NME/NM23/NDPK and Histidine Phosphorylation


Abstract
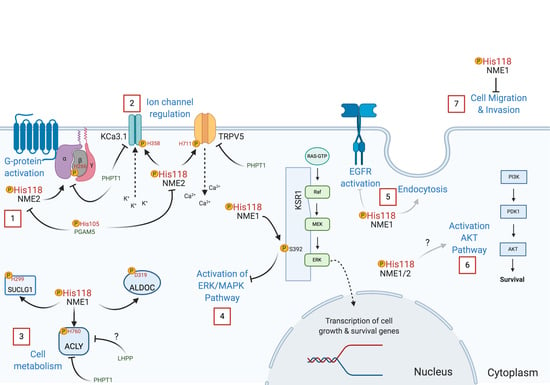
:
1. Histidine Kinase in Mammals: from Myth to Reality?
1.1. Complexity of NME Genes and Functions
1.2. The Resurgence of Histidine Phophorylation
1.3. The Pros and Cons of Histidine Kinase Potential of NME
2. NDPK and Protein Kinase Activity: from Structure to Function
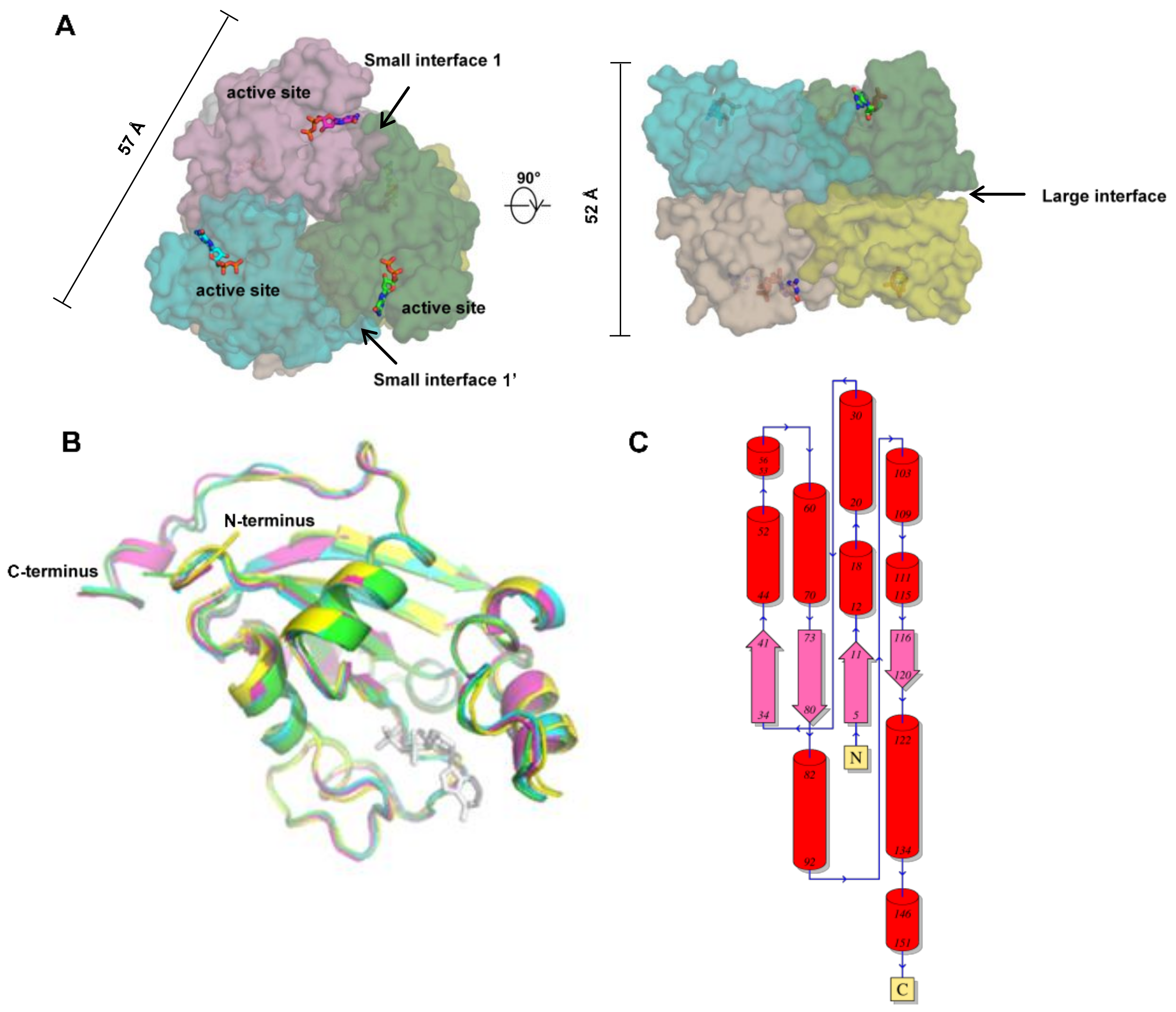
2.1. General Architecture of Human NMEs
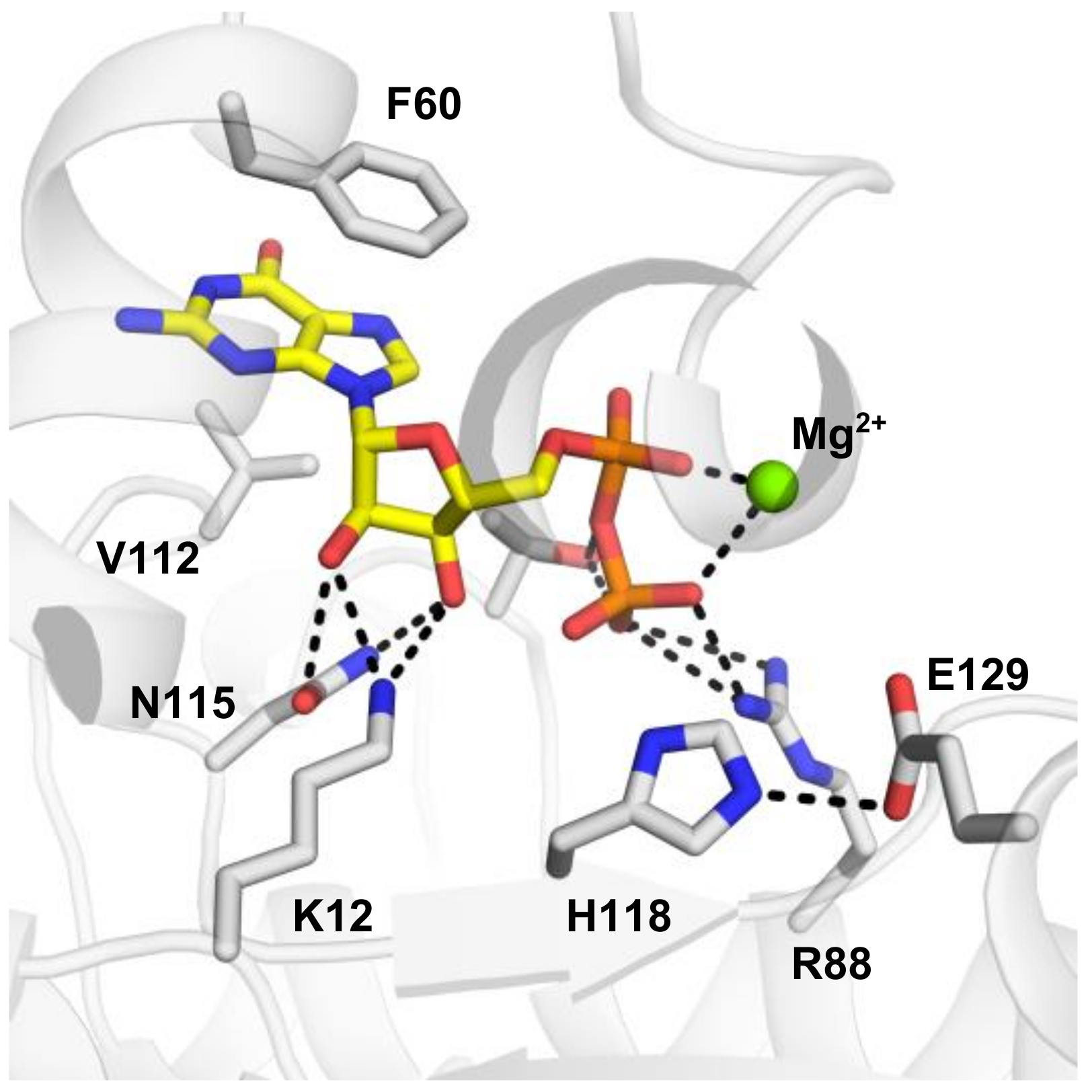
2.2. Structural Basics of the Catalysis of Human NMEs
2.3. Assembly and Oligomerization of Human NMEs
3. NME as a Protein Histidine Kinase
3.1. NME Binding Partners
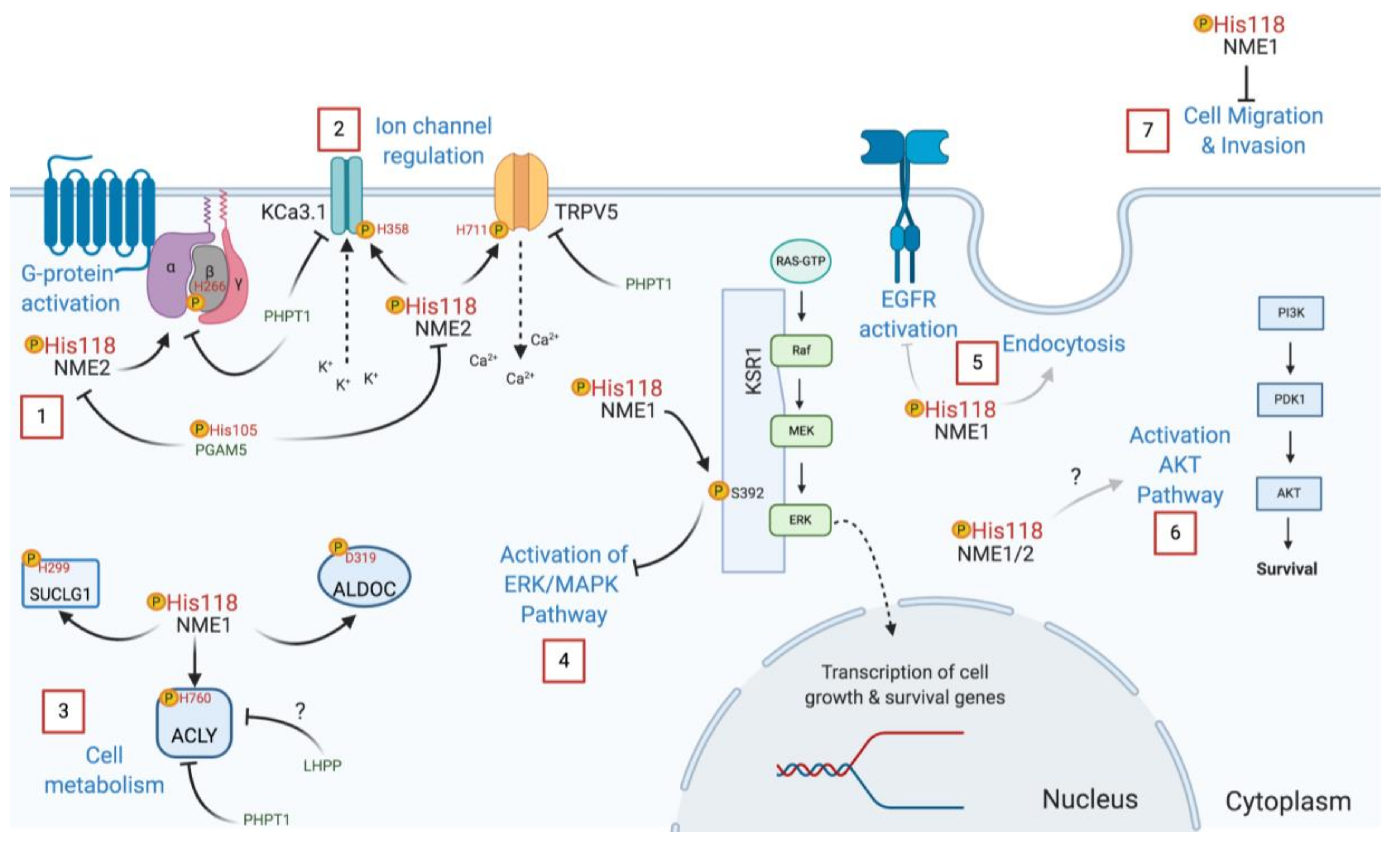
3.2. NME Protein Histidine Kinase Functions as Revealed by Mutagenesis
3.3. Histidine Phosphatases
4. Role of NME Protein Histidine Phosphorylation Function in Cell Signaling
4.1. NME Histidine Phosphorylation Signaling in Tumor Metastasis Suppression
4.2. NME Histidine Phosphorylation Function in Development and other Biological Processes
4.3. Detecting Histidine Phosphorylated NME in the Cell
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| NDPK | Nucleoside diphosphate kinase |
| NTPs | Nucleoside triphosphates |
| NDPs | Nucleoside diphosphates |
| PDB | Protein Data Bank |
| TCS | Two-component system |
| KSR | Kinase suppressor of Ras |
| MSS | Metastasis Suppressor Signature |
| MLC | Myosin light chain |
| pNME | Phosphorylated NME |
| GDP | Guanosine diphosphate |
| CAMKII | Calcium/Calmodulin-dependent kinase II |
| TRPV5 | Transient receptor potential cation channel subfamily V member 5 |
| GNB1 | Guanine nucleotide-binding protein G(I)/G(S)/G(T) subunit beta-1 |
| KCa3.1 | Intermediate conductance calcium-activated potassium channel protein 4 |
| ACLY | ATP-citrate synthase |
| SUCLG1 | Succinyl-CoA synthetase |
| KSR1 | Kinase suppressor of Ras 1 |
| ANXA1 | Annexin A1 |
| ALDOC | Aldolase C |
| PGAM5 | PGAM Family Member 5 |
| PHPT1 | Phosphohistidine Phosphatase 1 |
| LHPP | Phospholysine phosphohistidine inorganic pyrophosphate phosphatase |
| GWAS | Genome-wide association studies |
References
- Berg, P.; Joklik, W.K. Transphosphorylation between nucleoside polyphosphates. Nature 1953, 172, 1008–1009. [Google Scholar] [CrossRef]
- Krebs, H.A.; Hems, R. Some reactions of adenosine and inosine phosphates in animal tissues. Biochim. Biophys. Acta 1953, 12, 172–180. [Google Scholar] [CrossRef]
- Boyer, P.D.; Deluca, M.; Ebner, K.E.; Hultquist, D.E.; Peter, J.B. Identification of phosphohistidine in digests from a probable intermediate of oxidative phosphorylation. J. Biol. Chem. 1962, 237, PC3306–PC3308. [Google Scholar] [PubMed]
- Wålinder, O. Evidence of the presence of 1-phosphohistidine as the main phosphohistidine as the main phosphorylated component at the active site of bovine liver nucleoside diphosphate kinase. Acta Chem. Scand. 1969, 23, 339–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wålinder, O. Identification of a phosphate-incorporating protein from bovine liver as nucleoside diphosphate kinase and isolation of 1-32P-phosphohistidine, 3-32P-phosphohistidine, and N-epsilon-32P-phospholysine from erythrocytic nucleoside diphosphate kinase, incubated with adenosine triphosphate-32P. J. Biol. Chem. 1968, 243, 3947–3952. [Google Scholar] [PubMed]
- Norman, A.W.; Wedding, R.T.; Black, M.K. Detection of phosphohistidine in nucleoside diphosphokinase isolated from Jerusalem artichoke mitochondria. Biochem. Biophys. Res. Commun. 1965, 20, 703–709. [Google Scholar] [CrossRef]
- Edlund, B.; Rask, L.; Olsson, P.; Wålinder, O.; Zetterqvist, O.; Engström, L. Preparation of crystalline nucleoside diphosphate kinase from baker’s yeast and identification of 1-[32P]phosphohistidine as the main phosphorylated product of an alkaline hydrolysate of enzyme incubated with adenosine [32P]triphosphate. Eur. J. Biochem. 1969, 9, 451–455. [Google Scholar] [CrossRef]
- Attwood, P.V. P-N bond protein phosphatases. Biochim. Biophys. Acta 2013, 1834, 470–478. [Google Scholar] [CrossRef]
- Adam, K.; Hunter, T. Histidine kinases and the missing phosphoproteome from prokaryotes to eukaryotes. Lab. Investig. 2018, 98, 233–247. [Google Scholar] [CrossRef] [Green Version]
- Boissan, M.; Dabernat, S.; Peuchant, E.; Schlattner, U.; Lascu, I.; Lacombe, M.L. The mammalian Nm23/NDPK family: from metastasis control to cilia movement. Mol. Cell. Biochem. 2009, 329, 51–62. [Google Scholar] [CrossRef]
- Yoon, J.-H.; Singh, P.; Lee, D.-H.; Qiu, J.; Cai, S.; O’Connor, T.R.; Chen, Y.; Shen, B.; Pfeifer, G.P. Characterization of the 3’ --> 5’ exonuclease activity found in human nucleoside diphosphate kinase 1 (NDK1) and several of its homologues. Biochemistry 2005, 44, 15774–15786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Choi, Y.-K.; Qi, R.Z. NME7 is a functional component of the γ-tubulin ring complex. Mol. Biol. Cell 2014, 25, 2017–2025. [Google Scholar] [CrossRef] [PubMed]
- Tsuiki, H.; Nitta, M.; Furuya, A.; Hanai, N.; Fujiwara, T.; Inagaki, M.; Kochi, M.; Ushio, Y.; Saya, H.; Nakamura, H. A novel human nucleoside diphosphate (NDP) kinase, Nm23-H6, localizes in mitochondria and affects cytokinesis. J. Cell. Biochem. 1999, 76, 254–269. [Google Scholar] [CrossRef]
- Perina, D.; Bosnar, M.H.; Bago, R.; Mikoč, A.; Harcet, M.; Deželjin, M.; Cetković, H. Sponge non-metastatic group I Nme gene/protein - structure and function is conserved from sponges to humans. BMC Evol. Biol. 2011, 11, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morera, S.; Lascu, I.; Dumas, C.; LeBras, G.; Briozzo, P.; Veron, M.; Janin, J. Adenosine 5’-diphosphate binding and the active site of nucleoside diphosphate kinase. Biochemistry 1994, 33, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Giraud, M.-F.; Georgescauld, F.; Lascu, I.; Dautant, A. Crystal structures of S120G mutant and wild type of human nucleoside diphosphate kinase A in complex with ADP. J. Bioenerg. Biomembr. 2006, 38, 261–264. [Google Scholar] [CrossRef] [PubMed]
- Cuello, F.; Schulze, R.A.; Heemeyer, F.; Meyer, H.E.; Lutz, S.; Jakobs, K.H.; Niroomand, F.; Wieland, T. Activation of heterotrimeric G proteins by a high energy phosphate transfer via nucleoside diphosphate kinase (NDPK) B and Gbeta subunits. Complex formation of NDPK B with Gbeta gamma dimers and phosphorylation of His-266 in Gbeta. J. Biol. Chem. 2003, 278, 7220–7226. [Google Scholar] [CrossRef] [Green Version]
- Wagner, P.D.; Vu, N.D. Phosphorylation of geranyl and farnesyl pyrophosphates by Nm23 proteins/nucleoside diphosphate kinases. J. Biol. Chem. 2000, 275, 35570–35576. [Google Scholar] [CrossRef] [Green Version]
- Ma, D.; McCorkle, J.R.; Kaetzel, D.M. The metastasis suppressor NM23-H1 possesses 3’-5’ exonuclease activity. J. Biol. Chem. 2004, 279, 18073–18084. [Google Scholar] [CrossRef] [Green Version]
- Engel, M.; Véron, M.; Theisinger, B.; Lacombe, M.L.; Seib, T.; Dooley, S.; Welter, C. A novel serine/threonine-specific protein phosphotransferase activity of Nm23/nucleoside-diphosphate kinase. Eur. J. Biochem. 1995, 234, 200–207. [Google Scholar] [CrossRef]
- Lecroisey, A.; Lascu, I.; Bominaar, A.; Véron, M.; Delepierre, M. Phosphorylation mechanism of nucleoside diphosphate kinase: 31P-nuclear magnetic resonance studies. Biochemistry 1995, 34, 12445–12450. [Google Scholar] [CrossRef] [PubMed]
- Lapek, J.D.; Tombline, G.; Friedman, A.E. Mass spectrometry detection of histidine phosphorylation on NM23-H1. J. Proteome Res. 2011, 10, 751–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boissan, M.; Schlattner, U.; Lacombe, M.L. The NDPK/NME superfamily: state of the art. Lab. Investig. 2018, 98, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Steeg, P.S.; Bevilacqua, G.; Kopper, L.; Thorgeirsson, U.P.; Talmadge, J.E.; Liotta, L.A.; Sobel, M.E. Evidence for a novel gene associated with low tumor metastatic potential. J. Natl. Cancer Inst. 1988, 80, 200–204. [Google Scholar] [CrossRef] [PubMed]
- Boissan, M.; Lacombe, M.L. Nm23/NDP kinases in hepatocellular carcinoma. J. Bioenerg. Biomembr. 2006, 38, 169–175. [Google Scholar] [CrossRef]
- Krishnan, K.S.; Rikhy, R.; Rao, S.; Shivalkar, M.; Mosko, M.; Narayanan, R.; Etter, P.; Estes, P.S.; Ramaswami, M. Nucleoside diphosphate kinase, a source of GTP, is required for dynamin-dependent synaptic vesicle recycling. Neuron 2001, 30, 197–210. [Google Scholar] [CrossRef] [Green Version]
- Di, L.; Srivastava, S.; Zhdanova, O.; Sun, Y.; Li, Z.; Skolnik, E.Y. Nucleoside diphosphate kinase B knock-out mice have impaired activation of the K+ channel KCa3.1, resulting in defective T cell activation. J. Biol. Chem. 2010, 285, 38765–38771. [Google Scholar] [CrossRef] [Green Version]
- Boissan, M.; Lacombe, M.-L. Learning about the functions of NME/NM23: lessons from knockout mice to silencing strategies. Naunyn Schmiedeberg’s Arch. Pharm. 2011, 384, 421–431. [Google Scholar] [CrossRef]
- Sickmann, A.; Meyer, H.E. Phosphoamino acid analysis. Proteomics 2001, 1, 200–206. [Google Scholar] [CrossRef]
- Fuhs, S.R.; Hunter, T. pHisphorylation: the emergence of histidine phosphorylation as a reversible regulatory modification. Curr. Opin. Cell Biol. 2017, 45, 8–16. [Google Scholar] [CrossRef]
- Besant, P.G.; Attwood, P.V. Mammalian histidine kinases. Biochim. Biophys. Acta 2005, 1754, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Hultquist, D.E. The preparation and characterization of phosphorylated derivatives of histidine. Biochim. Biophys. Acta 1968, 153, 329–340. [Google Scholar] [CrossRef] [Green Version]
- Attwood, P.V.; Piggott, M.J.; Zu, X.L.; Besant, P.G. Focus on phosphohistidine. Amino Acids 2007, 32, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Makwana, M.V.; Muimo, R.; Jackson, R.F. Advances in development of new tools for the study of phosphohistidine. Lab. Investig. A J. Tech. Methods Pathol. 2017, 98, 291–303. [Google Scholar] [CrossRef] [Green Version]
- Kee, J.M.; Villani, B.; Carpenter, L.R.; Muir, T.W. Development of stable phosphohistidine analogues. J. Am. Chem. Soc. 2010, 132, 14327–14329. [Google Scholar] [CrossRef] [Green Version]
- Kee, J.M.; Oslund, R.C.; Perlman, D.H.; Muir, T.W. A pan-specific antibody for direct detection of protein histidine phosphorylation. Nat. Chem. Biol. 2013, 9, 416–421. [Google Scholar] [CrossRef] [Green Version]
- Fuhs, S.R.; Meisenhelder, J.; Aslanian, A.; Ma, L.; Zagorska, A.; Stankova, M.; Binnie, A.; Al-Obeidi, F.; Mauger, J.; Lemke, G.; et al. Monoclonal 1- and 3-phosphohistidine antibodies: new tools to study histidine phosphorylation. Cell 2015, 162, 198–210. [Google Scholar] [CrossRef] [Green Version]
- Tomlinson, L.J.; Clubbs Coldron, A.K.M.; Eyers, P.A.; Eyers, C.E. Determination of phosphohistidine stoichiometry in histidine kinases by intact mass spectrometry. In Histidine Phosphorylation: Methods and Protocols; Eyers, C.E., Ed.; Springer: New York, NY, USA, 2020; pp. 83–91. [Google Scholar]
- Luhtala, N.; Hunter, T. Immunohistochemistry (IHC): Chromogenic detection of 3-phosphohistidine proteins in formaldehyde-fixed, frozen mouse liver tissue sections. In Histidine Phosphorylation: Methods and Protocols; Eyers, C.E., Ed.; Springer: New York, NY, USA, 2020; pp. 193–208. [Google Scholar]
- Kalagiri, R.; Adam, K.; Hunter, T. Empirical evidence of cellular histidine phosphorylation by immunoblotting using pHis mAbs. In Histidine Phosphorylation: Methods and Protocols; Eyers, C.E., Ed.; Springer: New York, NY, USA, 2020; pp. 181–191. [Google Scholar]
- Clubbs Coldron, A.K.M.; Byrne, D.P.; Eyers, P.A. Analysis of 1- and 3-phosphohistidine (pHis) protein modification using model enzymes expressed in bacteria. In Histidine Phosphorylation: Methods and Protocols; Eyers, C.E., Ed.; Springer: New York, NY, USA, 2020; pp. 63–81. [Google Scholar]
- Attwood, P.V. A quantitative method for the measurement of protein histidine phosphorylation. In Histidine Phosphorylation: Methods and Protocols; Eyers, C.E., Ed.; Springer: New York, NY, USA, 2020; pp. 51–61. [Google Scholar]
- Adam, K.; Hunter, T. Subcellular localization of histidine phosphorylated proteins through indirect immunofluorescence. In Histidine Phosphorylation: Methods and Protocols; Eyers, C.E., Ed.; Springer: New York, NY, USA, 2020; Volume 2077, pp. 209–224. [Google Scholar]
- Fischer, J.T.; Heckler, I.; Boon, E.M. SDS-PAGE and dot blot autoradiography: tools for quantifying histidine kinase autophosphorylation. In Histidine Phosphorylation: Methods and Protocols; Eyers, C.E., Ed.; Springer: New York, NY, USA, 2020; pp. 37–49. [Google Scholar]
- Potel, C.M.; Lin, M.-H.; Heck, A.J.R.; Lemeer, S. Widespread bacterial protein histidine phosphorylation revealed by mass spectrometry-based proteomics. Nat. Methods 2018, 15, 187–190. [Google Scholar] [CrossRef]
- Oslund, R.C.; Kee, J.-M.; Couvillon, A.D.; Bhatia, V.N.; Perlman, D.H.; Muir, T.W. A phosphohistidine proteomics strategy based on elucidation of a unique gas-phase phosphopeptide fragmentation mechanism. J. Am. Chem. Soc. 2014, 136, 12899–12911. [Google Scholar] [CrossRef] [Green Version]
- Kleinnijenhuis, A.J.; Kjeldsen, F.; Kallipolitis, B.; Haselmann, K.F.; Jensen, O.N. Analysis of histidine phosphorylation using tandem MS and ion-electron reactions. Anal. Chem. 2007, 79, 7450–7456. [Google Scholar] [CrossRef]
- Himmel, S.; Wolff, S.; Becker, S.; Lee, D.; Griesinger, C. Detection and identification of protein-phosphorylation sites in histidines through HNP correlation patterns. Angew. Chem. (Int. Ed. Engl.) 2010, 49, 8971–8974. [Google Scholar] [CrossRef] [PubMed]
- Hardman, G.; Eyers, C.E. High-throughput characterization of histidine phosphorylation sites using UPAX and tandem mass spectrometry. In Histidine Phosphorylation: Methods and Protocols; Eyers, C.E., Ed.; Springer: New York, NY, USA, 2020; pp. 225–235. [Google Scholar]
- Attwood, P.V.; Muimo, R. The actions of NME1/NDPK-A and NME2/NDPK-B as protein kinases. Lab. Investig. 2018, 98, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Park, H.; Egger, L.A.; Inouye, M. Nucleoside-diphosphate kinase-mediated signal transduction via histidyl-aspartyl phosphorelay systems in Escherichia coli. J. Biol. Chem. 1996, 271, 32886–32893. [Google Scholar] [CrossRef] [Green Version]
- Wagner, P.D.; Steeg, P.S.; Vu, N.D. Two-component kinase-like activity of nm23 correlates with its motility-suppressing activity. Proc. Natl. Acad. Sci. USA 1997, 94, 9000–9005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freije, J.M.; Blay, P.; MacDonald, N.J.; Manrow, R.E.; Steeg, P.S. Site-directed mutation of Nm23-H1. Mutations lacking motility suppressive capacity upon transfection are deficient in histidine-dependent protein phosphotransferase pathways in vitro. J. Biol. Chem. 1997, 272, 5525–5532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartsough, M.T.; Morrison, D.K.; Salerno, M.; Palmieri, D.; Ouatas, T.; Mair, M.; Patrick, J.; Steeg, P.S. Nm23-H1 metastasis suppressor phosphorylation of kinase suppressor of Ras via a histidine protein kinase pathway. J. Biol. Chem. 2002, 277, 32389–32399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wieland, T.; Hippe, H.-J.; Ludwig, K.; Zhou, X.-B.; Korth, M.; Klumpp, S. Reversible histidine phosphorylation in mammalian cells: a teeter-totter formed by nucleoside diphosphate kinase and protein histidine phosphatase 1. Methods Enzym. 2010, 471, 379–402. [Google Scholar]
- Wagner, P.D.; Vu, N.D. Phosphorylation of ATP-citrate lyase by nucleoside diphosphate kinase. J. Biol. Chem. 1995, 270, 21758–21764. [Google Scholar] [CrossRef] [Green Version]
- Klumpp, S.; Bechmann, G.; Maurer, A.; Selke, D.; Krieglstein, J. ATP-citrate lyase as a substrate of protein histidine phosphatase in vertebrates. Biochem. Biophys. Res. Commun. 2003, 306, 110–115. [Google Scholar] [CrossRef]
- Wagner, P.D.; Vu, N.D. Histidine to aspartate phosphotransferase activity of nm23 proteins: phosphorylation of aldolase C on Asp-319. Biochem. J. 2000, 346 Pt 3, 623–630. [Google Scholar] [CrossRef]
- Hippe, H.J.; Abu-Taha, I.; Wolf, N.M.; Katus, H.A.; Wieland, T. Through scaffolding and catalytic actions nucleoside diphosphate kinase B differentially regulates basal and beta-adrenoceptor-stimulated cAMP synthesis. Cell. Signal. 2011, 23, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Panda, S.; Li, Z.; Fuhs, S.R.; Hunter, T.; Thiele, D.J.; Hubbard, S.R.; Skolnik, E.Y. Histidine phosphorylation relieves copper inhibition in the mammalian potassium channel KCa3.1. Elife 2016, 5, e16093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, S.; Li, Z.; Ko, K.; Choudhury, P.; Albaqumi, M.; Johnson, A.K.; Yan, Y.; Backer, J.M.; Unutmaz, D.; Coetzee, W.A.; et al. Histidine phosphorylation of the potassium channel KCa3.1 by nucleoside diphosphate kinase B is required for activation of KCa3.1 and CD4 T cells. Mol. Cell 2006, 24, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Kim, H.S.; Lee, J.S.; Park, J.; Shin, S.C.; Song, S.; Lee, E.; Choi, J.E.; Suh, J.W.; Lee, H.; et al. Small molecule activator of Nm23/NDPK as an inhibitor of metastasis. Sci. Rep. 2018, 8, 10909. [Google Scholar] [CrossRef] [Green Version]
- Bamdad, C.; Smagghe, B. NME Inhibitors and Methods of Using NME Inhibitors. U.S. Patent US20160326263A1, 10 November 2016. [Google Scholar]
- Webb, P.A.; Perisic, O.; Mendola, C.E.; Backer, J.M.; Williams, R.L. The crystal structure of a human nucleoside diphosphate kinase, NM23-H2. J. Mol. Biol. 1995, 251, 574–587. [Google Scholar] [CrossRef]
- Min, K.; Song, H.K.; Chang, C.; Kim, S.Y.; Lee, K.-J.; Suh, S.W. Crystal structure of human nucleoside diphosphate kinase A, a metastasis suppressor. Proteins: Struct. Funct. Bioinform. 2002, 46, 340–342. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Gallois-Montbrun, S.; Schneider, B.; Véron, M.; Moréra, S.; Deville-Bonne, D.; Janin, J. Nucleotide binding to nucleoside diphosphate kinases: X-ray structure of human NDPK-A in complex with ADP and comparison to protein kinases. J. Mol. Biol. 2003, 332, 915–926. [Google Scholar] [CrossRef]
- Kim, M.S.; Jeong, J.; Jeong, J.; Shin, D.H.; Lee, K.J. Structure of Nm23-H1 under oxidative conditions. Acta Cryst. D Biol. Cryst. 2013, 69, 669–680. [Google Scholar] [CrossRef]
- Mortenson, D.E.; Brighty, G.J.; Plate, L.; Bare, G.; Chen, W.; Li, S.; Wang, H.; Cravatt, B.F.; Forli, S.; Powers, E.T.; et al. “Inverse drug discovery” strategy to identify proteins that are targeted by latent electrophiles as exemplified by aryl fluorosulfates. J. Am. Chem. Soc. 2018, 140, 200–210. [Google Scholar] [CrossRef]
- Morera, S.; Lacombe, M.L.; Xu, Y.; LeBras, G.; Janin, J. X-ray structure of human nucleoside diphosphate kinase B complexed with GDP at 2 A resolution. Structure 1995, 3, 1307–1314. [Google Scholar] [CrossRef]
- Dexheimer, T.S.; Carey, S.S.; Zuohe, S.; Gokhale, V.M.; Hu, X.; Murata, L.B.; Maes, E.M.; Weichsel, A.; Sun, D.; Meuillet, E.J.; et al. NM23-H2 may play an indirect role in transcriptional activation of c-myc gene expression but does not cleave the nuclease hypersensitive element III(1). Mol. Cancer 2009, 8, 1363–1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milon, L.; Meyer, P.; Chiadmi, M.; Munier, A.; Johansson, M.; Karlsson, A.; Lascu, I.; Capeau, J.; Janin, J.; Lacombe, M.L. The human nm23-H4 gene product is a mitochondrial nucleoside diphosphate kinase. J. Biol. Chem. 2000, 275, 14264–14272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desvignes, T.; Pontarotti, P.; Fauvel, C.; Bobe, J. Nme protein family evolutionary history, a vertebrate perspective. Bmc Evol. Biol. 2009, 9, 256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puts, G.S.; Leonard, M.K.; Pamidimukkala, N.V.; Snyder, D.E.; Kaetzel, D.M. Nuclear functions of NME proteins. Lab. Investig. A J. Tech. Methods Pathol. 2018, 98, 211–218. [Google Scholar] [CrossRef]
- Postel, E.H.; Abramczyk, B.A.; Gursky, S.K.; Xu, Y. Structure-based mutational and functional analysis identify human NM23-H2 as a multifunctional enzyme. Biochemistry 2002, 41, 6330–6337. [Google Scholar] [CrossRef]
- Janin, J.; Dumas, C.; Moréra, S.; Xu, Y.; Meyer, P.; Chiadmi, M.; Cherfils, J. Three-dimensional structure of nucleoside diphosphate kinase. J. Bioenerg. Biomembr. 2000, 32, 215–225. [Google Scholar] [CrossRef]
- Wang, H.; Bao, R.; Jiang, C.; Yang, Z.; Zhou, C.-Z.; Chen, Y. Structure of Ynk1 from the yeast Saccharomyces cerevisiae. Acta Crystallogr. Sect. F 2008, 64, 572–576. [Google Scholar] [CrossRef] [Green Version]
- Hamby, C.V.; Abbi, R.; Prasad, N.; Stauffer, C.; Thomson, J.; Mendola, C.E.; Sidorov, V.; Backer, J.M. Expression of a catalytically inactive H118Y mutant of nm23-H2 suppresses the metastatic potential of line IV Cl 1 human melanoma cells. Int. J. Cancer. J. Int. Du Cancer 2000, 88, 547–553. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]
- Song, E.J.; Kim, Y.S.; Chung, J.Y.; Kim, E.; Chae, S.K.; Lee, K.J. Oxidative modification of nucleoside diphosphate kinase and its identification by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Biochemistry 2000, 39, 10090–10097. [Google Scholar] [CrossRef]
- Peuchant, E.; Bats, M.-L.; Moranvillier, I.; Lepoivre, M.; Guitton, J.; Wendum, D.; Lacombe, M.-L.; Moreau-Gaudry, F.; Boissan, M.; Dabernat, S. Metastasis suppressor NM23 limits oxidative stress in mammals by preventing activation of stress-activated protein kinases/JNKs through its nucleoside diphosphate kinase activity. Faseb J. 2017, 31, 1531–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abu-Taha, I.H.; Vettel, C.; Wieland, T. Targeting altered Nme heterooligomerization in disease? Oncotarget 2018, 9, 1492–1493. [Google Scholar] [CrossRef] [PubMed]
- Gilles, A.M.; Presecan, E.; Vonica, A.; Lascu, I. Nucleoside diphosphate kinase from human erythrocytes. Structural characterization of the two polypeptide chains responsible for heterogeneity of the hexameric enzyme. J. Biol. Chem. 1991, 266, 8784–8789. [Google Scholar] [PubMed]
- Radić, M.; Šoštar, M.; Weber, I.; Ćetković, H.; Slade, N.; Herak Bosnar, M. The subcellular localization and oligomerization preferences of NME1/NME2 upon radiation-induced DNA damage. Int. J. Mol. Sci. 2020, 21, 2363. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.W.; Wang, H.L.; Huang, C.W.; Huang, C.Y.; Lim, W.K.; Tu, I.C.; Koorapati, A.; Hsieh, S.T.; Kan, H.W.; Tzeng, S.R.; et al. Two separate functions of NME3 critical for cell survival underlie a neurodegenerative disorder. Proc. Natl. Acad. Sci. USA 2019, 116, 566–574. [Google Scholar] [CrossRef] [Green Version]
- Willett, J.W.; Crosson, S. Atypical modes of bacterial histidine kinase signaling. Mol. Microbiol. 2017, 103, 197–202. [Google Scholar] [CrossRef] [Green Version]
- Marino, N.; Marshall, J.C.; Steeg, P.S. Protein-protein interactions: a mechanism regulating the anti-metastatic properties of Nm23-H1. Naunyn Schmiedebergs Arch. Pharm. 2011, 384, 351–362. [Google Scholar] [CrossRef]
- Cai, X.; Srivastava, S.; Surindran, S.; Li, Z.; Skolnik, E.Y. Regulation of the epithelial Ca(2)(+) channel TRPV5 by reversible histidine phosphorylation mediated by NDPK-B and PHPT1. Mol. Biol. Cell 2014, 25, 1244–1250. [Google Scholar] [CrossRef]
- Bridger, W. Contribution of subunit interactions to the effectiveness of catalysis by succinyl coenzyme A synthetase. Curr. Top. Cell. Regul. 1984, 24, 345–355. [Google Scholar]
- Muimo, R.; Hornickova, Z.; Riemen, C.E.; Gerke, V.; Matthews, H.; Mehta, A. Histidine phosphorylation of annexin I in airway epithelia. J. Biol. Chem. 2000, 275, 36632–36636. [Google Scholar] [CrossRef] [Green Version]
- Wieland, T.; Attwood, P.V. Alterations in reversible protein histidine phosphorylation as intracellular signals in cardiovascular disease. Front. Pharm. 2015, 6, 173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pamidimukkala, N.V.; Leonard, M.K.; Snyder, D.; McCorkle, J.R.; Kaetzel, D.M. Metastasis suppressor NME1 directly activates transcription of the ALDOC gene in melanoma cells. Anticancer Res. 2018, 38, 6059–6068. [Google Scholar] [CrossRef] [PubMed]
- Leonard, M.K.; McCorkle, J.R.; Snyder, D.E.; Novak, M.; Zhang, Q.; Shetty, A.C.; Mahurkar, A.A.; Kaetzel, D.M. Identification of a gene expression signature associated with the metastasis suppressor function of NME1: prognostic value in human melanoma. Lab. Investig. 2018, 98, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Potel, C.M.; Fasci, D.; Heck, A.J.R. Mix and match of the tumor metastasis suppressor Nm23 protein isoforms in vitro and in vivo. Febs J. 2018, 285, 2856–2868. [Google Scholar] [CrossRef] [Green Version]
- Hardman, G.; Perkins, S.; Brownridge, P.J.; Clarke, C.J.; Byrne, D.P.; Campbell, A.E.; Kalyuzhnyy, A.; Myall, A.; Eyers, P.A.; Jones, A.R.; et al. Strong anion exchange-mediated phosphoproteomics reveals extensive human non-canonical phosphorylation. Embo J. 2019, 38, e100847. [Google Scholar] [CrossRef]
- Adam, K.; Fuhs, S.; Meisenhelder, J.; Aslanian, A.; Diedrich, J.; Moresco, J.; La Clair, J.; Yates, J., III; Hunter, T. A non-acidic method using hydroxyapatite and phosphohistidine monoclonal antibodies allows enrichment of phosphopeptides containing non-conventional phosphorylations for mass spectrometry analysis. BioRxiv 2019. BioRxiv:691352. [Google Scholar]
- Otsuki, Y.; Tanaka, M.; Yoshii, S.; Kawazoe, N.; Nakaya, K.; Sugimura, H. Tumor metastasis suppressor nm23H1 regulates Rac1 GTPase by interaction with Tiam1. Proc. Natl. Acad. Sci. USA 2001, 98, 4385–4390. [Google Scholar] [CrossRef] [Green Version]
- Murakami, M.; Meneses, P.I.; Knight, J.S.; Lan, K.; Kaul, R.; Verma, S.C.; Robertson, E.S. Nm23-H1 modulates the activity of the guanine exchange factor Dbl-1. Int. J. Cancer 2008, 123, 500–510. [Google Scholar] [CrossRef]
- Marino, N.; Marshall, J.C.; Collins, J.W.; Zhou, M.; Qian, Y.; Veenstra, T.; Steeg, P.S. Nm23-h1 binds to gelsolin and inactivates its actin-severing capacity to promote tumor cell motility and metastasis. Cancer Res. 2013, 73, 5949–5962. [Google Scholar] [CrossRef] [Green Version]
- D’Angelo, A.; Garzia, L.; Andre, A.; Carotenuto, P.; Aglio, V.; Guardiola, O.; Arrigoni, G.; Cossu, A.; Palmieri, G.; Aravind, L.; et al. Prune cAMP phosphodiesterase binds nm23-H1 and promotes cancer metastasis. Cancer Cell 2004, 5, 137–149. [Google Scholar] [CrossRef] [Green Version]
- Garzia, L.; D’Angelo, A.; Amoresano, A.; Knauer, S.K.; Cirulli, C.; Campanella, C.; Stauber, R.H.; Steegborn, C.; Iolascon, A.; Zollo, M. Phosphorylation of nm23-H1 by CKI induces its complex formation with h-prune and promotes cell motility. Oncogene 2008, 27, 1853–1864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boissan, M.; Montagnac, G.; Shen, Q.; Griparic, L.; Guitton, J.; Romao, M.; Sauvonnet, N.; Lagache, T.; Lascu, I.; Raposo, G.; et al. Membrane trafficking. Nucleoside diphosphate kinases fuel dynamin superfamily proteins with GTP for membrane remodeling. Science 2014, 344, 1510–1515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, I.; Gril, B.; Steeg, P.S. Metastasis suppressors NME1 and NME2 promote dynamin 2 oligomerization and regulate tumor cell endocytosis, motility, and metastasis. Cancer Res. 2019, 79, 4689–4702. [Google Scholar] [CrossRef]
- Matyasi, B.; Farkas, Z.; Kopper, L.; Sebestyen, A.; Boissan, M.; Mehta, A.; Takacs-Vellai, K. The function of NM23-H1/NME1 and its homologs in major processes linked to metastasis. Pathol. Oncol. Res. 2020, 26, 49–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romani, P.; Ignesti, M.; Gargiulo, G.; Hsu, T.; Cavaliere, V. Extracellular NME proteins: A player or a bystander? Lab. Investig. A J. Tech. Methods Pathol. 2017, 98, 248–257. [Google Scholar] [CrossRef] [Green Version]
- Lacombe, M.L.; Tokarska-Schlattner, M.; Boissan, M.; Schlattner, U. The mitochondrial nucleoside diphosphate kinase (NDPK-D/NME4), a moonlighting protein for cell homeostasis. Lab. Investig. 2018, 98, 582–588. [Google Scholar] [CrossRef] [Green Version]
- Bunce, C.M.; Khanim, F.L. The ‘known-knowns’, and ‘known-unknowns’ of extracellular Nm23-H1/NDPK proteins. Lab. Investig. 2018, 98, 602–608. [Google Scholar] [CrossRef]
- Vlatković, N.; Chang, S.-H.; Boyd, M.T. Janus-faces of NME-oncoprotein interactions. Naunyn Schmiedeberg’s Arch. Pharm. 2015, 388, 175–187. [Google Scholar] [CrossRef]
- Tokarska-Schlattner, M.; Boissan, M.; Munier, A.; Borot, C.; Mailleau, C.; Speer, O.; Schlattner, U.; Lacombe, M.L. The nucleoside diphosphate kinase D (NM23-H4) binds the inner mitochondrial membrane with high affinity to cardiolipin and couples nucleotide transfer with respiration. J. Biol. Chem. 2008, 283, 26198–26207. [Google Scholar] [CrossRef] [Green Version]
- Kowluru, A.; Tannous, M.; Chen, H.Q. Localization and characterization of the mitochondrial isoform of the nucleoside diphosphate kinase in the pancreatic beta cell: evidence for its complexation with mitochondrial succinyl-CoA synthetase. Arch. Biochem. Biophys. 2002, 398, 160–169. [Google Scholar] [CrossRef]
- Besse, A.; Wu, P.; Bruni, F.; Donti, T.; Graham, B.H.; Craigen, W.J.; McFarland, R.; Moretti, P.; Lalani, S.; Scott, K.L.; et al. The GABA transaminase, ABAT, is essential for mitochondrial nucleoside metabolism. Cell Metab 2015, 21, 417–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlattner, U.; Tokarska-Schlattner, M.; Ramirez, S.; Tyurina, Y.Y.; Amoscato, A.A.; Mohammadyani, D.; Huang, Z.; Jiang, J.; Yanamala, N.; Seffouh, A.; et al. Dual function of mitochondrial Nm23-H4 protein in phosphotransfer and intermembrane lipid transfer: a cardiolipin-dependent switch. J. Biol. Chem. 2013, 288, 111–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mochizuki, T.; Bilitou, A.; Waters, C.T.; Hussain, K.; Zollo, M.; Ohnuma, S. Xenopus NM23-X4 regulates retinal gliogenesis through interaction with p27Xic1. Neural Dev. 2009, 4, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Liu, L.Z.; Deng, X.F.; Timmons, L.; Hersperger, E.; Steeg, P.S.; Veron, M.; Shearn, A. The enzymatic activity of Drosophila AWD/NDP kinase is necessary but not sufficient for its biological function. Dev. Biol. 1996, 177, 544–557. [Google Scholar] [CrossRef] [Green Version]
- Khan, I.; Steeg, P.S. The relationship of NM23 (NME) metastasis suppressor histidine phosphorylation to its nucleoside diphosphate kinase, histidine protein kinase and motility suppression activities. Oncotarget 2018, 9, 10185–10202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timmons, L.; Xu, J.; Hersperger, G.; Deng, X.F.; Shearn, A. Point mutations in awdKpn which revert the prune/Killer of prune lethal interaction affect conserved residues that are involved in nucleoside diphosphate kinase substrate binding and catalysis. J. Biol. Chem. 1995, 270, 23021–23030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biggs, J.; Tripoulas, N.; Hersperger, E.; Dearolf, C.; Shearn, A. Analysis of the lethal interaction between the prune and Killer of prune mutations of Drosophila. Genes Dev. 1988, 2, 1333–1343. [Google Scholar] [CrossRef] [Green Version]
- Chiadmi, M.; Morera, S.; Lascu, I.; Dumas, C.; Le Bras, G.; Veron, M.; Janin, J. Crystal structure of the Awd nucleotide diphosphate kinase from Drosophila. Structure 1993, 1, 283–293. [Google Scholar] [CrossRef]
- Royer, L.; Shangraw, K.; Herzog, J.; Pouvreau, S.; Marr, M.; Paradis, S. The metastasis suppressor protein Nme1 is a concentration-dependent modulator of Ca2+/Calmodulin-dependent protein kinase II. Biochemistry 2019, 2710–2714. [Google Scholar] [CrossRef]
- Tan, C.Y.; Chang, C.L. NDPKA is not just a metastasis suppressor - be aware of its metastasis-promoting role in neuroblastoma. Lab. Investig. 2018, 98, 219–227. [Google Scholar] [CrossRef]
- Panda, S.; Srivastava, S.; Li, Z.; Vaeth, M.; Fuhs, S.R.; Hunter, T.; Skolnik, E.Y. Identification of PGAM5 as a mammalian protein histidine phosphatase that plays a central role to negatively regulate CD4(+) T cells. Mol. Cell 2016, 63, 457–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, S.; Zhdanova, O.; Di, L.; Li, Z.; Albaqumi, M.; Wulff, H.; Skolnik, E.Y. Protein histidine phosphatase 1 negatively regulates CD4 T cells by inhibiting the K+ channel KCa3.1. Proc. Natl. Acad. Sci. USA 2008, 105, 14442–14446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klumpp, S.; Krieglstein, J. Reversible phosphorylation of histidine residues in proteins from vertebrates. Sci Signal 2009, 2, pe13. [Google Scholar] [CrossRef] [PubMed]
- Maurer, A.; Wieland, T.; Meissl, F.; Niroomand, F.; Mehringer, R.; Krieglstein, J.; Klumpp, S. The beta-subunit of G proteins is a substrate of protein histidine phosphatase. Biochem. Biophys. Res. Commun. 2005, 334, 1115–1120. [Google Scholar] [CrossRef]
- Srivastava, S.; Li, Z.; Soomro, I.; Sun, Y.; Wang, J.; Bao, L.; Coetzee, W.A.; Stanley, C.A.; Li, C.; Skolnik, E.Y. Regulation of KATP channel trafficking in pancreatic beta-cells by protein histidine phosphorylation. Diabetes 2018, 67, 849–860. [Google Scholar] [CrossRef] [Green Version]
- Seal, U.S.; Binkley, F. An inorganic pyrophosphatase of swine brain. J. Biol. Chem. 1957, 228, 193–199. [Google Scholar]
- Hindupur, S.K.; Colombi, M.; Fuhs, S.R.; Matter, M.S.; Guri, Y.; Adam, K.; Cornu, M.; Piscuoglio, S.; Ng, C.K.Y.; Betz, C.; et al. The protein histidine phosphatase LHPP. is a tumour suppressor. Nature 2018, 555, 678–682. [Google Scholar] [CrossRef]
- Hou, B.; Li, W.; Li, J.; Ma, J.; Xia, P.; Liu, Z.; Zeng, Q.; Zhang, X.; Chang, D. Tumor suppressor LHPP. regulates the proliferation of colorectal cancer cells via the PI3K/AKT pathway. Oncol. Rep. 2020, 43, 536–548. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, X.; Zhou, X.; Zhang, X. LHPP. suppresses bladder cancer cell proliferation and growth via inactivating AKT/p65 signaling pathway. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.; Chen, Y.; Zhu, J. LHPP. suppresses proliferation, migration, and invasion and promotes apoptosis in pancreatic cancer. Biosci. Rep. 2020, 40. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Qian, K.; Guo, K.; Chen, L.; Xiang, J.; Li, D.; Wu, Y.; Ji, Q.; Sun, T.; Wang, Z. LHPP. inhibits cell growth and migration and triggers autophagy in papillary thyroid cancer by regulating the AKT/AMPK/mTOR signaling pathway. Acta Biochim Biophys Sin 2020, 52, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Dai, X.; Chen, H.; Fang, C.; Chen, J.; Sun, L. Down-regulation of LHPP. in cervical cancer influences cell proliferation, metastasis and apoptosis by modulating AKT. Biochem. Biophys. Res. Commun. 2018, 503, 1108–1114. [Google Scholar] [CrossRef] [PubMed]
- Adam, K.; Lesperance, J.; Hunter, T.; Zage, P.E. The potential functional roles of NME1 histidine kinase activity in neuroblastoma pathogenesis. Int. J. Mol. Sci. 2020, 21, 3319. [Google Scholar] [CrossRef] [PubMed]
- Neff, C.D.; Abkevich, V.; Packer, J.C.; Chen, Y.; Potter, J.; Riley, R.; Davenport, C.; DeGrado Warren, J.; Jammulapati, S.; Bhathena, A.; et al. Evidence for HTR1A and LHPP. as interacting genetic risk factors in major depression. Mol. Psychiatry 2009, 14, 621–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, L.; Gong, X.; Tang, Y.; Kong, L.; Chang, M.; Geng, H.; Xu, K.; Wang, F. Relationship between the LHPP. gene polymorphism and resting-state brain activity in major depressive disorder. Neural Plast. 2016, 2016, 9162590. [Google Scholar] [CrossRef] [Green Version]
- Cui, L.; Wang, F.; Yin, Z.; Chang, M.; Song, Y.; Wei, Y.; Lv, J.; Zhang, Y.; Tang, Y.; Gong, X.; et al. Effects of the LHPP. gene polymorphism on the functional and structural changes of gray matter in major depressive disorder. Quant Imaging Med. Surg. 2020, 10, 257–268. [Google Scholar] [CrossRef]
- Polimanti, R.; Wang, Q.; Meda, S.A.; Patel, K.T.; Pearlson, G.D.; Zhao, H.; Farrer, L.A.; Kranzler, H.R.; Gelernter, J. The interplay between risky sexual behaviors and alcohol dependence: genome-wide association and neuroimaging support for LHPP. as a risk gene. Neuropsychopharmacology 2017, 42, 598–605. [Google Scholar] [CrossRef] [Green Version]
- Gohla, A. Do metabolic HAD phosphatases moonlight as protein phosphatases? Biochim Biophys Acta Mol. Cell Res. 2019, 1866, 153–166. [Google Scholar] [CrossRef]
- Back, S.H.; Adapala, N.S.; Barbe, M.F.; Carpino, N.C.; Tsygankov, A.Y.; Sanjay, A. TULA-2, a novel histidine phosphatase, regulates bone remodeling by modulating osteoclast function. Cell. Mol. Life Sci. 2013, 70, 1269–1284. [Google Scholar] [CrossRef] [Green Version]
- Boissan, M.; De Wever, O.; Lizarraga, F.; Wendum, D.; Poincloux, R.; Chignard, N.; Desbois-Mouthon, C.; Dufour, S.; Nawrocki-Raby, B.; Birembaut, P.; et al. Implication of metastasis suppressor NM23-H1 in maintaining adherens junctions and limiting the invasive potential of human cancer cells. Cancer Res. 2010, 70, 7710–7722. [Google Scholar] [CrossRef] [Green Version]
- Howlett, A.R.; Petersen, O.W.; Steeg, P.S.; Bissell, M.J. A novel function for the nm23-H1 gene: overexpression in human breast carcinoma cells leads to the formation of basement membrane and growth arrest. J. Natl. Cancer Inst. 1994, 86, 1838–1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, R.; Meng, J.; Shi, Q.; Dai, X.X.; Chen, J.H.; Lei, Y.J.; Shan, B.; Gao, C.; Chu, Y.L.; Dong, X.P. Expressions of nucleoside diphosphate kinase (nm23) in tumor tissues are related with metastasis and length of survival of patients with hepatocellular carcinoma. BiomEd. Environ. Sci. 2010, 23, 267–272. [Google Scholar] [CrossRef]
- Hartsough, M.T.; Steeg, P.S. Nm23/nucleoside diphosphate kinase in human cancers. J. Bioenerg. Biomembr. 2000, 32, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, M.; Zhang, C.; Zhang, J.; Li, G.; Zhang, Z.; He, X.; Fan, M. Prognostic value and clinicopathologic significance of nm23 in various cancers: a systematic review and meta-analysis. Int. J. Surg. 2018, 60, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Guo, X.; Zheng, B.; Han, W.; Chen, X.; Zhu, J.; Xie, B.; Liu, J.; Luan, X.; Yan, Y.; et al. Correlation between NM23 protein overexpression and prognostic value and clinicopathologic features of ovarian cancer: a meta-analysis. Arch. Gynecol. Obs. 2018, 297, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Harlozinska, A.; Bar, J.K.; Gerber, J. nm23 expression in tissue sections and tumor effusion cells of ovarian neoplasms. Int. J. Cancer 1996, 69, 415–419. [Google Scholar] [CrossRef]
- Niitsu, N.; Nakamine, H.; Okamoto, M.; Akamatsu, H.; Honma, Y.; Higashihara, M.; Okabe-Kado, J.; Hirano, M.; Adult Lymphoma Treatment Study Group, A. Expression of nm23-H1 is associated with poor prognosis in peripheral T-cell lymphoma. Br. J. Haematol. 2003, 123, 621–630. [Google Scholar] [CrossRef]
- Andolfo, I.; De Martino, D.; Liguori, L.; Petrosino, G.; Troncone, G.; Tata, N.; Galasso, A.; Roma, C.; Chiancone, F.; Zarrilli, S.; et al. Correlation of NM23-H1 cytoplasmic expression with metastatic stage in human prostate cancer tissue. Naunyn Schmiedebergs Arch. Pharm. 2011, 384, 489–498. [Google Scholar] [CrossRef] [Green Version]
- Roy, F.; Laberge, G.; Douziech, M.; Ferland-McCollough, D.; Therrien, M. KSR is a scaffold required for activation of the ERK/MAPK module. Genes Dev. 2002, 16, 427–438. [Google Scholar] [CrossRef] [Green Version]
- Morrison, D.K. KSR: A MAPK scaffold of the Ras pathway? J. Cell Sci. 2001, 114, 1609–1612. [Google Scholar]
- Alexa, A.; Varga, J.; Remenyi, A. Scaffolds are ‘active’ regulators of signaling modules. Febs J. 2010, 277, 4376–4382. [Google Scholar] [CrossRef] [Green Version]
- Masoudi, N.; Fancsalszky, L.; Pourkarimi, E.; Vellai, T.; Alexa, A.; Remenyi, A.; Gartner, A.; Mehta, A.; Takacs-Vellai, K. The NM23-H1/H2 homolog NDK-1 is required for full activation of Ras signaling in C. elegans. Development 2013, 140, 3486–3495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenmann, D.M.; Kim, S.K. Protruding vulva mutants identify novel loci and Wnt signaling factors that function during Caenorhabditis elegans vulva development. Genetics 2000, 156, 1097–1116. [Google Scholar] [PubMed]
- Salerno, M.; Palmieri, D.; Bouadis, A.; Halverson, D.; Steeg, P.S. Nm23-H1 metastasis suppressor expression level influences the binding properties, stability, and function of the kinase suppressor of Ras1 (KSR1) Erk scaffold in breast carcinoma cells. Mol. Cell. Biol. 2005, 25, 1379–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witkiewicz, A.K.; Nguyen, K.H.; Dasgupta, A.; Kennedy, E.P.; Yeo, C.J.; Lisanti, M.P.; Brody, J.R. Co-expression of fatty acid synthase and caveolin-1 in pancreatic ductal adenocarcinoma: implications for tumor progression and clinical outcome. Cell Cycle 2008, 7, 3021–3025. [Google Scholar] [CrossRef]
- Blum, R.; Kloog, Y. Metabolism addiction in pancreatic cancer. Cell Death Dis. 2014, 5, e1065. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, E.; Ota, T.; Tsukuda, K.; Okita, A.; Matsuoka, K.; Murakami, M.; Doihara, H.; Shimizu, N. nm23-H1 reduces in vitro cell migration and the liver metastatic potential of colon cancer cells by regulating myosin light chain phosphorylation. Int. J. Cancer 2004, 108, 207–211. [Google Scholar] [CrossRef]
- Zhao, R.; Gong, L.; Li, L.; Guo, L.; Zhu, D.; Wu, Z.; Zhou, Q. nm23-H1 is a negative regulator of TGF-beta1-dependent induction of epithelial-mesenchymal transition. Exp. Cell Res. 2013, 319, 740–749. [Google Scholar] [CrossRef]
- Ferrucci, V.; de Antonellis, P.; Pennino, F.P.; Asadzadeh, F.; Virgilio, A.; Montanaro, D.; Galeone, A.; Boffa, I.; Pisano, I.; Scognamiglio, I.; et al. Metastatic group 3 medulloblastoma is driven by PRUNE1 targeting NME1-TGF-beta-OTX2-SNAIL via PTEN inhibition. Brain 2018, 141, 1300–1319. [Google Scholar] [CrossRef]
- Marshall, J.C.; Collins, J.W.; Nakayama, J.; Horak, C.E.; Liewehr, D.J.; Steinberg, S.M.; Albaugh, M.; Vidal-Vanaclocha, F.; Palmieri, D.; Barbier, M.; et al. Effect of inhibition of the lysophosphatidic acid receptor 1 on metastasis and metastatic dormancy in breast cancer. J. Natl. Cancer Inst. 2012, 104, 1306–1319. [Google Scholar] [CrossRef]
- Marino, N.; Nakayama, J.; Collins, J.W.; Steeg, P.S. Insights into the biology and prevention of tumor metastasis provided by the Nm23 metastasis suppressor gene. Cancer Metastasis Rev. 2012, 31, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Okabe-Kado, J.; Hagiwara-Watanabe, Y.; Niitsu, N.; Kasukabe, T.; Kaneko, Y. NM23 downregulation and lysophosphatidic acid receptor EDG2/lpa1 upregulation during myeloid differentiation of human leukemia cells. Leuk. Res. 2018, 66, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Tran, A.T.; Chapman, E.M.; Flamand, M.N.; Yu, B.; Krempel, S.J.; Duchaine, T.F.; Eroglu, M.; Derry, W.B. MiR-35 buffers apoptosis thresholds in the C. elegans germline by antagonizing both MAPK and core apoptosis pathways. Cell Death Differ. 2019, 26, 2637–2651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Z.; Beresford, P.J.; Oh, D.Y.; Zhang, D.; Lieberman, J. Tumor suppressor NM23-H1 is a granzyme A-activated DNase during CTL-mediated apoptosis, and the nucleosome assembly protein SET is its inhibitor. Cell 2003, 112, 659–672. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, D.; Beresford, P.J.; Zhu, P.; Zhang, D.; Sung, J.S.; Demple, B.; Perrino, F.W.; Lieberman, J. The exonuclease TREX1 is in the SET complex and acts in concert with NM23-H1 to degrade DNA during granzyme A-mediated cell death. Mol. Cell 2006, 23, 133–142. [Google Scholar] [CrossRef]
- Fancsalszky, L.; Monostori, E.; Farkas, Z.; Pourkarimi, E.; Masoudi, N.; Hargitai, B.; Bosnar, M.H.; Dezeljin, M.; Zsakai, A.; Vellai, T.; et al. NDK-1, the homolog of NM23-H1/H2 regulates cell migration and apoptotic engulfment in C. elegans. PLoS ONE 2014, 9, e92687. [Google Scholar] [CrossRef] [Green Version]
- Farkas, Z.; Petric, M.; Liu, X.; Herit, F.; Rajnavolgyi, E.; Szondy, Z.; Budai, Z.; Orban, T.I.; Sandor, S.; Mehta, A.; et al. The nucleoside diphosphate kinase NDK-1/NME1 promotes phagocytosis in concert with DYN-1/Dynamin. Faseb J. 2019, 33, 11606–11614. [Google Scholar] [CrossRef] [Green Version]
- Chang, Z.F. NME3 is critical for maintining genome stability and impacts tumor metastasis in breast cancer. In Proceedings of the SYMER: A Systems Approach to New Paradigms in Metabolic and Epigenetic Regulation, Annecy, France, 6–9 October 2019. [Google Scholar]
- Takacs-Vellai, K.; Vellai, T.; Farkas, Z.; Mehta, A. Nucleoside diphosphate kinases (NDPKs) in animal development. Cell. Mol. Life Sci. 2015, 72, 1447–1462. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, S.; Di, L.; Zhdanova, O.; Li, Z.; Vardhana, S.; Wan, Q.; Yan, Y.; Varma, R.; Backer, J.; Wulff, H.; et al. The class II phosphatidylinositol 3 kinase C2beta is required for the activation of the K+ channel KCa3.1 and CD4 T-cells. Mol. Biol. Cell 2009, 20, 3783–3791. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, S.; Cai, X.; Li, Z.; Sun, Y.; Skolnik, E.Y. Phosphatidylinositol-3-kinase C2beta and TRIM27 function to positively and negatively regulate IgE receptor activation of mast cells. Mol. Cell. Biol. 2012, 32, 3132–3139. [Google Scholar] [CrossRef] [Green Version]
- Hoenderop, J.G.; van Leeuwen, J.P.; van der Eerden, B.C.; Kersten, F.F.; van der Kemp, A.W.; Merillat, A.M.; Waarsing, J.H.; Rossier, B.C.; Vallon, V.; Hummler, E.; et al. Renal Ca2+ wasting, hyperabsorption, and reduced bone thickness in mice lacking TRPV5. J. Clin. Investig. 2003, 112, 1906–1914. [Google Scholar] [CrossRef] [PubMed]
- Hippe, H.J.; Luedde, M.; Lutz, S.; Koehler, H.; Eschenhagen, T.; Frey, N.; Katus, H.A.; Wieland, T.; Niroomand, F. Regulation of cardiac cAMP synthesis and contractility by nucleoside diphosphate kinase B/G protein beta gamma dimer complexes. Circ. Res. 2007, 100, 1191–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hippe, H.J.; Lutz, S.; Cuello, F.; Knorr, K.; Vogt, A.; Jakobs, K.H.; Wieland, T.; Niroomand, F. Activation of heterotrimeric G proteins by a high energy phosphate transfer via nucleoside diphosphate kinase (NDPK) B and Gbeta subunits. Specific activation of Gsalpha by an NDPK B.Gbetagamma complex in H10 cells. J. Biol. Chem. 2003, 278, 7227–7233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dearolf, C.R.; Tripoulas, N.; Biggs, J.; Shearn, A. Molecular consequences of awdb3, a cell-autonomous lethal mutation of Drosophila induced by hybrid dysgenesis. Dev. Biol. 1988, 129, 169–178. [Google Scholar] [CrossRef]
- Nallamothu, G.; Woolworth, J.A.; Dammai, V.; Hsu, T. Awd, the homolog of metastasis suppressor gene Nm23, regulates Drosophila epithelial cell invasion. Mol. Cell. Biol. 2008, 28, 1964–1973. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Gross, S.; Wolf, N.M.; Butenschon, V.M.; Qiu, Y.; Devraj, K.; Liebner, S.; Kroll, J.; Skolnik, E.Y.; Hammes, H.P.; et al. Nucleoside diphosphate kinase B regulates angiogenesis through modulation of vascular endothelial growth factor receptor type 2 and endothelial adherens junction proteins. Arter. Thromb. Vasc. Biol. 2014, 34, 2292–2300. [Google Scholar] [CrossRef] [Green Version]
- Arnaud-Dabernat, S.; Bourbon, P.M.; Dierich, A.; Le Meur, M.; Daniel, J.Y. Knockout mice as model systems for studying nm23/NDP kinase gene functions. Application to the nm23-M1 gene. J. Bioenerg. Biomembr. 2003, 35, 19–30. [Google Scholar] [CrossRef]
- Postel, E.H.; Wohlman, I.; Zou, X.; Juan, T.; Sun, N.; D’Agostin, D.; Cuellar, M.; Choi, T.; Notterman, D.A.; La Perle, K.M. Targeted deletion of Nm23/nucleoside diphosphate kinase A and B reveals their requirement for definitive erythropoiesis in the mouse embryo. Dev. Dyn. 2009, 238, 775–787. [Google Scholar] [CrossRef]
- Deplagne, C.; Peuchant, E.; Moranvillier, I.; Dubus, P.; Dabernat, S. The anti-metastatic nm23-1 gene is needed for the final step of mammary duct maturation of the mouse nipple. PLoS ONE 2011, 6, e18645. [Google Scholar] [CrossRef] [Green Version]
- Vogel, P.; Read, R.W.; Hansen, G.M.; Payne, B.J.; Small, D.; Sands, A.T.; Zambrowicz, B.P. Congenital hydrocephalus in genetically engineered mice. Vet. Pathol. 2012, 49, 166–181. [Google Scholar] [CrossRef]
- Vogel, P.; Read, R.; Hansen, G.M.; Freay, L.C.; Zambrowicz, B.P.; Sands, A.T. Situs inversus in Dpcd/Poll-/-, Nme7-/-, and Pkd1l1-/- mice. Vet. Pathol. 2010, 47, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Vogel, P.; Hansen, G.; Fontenot, G.; Read, R. Tubulin tyrosine ligase-like 1 deficiency results in chronic rhinosinusitis and abnormal development of spermatid flagella in mice. Vet. Pathol. 2010, 47, 703–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunzelmann, K.; Mehta, A. CFTR: a hub for kinases and crosstalk of cAMP and Ca2+. Febs J. 2013, 280, 4417–4429. [Google Scholar] [CrossRef] [PubMed]
- King, J.D., Jr.; Lee, J.; Riemen, C.E.; Neumann, D.; Xiong, S.; Foskett, J.K.; Mehta, A.; Muimo, R.; Hallows, K.R. Role of binding and nucleoside diphosphate kinase A in the regulation of the cystic fibrosis transmembrane conductance regulator by AMP-activated protein kinase. J. Biol. Chem. 2012, 287, 33389–33400. [Google Scholar] [CrossRef] [Green Version]
- Eshwaran, R. A sweet kiss of NDPK-B—Its involvement in glucose metabolism. In Proceedings of the SYMER: A Systems Approach to New Paradigms in Metabolic and Epigenetic Regulation, Annecy, France, 6–9 October 2019. [Google Scholar]
- Zollo, M. NME-1/Prune-1 at the interplay between cancer metastasis and neurodevelopment disorders. In Proceedings of the SYMER: A Systems Approach to New Paradigms in Metabolic and Epigenetic Regulation, Annecy, France, 6–9 October 2019. [Google Scholar]
- McAllister, T.E.; Nix, M.G.; Webb, M.E. Fmoc-chemistry of a stable phosphohistidine analogue. Chem. Commun. (Camb.) 2011, 47, 1297–1299. [Google Scholar] [CrossRef]
- Kee, J.M.; Oslund, R.C.; Couvillon, A.D.; Muir, T.W. A second-generation phosphohistidine analog for production of phosphohistidine antibodies. Org. Lett. 2015, 17, 187–189. [Google Scholar] [CrossRef] [Green Version]
- McAllister, T.E.; Hollins, J.J.; Webb, M.E. Prospects for stable analogues of phosphohistidine. Biochem. Soc. Trans. 2013, 41, 1072–1077. [Google Scholar] [CrossRef] [Green Version]






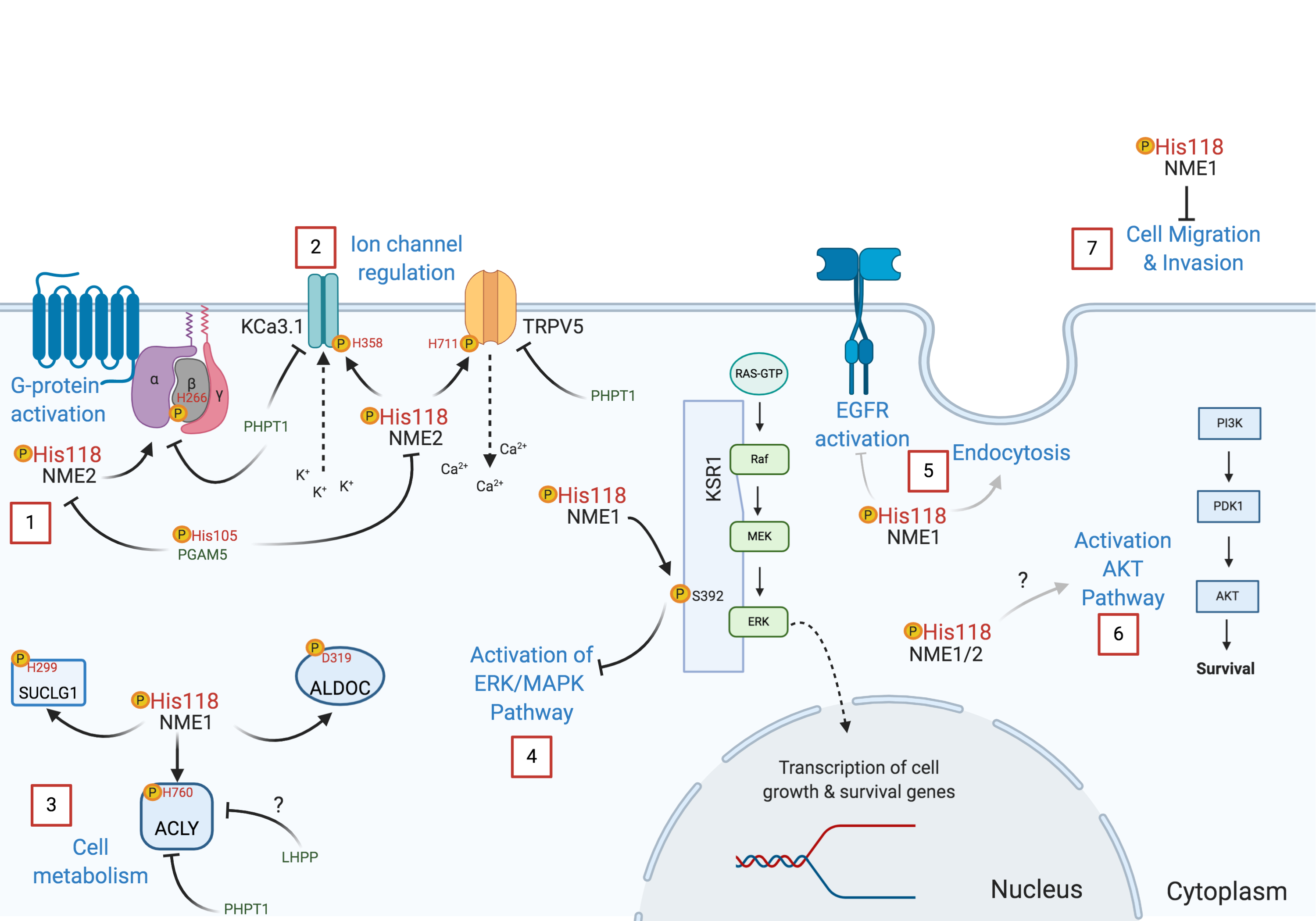{kind=link}
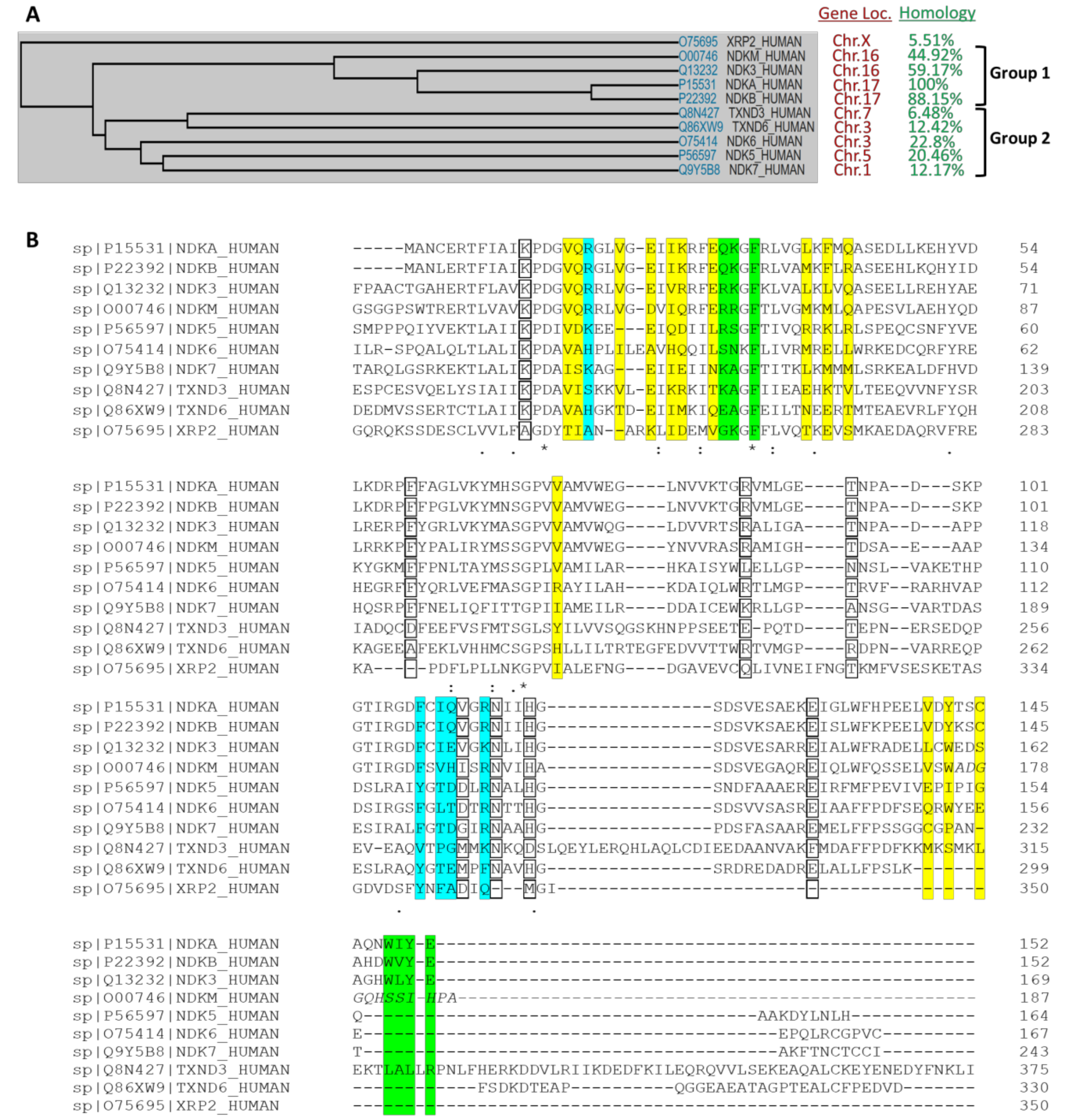{kind=link}
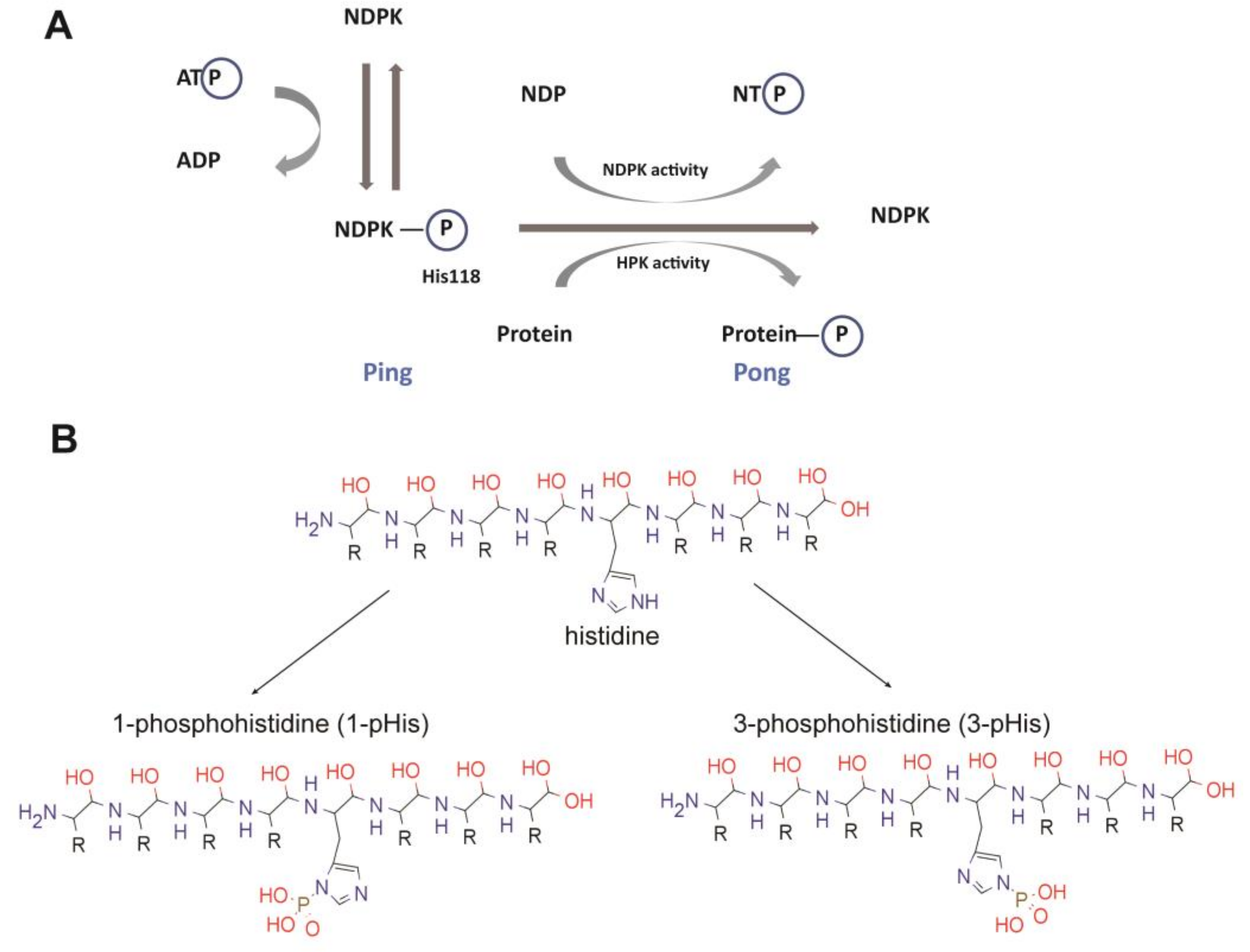{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB | Resolution | Ligand | |
|---|---|---|---|
| NME1 | 1JXV [65] | 2.2 | apo |
| NME1 | 1UCN [66] | 2 | complexed with ADP |
| NME1 | 2HVD [16] | 2.15 | complexed with ADP |
| NME1 | 2HVE [16] | 2.4 | S120G, complexed with ADP |
| NME1 | 3L7U [64] | 2.1 | apo |
| NME1 | 4ENO [67] | 2.8 | under oxidative conditions trigger allostery |
| NME1 | 5UI4 [68] | 2.75 | conjugated to imidazole fluorosulfate |
| NME2 | 1NSK [64] | 2.8 | apo |
| NME2 | 1NUE [69] | 2 | complexed with GDP |
| NME2 | 3BBB [70] | 1.3 | complexed with dinucleotide d(AG) |
| NME2 | 3BBC [70] | 1.7 | R88A |
| NME2 | 3BBF [70] | 1.7 | complexed with GDP |
| NME3 | 1ZS6 | 2.3 | complexed with ADP |
| NME4 | 1EHW [71] | 2.4 | apo |
| Large Interface | Small Interface 1 (Opposite to Active site) | Small Interface 1′ (on the Site of Active Site) | PDB ID | ||||
|---|---|---|---|---|---|---|---|
| Interface Area (Å2) | ∆G (kcal/mol) | Interface Area (Å2) | ∆G (kcal/mol) | Interface Area (Å2) | ∆G (kcal/mol) | ||
| NME1 | 1015 | −15.3 | 808 | −6.2 | 798 | −6.7 | 1UCN |
| NME2 | 1015 | −12.6 | 793 | −6.7 | 780 | −6.7 | 1NUE |
| NME3 | 1220 | −11.5 | 836 | −5.8 | 829 | −4.3 | 1ZS6 |
| NME4 | 998 | −15.6 | 520 | −1.4 | 509 | −0.7 | 1EHW |
| Gene | Uniprot ID | Name | pHis Site | pHis Sequence | N1 or N3 |
|---|---|---|---|---|---|
| NME1 | P15531 | Nucleoside diphosphate kinase A | H118 | NIIHGSD | 1-pHis |
| NME2 | P22392 | Nucleoside diphosphate kinase B | H118 | NIIHGSD | 1-pHis |
| TRPV5 | Q9NQA5 | Transient receptor potential cation channel subfamily V member 5 | H711 | TLGHLNL | 3-pHis |
| GNB1 | P62873 | Guanine nucleotide-binding protein G(I)/G(S)/G(T) subunit beta-1 | H266 | TYSHDNI | 3-pHis |
| KCa3.1 | O15554 | Intermediate conductance calcium-activated potassium channel protein 4 | H358 | RLKHRKL | 3-pHis |
| ACLY | P53396 | ATP-citrate synthase | H760 | QFGHAGA | 3-pHis |
| SUCLG1 | P53597 | Succinyl-CoA synthetase | H299 | RMGHAGA | 3-pHis |
| KSR1 | Q8IVT5 | Kinase suppressor of Ras 1 | S392 | RTESVPS | - |
| ANXA1 | P04083 | Annexin A1 | H246/H293? | YSKHDMN/ | ND |
| GTRHKAL | |||||
| ALDOC | P09972 | Aldolase C | D319 | GQRDNAG | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adam, K.; Ning, J.; Reina, J.; Hunter, T. NME/NM23/NDPK and Histidine Phosphorylation. Int. J. Mol. Sci. 2020, 21, 5848. https://doi.org/10.3390/ijms21165848
Adam K, Ning J, Reina J, Hunter T. NME/NM23/NDPK and Histidine Phosphorylation. International Journal of Molecular Sciences. 2020; 21(16):5848. https://doi.org/10.3390/ijms21165848
Chicago/Turabian StyleAdam, Kevin, Jia Ning, Jeffrey Reina, and Tony Hunter. 2020. "NME/NM23/NDPK and Histidine Phosphorylation" International Journal of Molecular Sciences 21, no. 16: 5848. https://doi.org/10.3390/ijms21165848
APA StyleAdam, K., Ning, J., Reina, J., & Hunter, T. (2020). NME/NM23/NDPK and Histidine Phosphorylation. International Journal of Molecular Sciences, 21(16), 5848. https://doi.org/10.3390/ijms21165848