DispHred: A Server to Predict pH-Dependent Order–Disorder Transitions in Intrinsically Disordered Proteins




Abstract
:
1. Introduction
2. Results
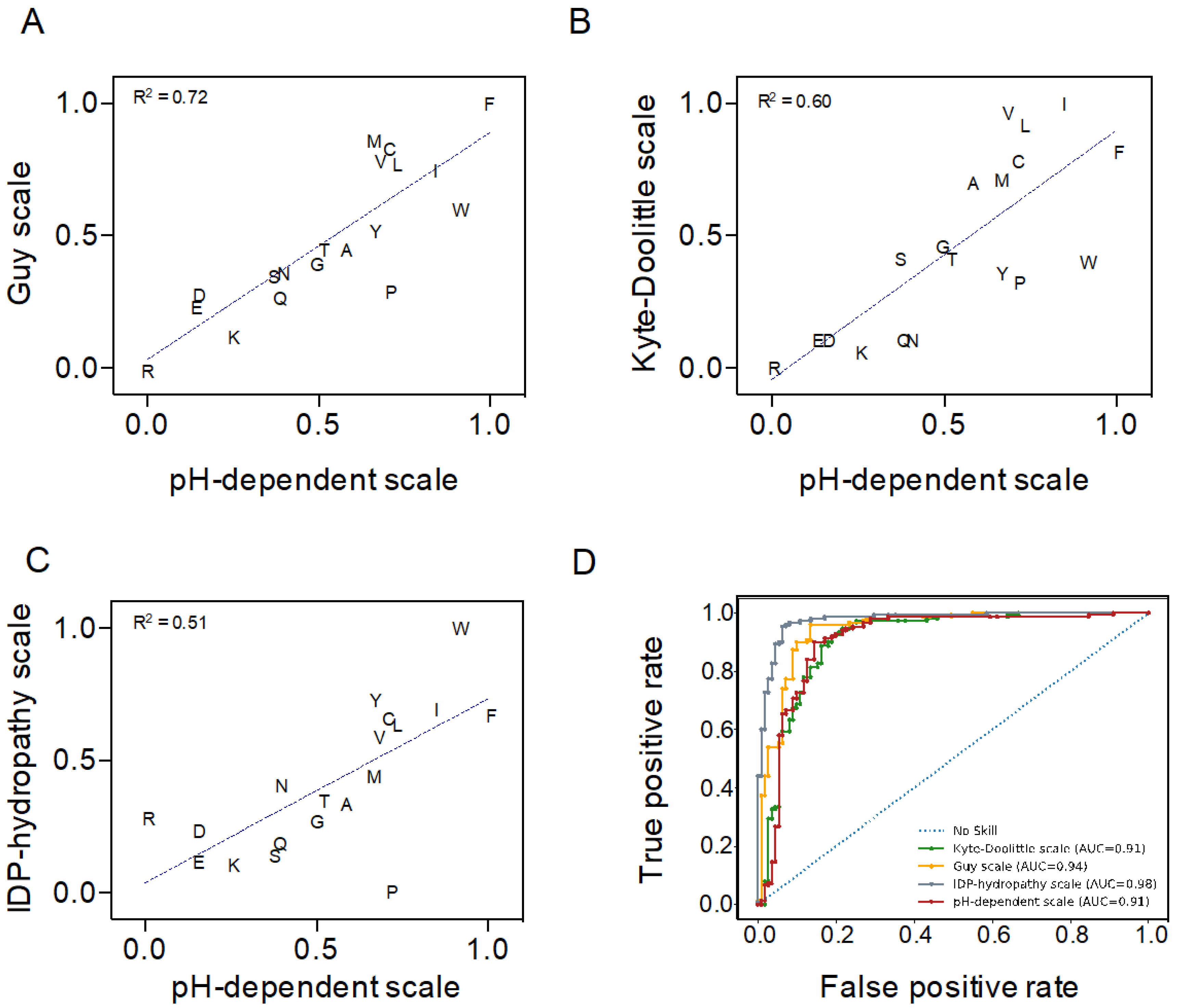
2.1. Validation of a pH-Dependent Hydropathy Scale for C–H Plot-Based Predictions
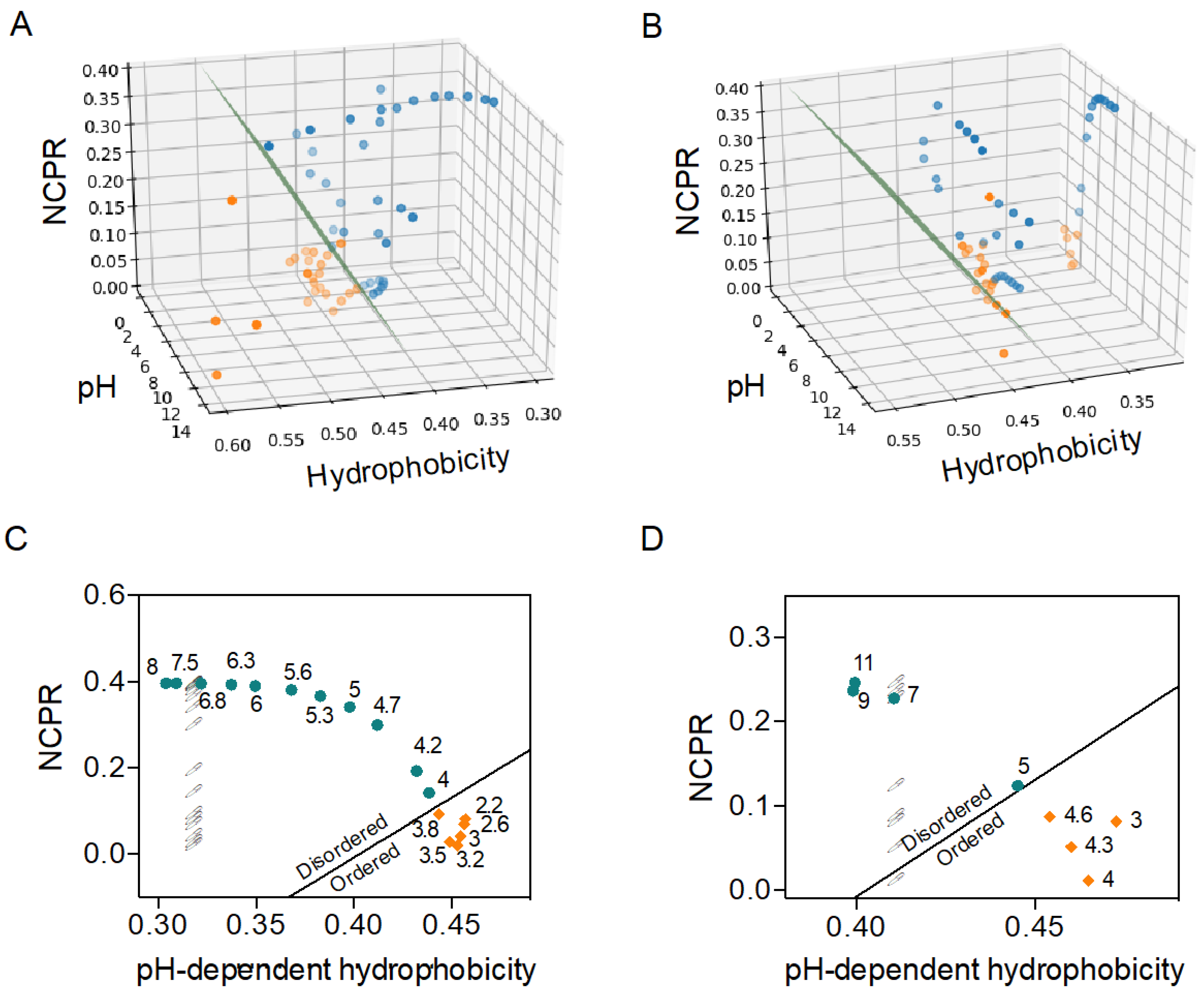
2.2. C–H Space Phase Diagram and Order–Disorder Boundary Condition Can Anticipate pH-Induced Order–Disorder Transition of IDPs
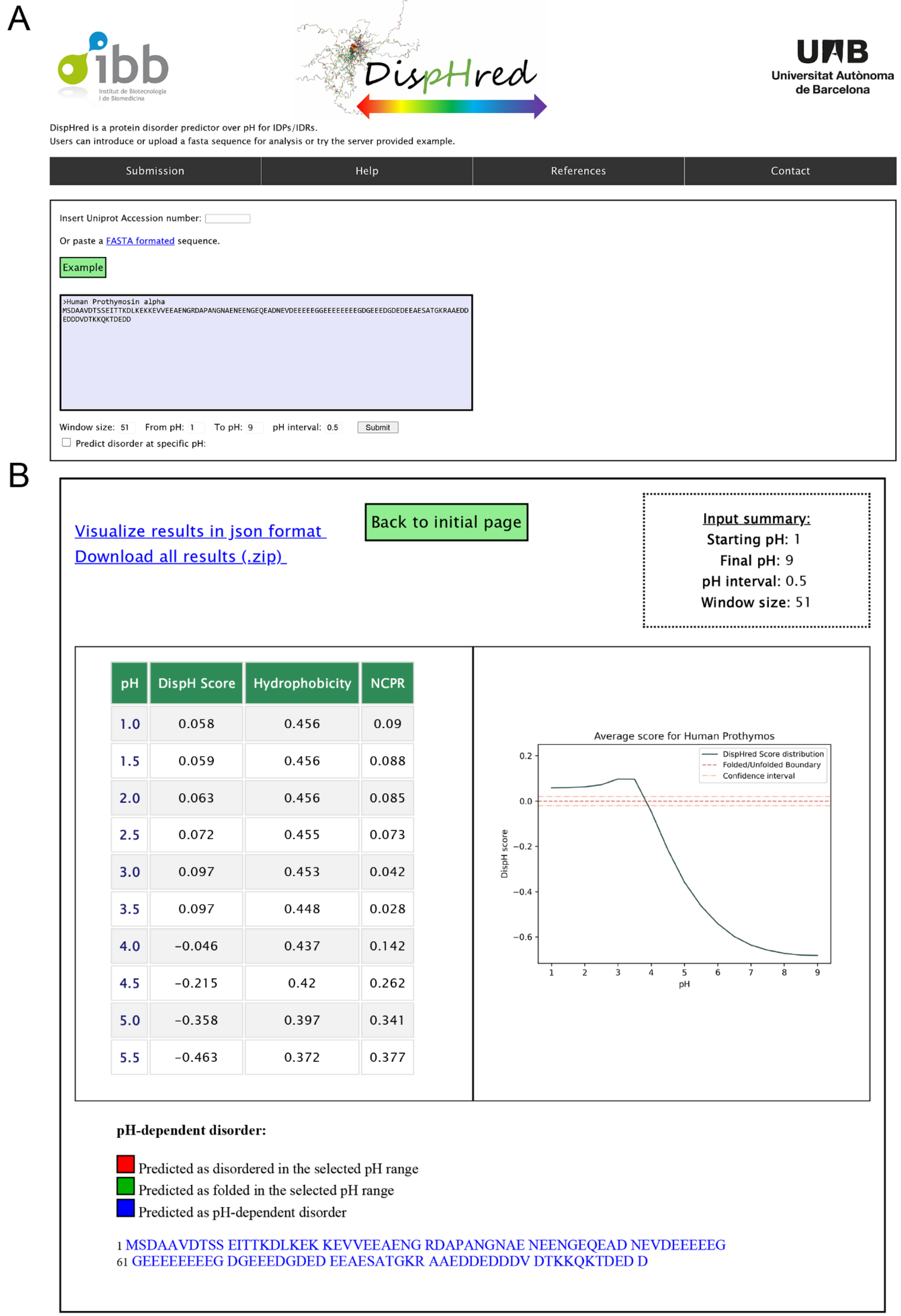
2.3. Rational and Implementation of DispHed, a pH-dependent Predictor of Sequence Disorder
3. Discussion
4. Materials and Methods
4.1. Data Collection
4.2. DispHred: Evaluation of Hydrophobicity and Charge as a Function of pH
4.3. Hydropathy Scales Performance Analysis at Neutral pH
4.4. Support Vector Machine Analysis
4.5. DispHred: Prediction of Sequence Disorder
4.6. Performance Analysis
4.7. DispHred Web-Server
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AUC | Area under the curve |
| C–H | Charge–hydropathy |
| IDP | Intrinsically disordered protein |
| NCPR | Net charge per residue |
| ROC | Receiver Operating Characteristic |
| SVM | Support Vector Machine |
| <H> | Mean hydrophobicity |
References
- Dunker, A.K.; Obradovic, Z. The protein trinity--linking function and disorder. Nat. Biotechnol. 2001, 19, 805–806. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, V.; Kulkarni, P. Intrinsically disordered proteins and phenotypic switching: Implications in cancer. Prog. Mol. Biol. Transl. Sci. 2019, 166, 63–84. [Google Scholar] [PubMed]
- Chen, J.; Kriwacki, R.W. Intrinsically Disordered Proteins: Structure, Function and Therapeutics. J. Mol. Biol. 2018, 430, 2275–2277. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef]
- Dyson, H.J. Making Sense of Intrinsically Disordered Proteins. Biophys. J. 2016, 110, 1013–1016. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N.; Gillespie, J.R.; Fink, A.L. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 2000, 41, 415–427. [Google Scholar] [CrossRef]
- Prilusky, J.; Felder, C.E.; Zeev-Ben-Mordehai, T.; Rydberg, E.H.; Man, O.; Beckmann, J.S.; Silman, I.; Sussman, J.L. FoldIndex: A simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics 2005, 21, 3435–3438. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Wang, K.; Liu, Y.; Xue, B.; Uversky, V.N.; Dunker, A.K. Predicting intrinsic disorder in proteins: An overview. Cell Res. 2009, 19, 929–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lieutaud, P.; Ferron, F.; Uversky, A.V.; Kurgan, L.; Uversky, V.N.; Longhi, S. How disordered is my protein and what is its disorder for? A guide through the “dark side” of the protein universe. Intrinsically Disord. Proteins 2016, 4, e1259708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dosztanyi, Z. Prediction of protein disorder based on IUPred. Protein Sci. 2018, 27, 331–340. [Google Scholar] [CrossRef] [Green Version]
- Schramm, A.; Lieutaud, P.; Gianni, S.; Longhi, S.; Bignon, C. InSiDDe: A Server for Designing Artificial Disordered Proteins. Int. J. Mol. Sci. 2017, 19, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harmon, T.S.; Crabtree, M.D.; Shammas, S.L.; Posey, A.E.; Clarke, J.; Pappu, R.V. GADIS: Algorithm for designing sequences to achieve target secondary structure profiles of intrinsically disordered proteins. Protein Eng. Des. Sel. 2016, 29, 339–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakob, U.; Kriwacki, R.; Uversky, V.N. Conditionally and transiently disordered proteins: Awakening cryptic disorder to regulate protein function. Chem. Rev. 2014, 114, 6779–6805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uversky, V.N. Intrinsically disordered proteins and their environment: Effects of strong denaturants, temperature, pH, counter ions, membranes, binding partners, osmolytes, and macromolecular crowding. Protein J. 2009, 28, 305–325. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.D.; Jelokhani-Niaraki, M. pH-induced changes in intrinsically disordered proteins. Methods Mol. Biol. 2012, 896, 223–231. [Google Scholar] [PubMed]
- Fonin, A.V.; Stepanenko, O.V.; Sitdikova, A.K.; Antifeeva, I.A.; Kostyleva, E.I.; Polyanichko, A.M.; Karasev, M.M.; Silonov, S.A.; Povarova, O.I.; Kuznetsova, I.M.; et al. Folding of poly-amino acids and intrinsically disordered proteins in overcrowded milieu induced by pH change. Int. J. Biol. Macromol. 2019, 125, 244–255. [Google Scholar] [CrossRef] [Green Version]
- Santos, J.; Iglesias, V.; Santos-Suarez, J.; Mangiagalli, M.; Brocca, S.; Pallares, I.; Ventura, S. pH-Dependent Aggregation in Intrinsically Disordered Proteins Is Determined by Charge and Lipophilicity. Cells 2020, 9, 145. [Google Scholar] [CrossRef] [Green Version]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef] [Green Version]
- Zamora, W.J.; Campanera, J.M.; Luque, F.J. Development of a Structure-Based, pH-Dependent Lipophilicity Scale of Amino Acids from Continuum Solvation Calculations. J. Phys. Chem. Lett. 2019, 10, 883–889. [Google Scholar] [CrossRef]
- Huang, F.; Oldfield, C.J.; Xue, B.; Hsu, W.L.; Meng, J.; Liu, X.; Shen, L.; Romero, P.; Uversky, V.N.; Dunker, A. Improving protein order-disorder classification using charge-hydropathy plots. BMC Bioinform. 2014, 15 (Suppl. 17), S4. [Google Scholar] [CrossRef] [Green Version]
- Guy, H.R. Amino acid side-chain partition energies and distribution of residues in soluble proteins. Biophys. J. 1985, 47, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Vapnik, V. Statistical Learning Theory; Wiley: Hoboken, NJ, USA, 1998; Volume 1, p. 768. [Google Scholar]
- Vapnik, V. The Nature of Statistical Learning Theory; Springer Science & Business Media: Berlin, Germany, 2013. [Google Scholar]
- Uversky, V.N.; Gillespie, J.R.; Millett, I.S.; Khodyakova, A.V.; Vasiliev, A.M.; Chernovskaya, T.V.; Vasilenko, R.N.; Kozlovskaya, G.D.; Dolgikh, D.A.; Fink, A.L.; et al. Natively unfolded human prothymosin alpha adopts partially folded collapsed conformation at acidic pH. Biochemistry 1999, 38, 15009–15016. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.Z.; Swaminathan, R. Structure and dynamics at N- and C-terminal regions of intrinsically disordered human c-Myc PEST degron reveal a pH-induced transition. Proteins 2020, 88, 889–909. [Google Scholar] [CrossRef] [PubMed]
- Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins and intrinsically disordered protein regions. Annu. Rev. Biochem. 2014, 83, 553–584. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P. Intrinsically disordered proteins: A 10-year recap. Trends Biochem. Sci. 2012, 37, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Babu, M.M.; Van der Lee, R.; de Groot, N.S.; Gsponer, J. Intrinsically disordered proteins: Regulation and disease. Curr. Opin. Struct. Biol. 2011, 21, 432–440. [Google Scholar] [CrossRef]
- Payliss, B.J.; Vogel, J.; Mittermaier, A.K. Side chain electrostatic interactions and pH-dependent expansion of the intrinsically disordered, highly acidic carboxyl-terminus of gamma-tubulin. Protein Sci. 2019, 28, 1095–1105. [Google Scholar] [CrossRef]
- Xue, B.; Dunbrack, R.L.; Williams, R.W.; Dunker, A.K.; Uversky, V.N. PONDR-FIT: A meta-predictor of intrinsically disordered amino acids. Biochim. Biophys. Acta 2010, 1804, 996–1010. [Google Scholar] [CrossRef] [Green Version]
- Garner, E.; Romero, P.; Dunker, A.K.; Brown, C.; Obradovic, Z. Predicting Binding Regions within Disordered Proteins. Genome Inform. Ser. Workshop Genome Inform. 1999, 10, 41–50. [Google Scholar]
- Iakoucheva, L.M.; Radivojac, P.; Brown, C.J.; O’Connor, T.R.; Sikes, J.G.; Obradovic, Z.; Dunker, A.K. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 2004, 32, 1037–1049. [Google Scholar] [CrossRef] [Green Version]
- Meng, F.; Uversky, V.N.; Kurgan, L. Comprehensive review of methods for prediction of intrinsic disorder and its molecular functions. Cell. Mol. Life Sci. 2017, 74, 3069–3090. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.J.; McGuffin, L.J.; Bryson, K.; Buxton, B.F.; Jones, D.T. The DISOPRED server for the prediction of protein disorder. Bioinformatics 2004, 20, 2138–2139. [Google Scholar] [CrossRef] [PubMed]
- Bardwell, J.C.; Jakob, U. Conditional disorder in chaperone action. Trends Biochem. Sci. 2012, 37, 517–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minde, D.P.; Halff, E.F.; Tans, S. Designing disorder: Tales of the unexpected tails. Intrinsically Disord. Proteins 2013, 1, e26790. [Google Scholar] [CrossRef] [PubMed]
- Hatos, A.; Hajdu-Soltesz, B.; Monzon, A.M.; Palopoli, N.; Alvarez, L.; Aykac-Fas, B.; Bassot, C.; Benitez, G.I.; Bevilacqua, M.; Chasapi, A.; et al. DisProt: Intrinsic protein disorder annotation in 2020. Nucleic Acids Res. 2020, 48, D269–D276. [Google Scholar] [CrossRef] [Green Version]
- Johansson, J.; Gudmundsson, G.H.; Rottenberg, M.E.; Berndt, K.D.; Agerberth, B. Conformation-dependent antibacterial activity of the naturally occurring human peptide LL-37. J. Biol. Chem. 1998, 273, 3718–3724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munoz, V.; Serrano, L. Elucidating the folding problem of helical peptides using empirical parameters. III. Temperature and pH dependence. J. Mol. Biol. 1995, 245, 297–308. [Google Scholar] [CrossRef] [Green Version]
- Munishkina, L.A.; Fink, A.L.; Uversky, V.N. Conformational prerequisites for formation of amyloid fibrils from histones. J. Mol. Biol. 2004, 342, 1305–1324. [Google Scholar] [CrossRef]
- Richardson, L.G.; Jelokhani-Niaraki, M.; Smith, M.D. The acidic domains of the Toc159 chloroplast preprotein receptor family are intrinsically disordered protein domains. BMC Biochem. 2009, 10, 35. [Google Scholar] [CrossRef] [Green Version]
- Carr, C.M.; Kim, P.S. A spring-loaded mechanism for the conformational change of influenza hemagglutinin. Cell 1993, 73, 823–832. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Chicco, D.; Jurman, G. The advantages of the Matthews correlation coefficient (MCC) over F1 score and accuracy in binary classification evaluation. BMC Genom. 2020, 21, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, J. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Measure | pH-Dependent Hydrophobicity | pH-Independent Hydrophobicity |
|---|---|---|
| Sensitivity | 1.00 | 1.00 |
| Specificity | 0.96 | 0.21 |
| Precision | 0.97 | 0.65 |
| False Discovery rate | 0.03 | 0.35 |
| Accuracy | 0.98 | 0.68 |
| F1 Score | 0.99 | 0.79 |
| Matthews Correlation Coefficient | 0.97 | 0.37 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santos, J.; Iglesias, V.; Pintado, C.; Santos-Suárez, J.; Ventura, S. DispHred: A Server to Predict pH-Dependent Order–Disorder Transitions in Intrinsically Disordered Proteins. Int. J. Mol. Sci. 2020, 21, 5814. https://doi.org/10.3390/ijms21165814
Santos J, Iglesias V, Pintado C, Santos-Suárez J, Ventura S. DispHred: A Server to Predict pH-Dependent Order–Disorder Transitions in Intrinsically Disordered Proteins. International Journal of Molecular Sciences. 2020; 21(16):5814. https://doi.org/10.3390/ijms21165814
Chicago/Turabian StyleSantos, Jaime, Valentín Iglesias, Carlos Pintado, Juan Santos-Suárez, and Salvador Ventura. 2020. "DispHred: A Server to Predict pH-Dependent Order–Disorder Transitions in Intrinsically Disordered Proteins" International Journal of Molecular Sciences 21, no. 16: 5814. https://doi.org/10.3390/ijms21165814
APA StyleSantos, J., Iglesias, V., Pintado, C., Santos-Suárez, J., & Ventura, S. (2020). DispHred: A Server to Predict pH-Dependent Order–Disorder Transitions in Intrinsically Disordered Proteins. International Journal of Molecular Sciences, 21(16), 5814. https://doi.org/10.3390/ijms21165814