of GSK-3beta, beta-catenin and TGF-beta1 signaling network to regulate myocardial fibrosis. J. Mol. Cell. Cardiol. 2017, 110, 109–120. [Google Scholar] [CrossRef]
- Horton, J.L.; Virag, J. Use of Multifactorial Treatments to Address the Challenge of Translating Experimental Myocardial Infarct Reduction Strategies. Int. J. Mol. Sci. 2019, 20, 1449. [Google Scholar] [CrossRef] [Green Version]
- Davidson, S.M.; Ferdinandy, P.; Andreadou, I.; Botker, H.E.; Heusch, G.; Ibanez, B.; Ovize, M.; Schulz, R.; Yellon, D.M.; Hausenloy, D.J.; et al. Multitarget strategies to reduce myocardial ischemia/reperfusion injury: JACC review topic of the week. J. Am. Coll. Cardiol. 2019, 73, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Janes, P.W.; Griesshaber, B.; Atapattu, L.; Nievergall, E.; Hii, L.L.; Mensinga, A.; Chheang, C.; Day, B.W.; Boyd, A.W.; Bastiaens, P.I.; et al. Eph receptor function is modulated by heterooligomerization of A and B type Eph receptors. J. Cell Biol. 2011, 195, 1033–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DuSablon, A.; Kent, S.; Coburn, A.; Virag, J. EphA2-receptor deficiency exacerbates myocardial infarction and reduces survival in hyperglycemic mice. Cardiovasc. Diabetol. 2014, 13, 114. [Google Scholar] [CrossRef] [Green Version]
- Virag, J.A.I.; Lust, R.M. Coronary Artery Ligation and Intramyocardial Injection in a Murine Model of Infarction. J. Vis. Exp. 2011, 7, e2581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.P.; Liu, Y.H.; Rhaleb, N.E.; Kurihara, N.; Kim, H.E.; Carretero, O.A. Echocardiographic assessment of cardiac function in conscious and anesthetized mice. Am. J. Physiol. 1999, 277, H1967–H1974. [Google Scholar] [CrossRef] [PubMed]
- Mor-Avi, V.; Lang, R.M.; Badano, L.P.; Belohlavek, M.; Cardim, N.M.; Derumeaux, G.; Galderisi, M.; Marwick, T.; Nagueh, S.F.; Sengupta, P.P.; et al. Current and evolving echocardiographic techniques for the quantitative evaluation of cardiac mechanics: ASE/EAE consensus statement on methodology and indications endorsed by the Japanese Society of Echocardiography. Eur. J. Echocardiogr. 2011, 12, 167–205. [Google Scholar] [CrossRef] [PubMed]
- Virag, J.A.; Rolle, M.L.; Reece, J.; Hardouin, S.; Feigl, E.O.; Murry, C.E. Fibroblast growth factor-2 regulates myocardial infarct repair: Effects on cell proliferation, scar contraction, and ventricular function. Am. J. Pathol. 2007, 171, 1431–1440. [Google Scholar] [CrossRef] [Green Version]
- Lennon, F.E.; Mirzapoiazova, T.; Mambetsariev, N.; Mambetsariev, B.; Salgia, R.; Singleton, P.A. Transactivation of the receptor-tyrosine kinase ephrin receptor A2 is required for the low molecular weight hyaluronan-mediated angiogenesis that is implicated in tumor progression. J. Biol. Chem. 2014, 289, 24043–24058. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Thibeault, S.L. Response of fibroblasts to transforming growth factor-beta1 on two-dimensional and in three-dimensional hyaluronan hydrogels. Tissue Eng. Part A 2012, 18, 2528–2538. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Morphometric Parameters | WT B6 Uninjured (n = 10) | EphA2-R Uninjured (n = 10) | WT B6 4wk (n = 5) | EphA2-R 4wk (n = 12) | WT B6 4wk + EA1 (n = 8) | EphA2-R 4wk + EA1 (n = 5) |
|---|---|---|---|---|---|---|
| LVIDd (mm) | 2.50 ± 0.10 | 2.82 ± 0.10 | 4.08 ± 0.30 * | 3.58 ± 0.32 * | 2.86 ± 0.08 | 3.64 ± 0.50 |
| LVIDs (mm) | 0.78 ± 0.06 | 1.51± 0.08 | 2.65 ± 0.27 * | 2.65 ± 0.29 * | 1.63 ± 0.08 | 2.66 ± 0.51 |
| AWT (mm) | 2.07 ± 0.09 | 2.07 ± 0.16 | 1.3 ± 0.12 | 2.34 ± 0.21 | 1.79 ± 0.11 | 1.71 ± 0.41 |
| SWT (mm) | 2.21 ± 0.19 | 2.02 ± 0.08 | 1.86 ± 0.23 | 2.18 ± 0.14 | 2.48 ± 0.13 | 2.74 ± 0.09 ‡ |
| LVVd (µL) | 23.10 ± 2.43 | 75.92 ± 13.31 | 75.92 ± 13.32 | 60.81 ± 14.1 | 31.30 ± 2.28 † | 62.26 ± 21.04 |
| LVVs (µL) | 1.21 ± 0.23 | 27.70 ± 7.18 | 27.70 ± 7.18 | 31.48 ± 9.24 | 7.77 ± 1.12 † | 32.59 ± 16.59 |
| LV Area (mm2) | 5.69 ± 0.9 | 6.07 ± 0.48 | 12.30 ±1.32 * | 7.85 ± 1.35 | 11.66 ± 2.22 | 12.44 ± 2.82 ‡ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Whitehurst, K.S.; Chan, V.A.; Estes, H.K.; Valsaraj, S.; Kent, S.; Sharma, U.M.; Chase, R.C.; Bhuiyan, M.; Virag, J.A.I. EphrinA1-Fc Attenuates Ventricular Remodeling and Dysfunction in Chronically Nonreperfused WT but not EphA2-R-M mice. Int. J. Mol. Sci. 2020, 21, 5811. https://doi.org/10.3390/ijms21165811
Whitehurst KS, Chan VA, Estes HK, Valsaraj S, Kent S, Sharma UM, Chase RC, Bhuiyan M, Virag JAI. EphrinA1-Fc Attenuates Ventricular Remodeling and Dysfunction in Chronically Nonreperfused WT but not EphA2-R-M mice. International Journal of Molecular Sciences. 2020; 21(16):5811. https://doi.org/10.3390/ijms21165811
Chicago/Turabian StyleWhitehurst, K’Shylah S., Victoria A. Chan, Heather K. Estes, Smrithi Valsaraj, Susan Kent, Uma M. Sharma, R. Christopher Chase, Maliha Bhuiyan, and Jitka A. I. Virag. 2020. "EphrinA1-Fc Attenuates Ventricular Remodeling and Dysfunction in Chronically Nonreperfused WT but not EphA2-R-M mice" International Journal of Molecular Sciences 21, no. 16: 5811. https://doi.org/10.3390/ijms21165811
APA StyleWhitehurst, K. S., Chan, V. A., Estes, H. K., Valsaraj, S., Kent, S., Sharma, U. M., Chase, R. C., Bhuiyan, M., & Virag, J. A. I. (2020). EphrinA1-Fc Attenuates Ventricular Remodeling and Dysfunction in Chronically Nonreperfused WT but not EphA2-R-M mice. International Journal of Molecular Sciences, 21(16), 5811. https://doi.org/10.3390/ijms21165811