New Anti SARS-Cov-2 Targets for Quinoline Derivatives Chloroquine and Hydroxychloroquine †




Abstract
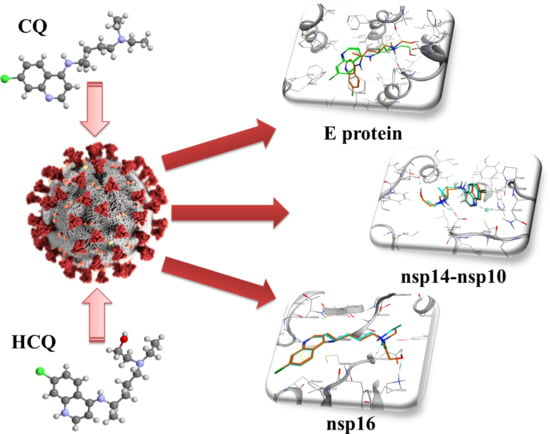
:
1. Introduction
2. Results and Discussion
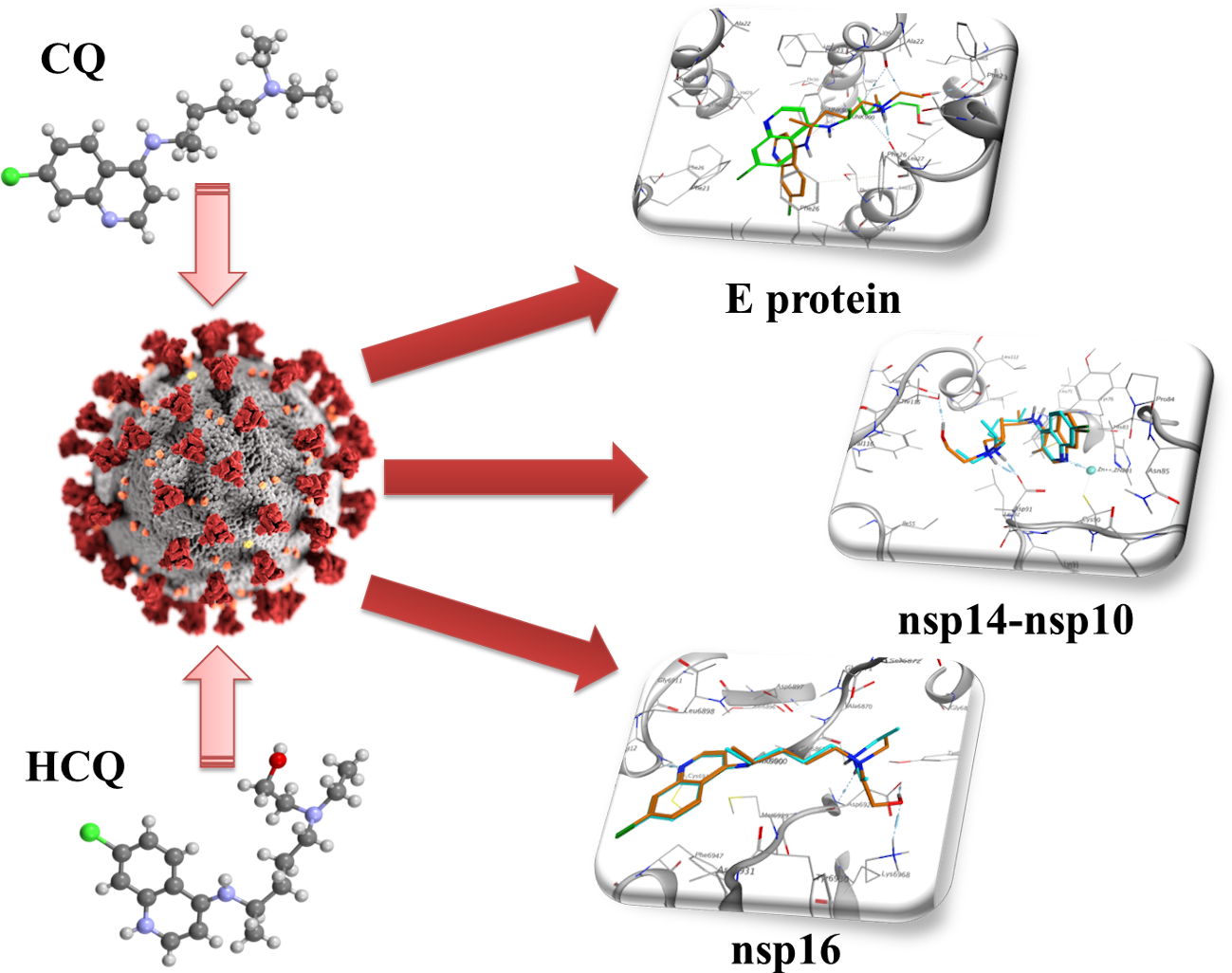
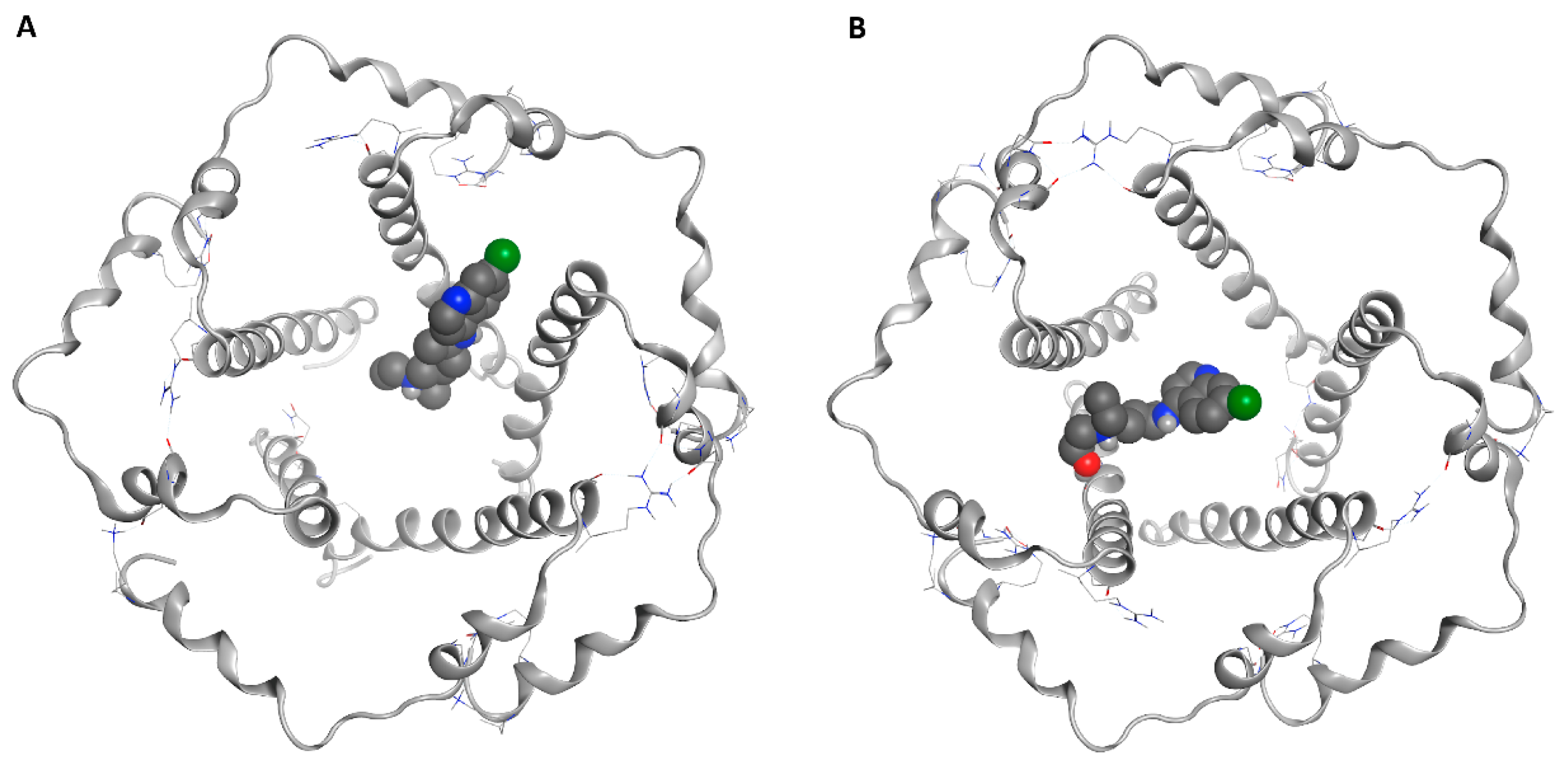
2.1. E Protein
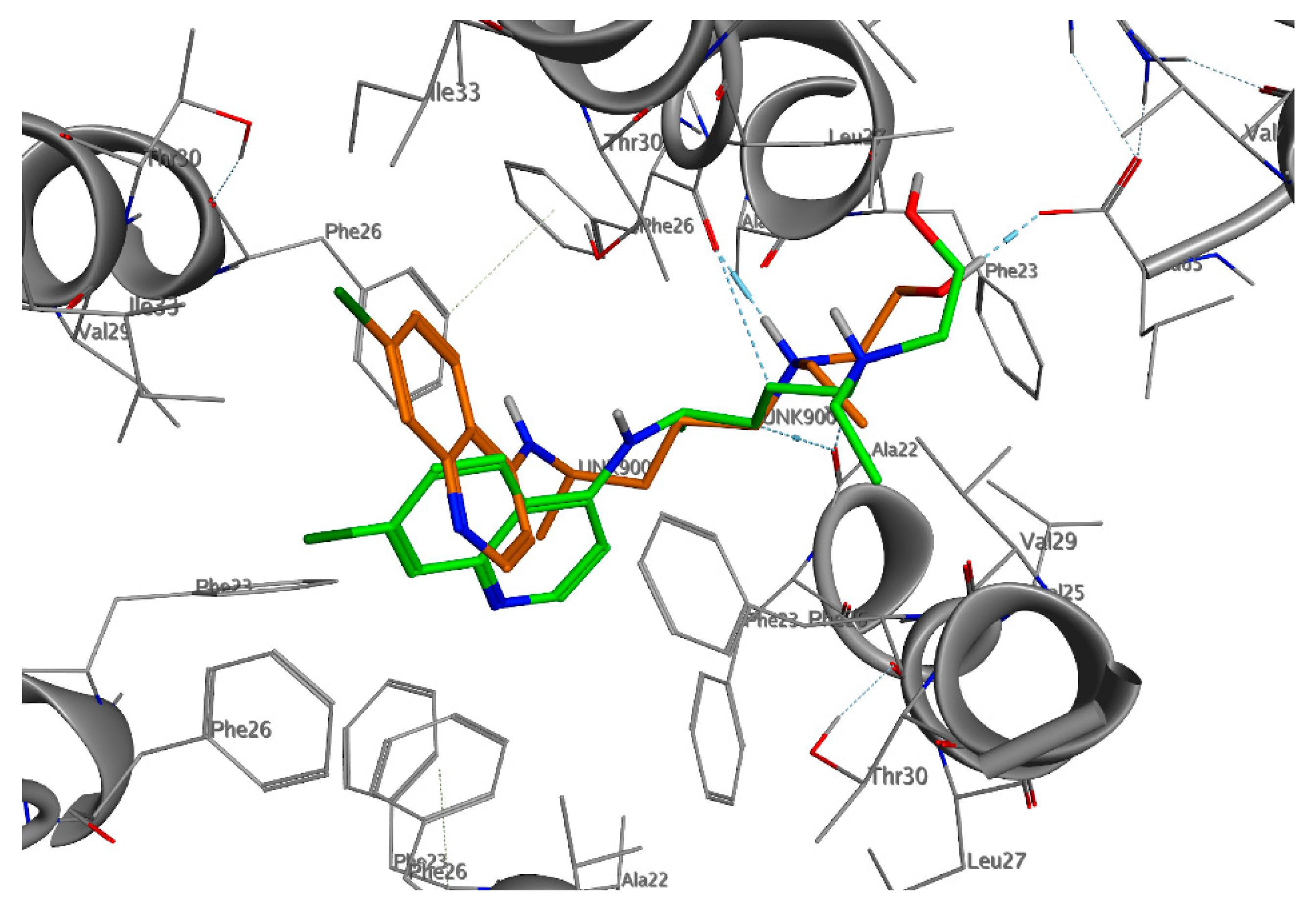
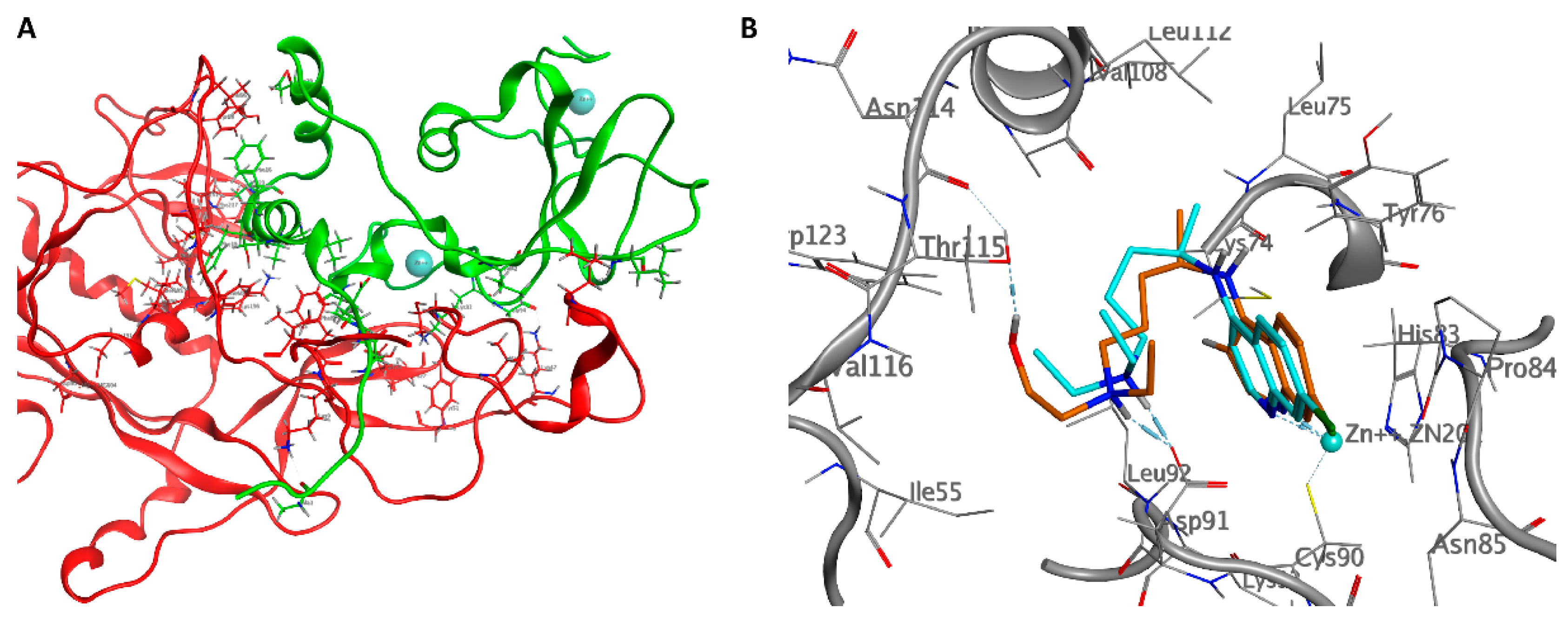
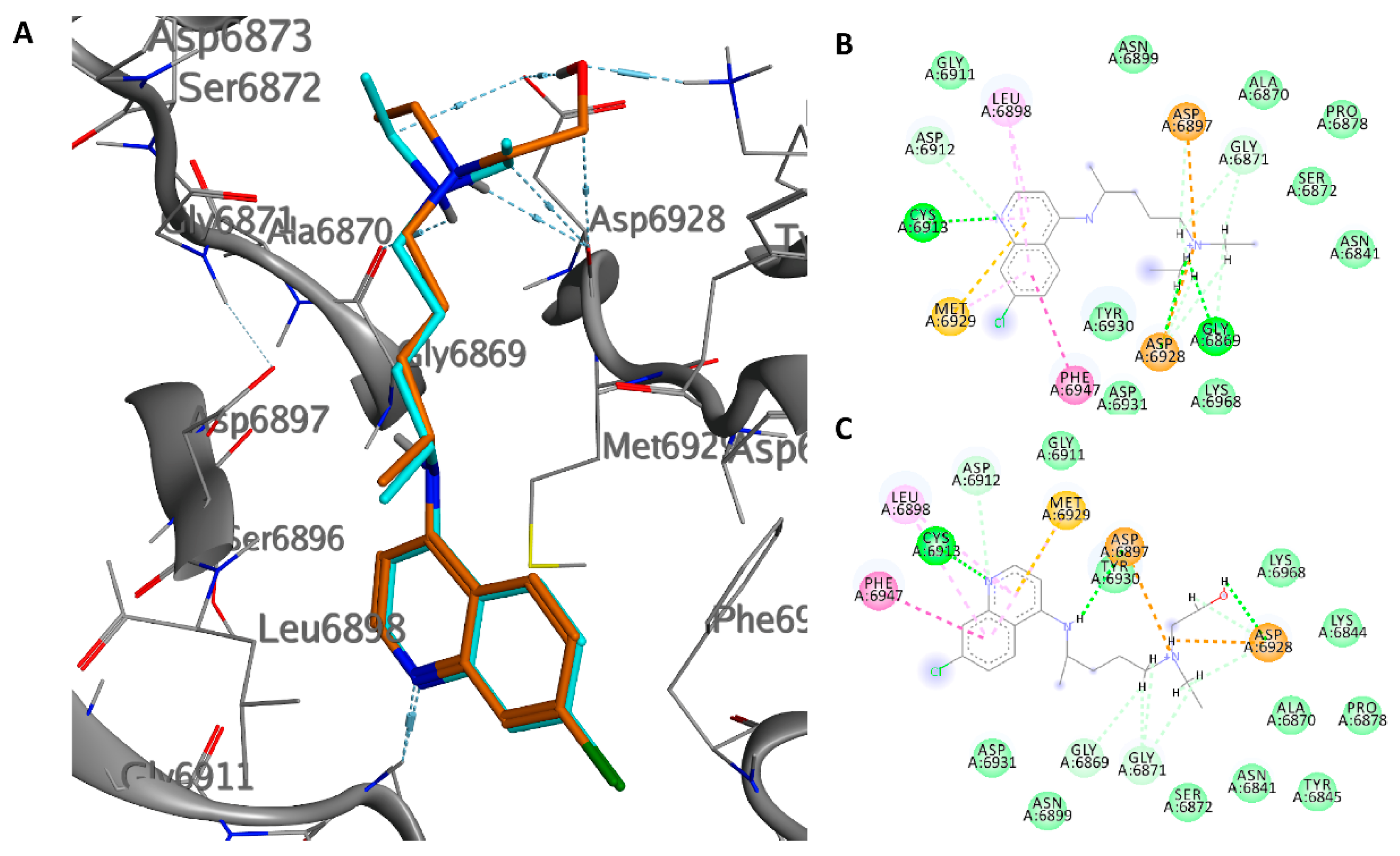
2.2. NSP10/NSP14
2.3. NSP10/NSP16
3. Materials and Methods
3.1. Structures Preparation and Minimization
3.2. Molecular Docking
3.3. Molecular Dynamics Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- de Wit, E.; van Doremalen, N.; Falzarano, D.; Munster, V.J. SARS and MERS: Recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016, 14, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Hui, D.S.; Memish, Z.A.; Zumla, A. Severe acute respiratory syndrome vs. the Middle East respiratory syndrome. Curr. Opin. Pulm. Med. 2014, 20, 233–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO. WHO Characterizes COVID-19 as a Pandemic; WHO: Geneva, Switzerland, 2020. [Google Scholar]
- Yu, W.B.; Tang, G.D.; Zhang, L.; Corlett, R.T. Decoding the evolution and transmissions of the novel pneumonia coronavirus (SARS-CoV-2 / HCoV-19) using whole genomic data. Zool. Res. 2020, 41, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malaiyan, J.; Arumugam, S.; Mohan, K.; Gomathi Radhakrishnan, G. An update on the origin of SARS-CoV-2: Despite closest identity, bat (RaTG13) and pangolin derived coronaviruses varied in the critical binding site and O-linked glycan residues. J. Med. Virol. 2020. [Google Scholar] [CrossRef]
- Tang, X.L.; Wu, C.C.; Li, X.; Song, Y.H.; Yao, X.M.; Wu, X.K.; Duan, Y.G.; Zhang, H.; Wang, Y.R.; Qian, Z.H.; et al. On the origin and continuing evolution of SARS-CoV-2. Natl. Sci. Rev. 2020, 7, 1012–1023. [Google Scholar] [CrossRef] [Green Version]
- Chaw, S.M.; Tai, J.H.; Chen, S.L.; Hsieh, C.H.; Chang, S.Y.; Yeh, S.H.; Yang, W.S.; Chen, P.J.; Wang, H.Y. The origin and underlying driving forces of the SARS-CoV-2 outbreak. J. Biomed. Sci. 2020, 27. [Google Scholar] [CrossRef]
- Rothan, H.A.; Byrareddy, S.N. The epidemiology and pathogenesis of coronavirus disease (COVID-19) outbreak. J. Autoimmun. 2020, 109, 102433. [Google Scholar] [CrossRef]
- Robinson, C.; Loeffelholz, M.J.; Pinsky, B.A. Clinical Virology Manual. In Clinical Virology Manual, 5th ed.; Loeffelholz, M.J., Ed.; ASM Press: Washington, DC, USA, 2016. [Google Scholar]
- Carriere, F.; Longhi, S.; Record, M. The endosomal lipid bis(monoacylglycero) phosphate as a potential key player in the mechanism of action of chloroquine against SARS-COV-2 and other enveloped viruses hijacking the endocytic pathway. Biochimie 2020. [Google Scholar] [CrossRef]
- Gao, Q. Pharmacological characteristics of chloroquine and suggestions for its use in treatment of coronavirus disease 2019 (COVID-19). Zhongguo Xue Xi Chong Bing Fang Zhi Za Zhi 2020, 32, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Beura, S.; Prabhakar, C. In-silico strategies for probing chloroquine based inhibitors against SARS-CoV-2. J. Biomol. Struct. Dyn. 2020, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Martinez, C.E.; Fernandes, R.M.; Hawcutt, D.B.; Sinha, I.P.; Pacheco, R.L. Efficacy, safety and cost-effectiveness of hydroxychloroquine in children with COVID-19: A call for evidence. Acta Paediatr. 2020. [Google Scholar] [CrossRef] [PubMed]
- D’Cruz, M. The ICMR bulletin on targeted hydroxychloroquine prophylaxis for Covid-19: Need to interpret with caution. Indian J. Med. Ethics 2020, 5, 100–102. [Google Scholar] [CrossRef] [PubMed]
- Coatney, G.R. Pitfalls in a discovery: The chronicle of chloroquine. Am. J. Trop. Med. Hyg. 1963, 12, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Radl, S. From chloroquine to antineoplastic drugs? the story of antibacterial quinolones. Arch. Pharm. (Weinh.) 1996, 329, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Loeb, F.; Clark, W.M.; Coatney, G.R.; Coggeshall, L.T.; Dieuaide, F.R.; Dochez, A.R.; Marshall, E.K., Jr.; Marvel, C.S.; McCay, O.R.; Sapero, J.J.; et al. ACTIVITY of a new antimalarial agent, chloroquine (SN 7618). J. Am. Med. Assoc. 1946, 130, 1069. [Google Scholar] [CrossRef]
- Halawani, A.; Baz, I.I.; Morcos, F. The antimalarial chloroquine-diphosphate (aralen). J. Egypt. Med. Assoc. 1947, 30, 128–136. [Google Scholar]
- Anonimous, TRIAL use of the new antimalarial drug chloroquine. Bull. US Army. Med. Dep. 1946, 6, 9–10.
- Anonimous, CHLOROQUINE-DIPHOSPHATE, a Newly-Standardized Antimalarial Drug. Bull. US Army. Med. Dep. 1947, 7, 834.
- Anonimous, CLINICAL evaluation of chloroquine. Bull U S Army. Med. Dep. 1947, 7, 835–837.
- Cowman, A.F.; Foote, S.J. Chemotherapy and Drug-Resistance in Malaria. Int. J. Parasitol. 1990, 20, 503–513. [Google Scholar] [CrossRef]
- Ciak, J.; Hahn, F.E. Chloroquine: Mode of action. Science 1966, 151, 347–349. [Google Scholar] [CrossRef] [PubMed]
- Whichard, L.P.; Washington, M.E.; Holbrook, D.J., Jr. The inhibition in vitro of bacterial DNA polymerases and RNA polymerase by antimalarial 8-aminoquinolines and by chloroquine. Biochim. Biophys. Acta 1972, 287, 52–67. [Google Scholar] [CrossRef]
- Marquez, V.E.; Cranston, J.W.; Ruddon, R.W.; Burckhalter, J.H. Binding to deoxyribonucleic acid and inhibition of ribonucleic acid polymerase by analogs of chloroquine. J. Med. Chem. 1974, 17, 856–862. [Google Scholar] [CrossRef] [PubMed]
- Conklin, K.A.; Heu, P.; Chou, S.C. The effects of antimalarial drugs on nucleic acid synthesis in vitro in Tetrahymena pyriformis. Mol. Pharm. 1973, 9, 304–310. [Google Scholar]
- Field, R.C.; Gibson, B.R.; Holbrook, D.J., Jr.; McCall, B.M. Inhibition of precursor incorporation into nucleic acids of mammalian tissues by antimalarial aminoquinolines. Br. J. Pharm. 1978, 62, 159–164. [Google Scholar] [CrossRef] [Green Version]
- Egan, T.J.; Marques, H.M. The role of haem in the activity of chloroquine and related antimalarial drugs. Coord. Chem. Rev. 1999, 190, 493–517. [Google Scholar] [CrossRef]
- Pareja-Coronel, A. Treatment of viral hepatitis with chloroquine. Am. J. Gastroenterol. 1963, 39, 288–298. [Google Scholar]
- Mallucci, L. Effect of chloroquine on lysosomes and on growth of mouse hepatitis virus (MHV-3). Virology 1966, 28, 355–362. [Google Scholar] [CrossRef]
- Yielding, K.L. Inhibition of the replication of a bacterial DNA virus by chloroquine and other 4-aminoquinoline drugs. Proc. Soc. Exp. Biol. Med. 1967, 125, 780–783. [Google Scholar] [CrossRef] [PubMed]
- Inglot, A.D. Comparison of the antiviral activity in vitro of some non-steroidal anti-inflammatory drugs. J. Gen. Virol. 1969, 4, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Durand, D.P.; Chalgren, S.D.; Franke, V. Effect of chloroquine on myxovirus replication. Antimicrob Agents Chemother (Bethesda) 1970, 10, 105–108. [Google Scholar] [PubMed]
- Pazmino, N.H.; Yuhas, J.M.; Tennant, R.W. Inhibition of murine RNA tumor virus replication and oncogenesis by chloroquine. Int. J. Cancer 1974, 14, 379–385. [Google Scholar] [CrossRef]
- Rolain, J.M.; Colson, P.; Raoult, D. Recycling of chloroquine and its hydroxyl analogue to face bacterial, fungal and viral infections in the 21st century. Int. J. Antimicrob. Agents 2007, 30, 297–308. [Google Scholar] [CrossRef]
- Al-Bari, M.A.A. Targeting endosomal acidification by chloroquine analogs as a promising strategy for the treatment of emerging viral diseases. Pharmacol. Res. Perspect. 2017, 5. [Google Scholar] [CrossRef]
- D’Alessandro, S.; Scaccabarozzi, D.; Signorini, L.; Perego, F.; Ilboudo, D.P.; Ferrante, P.; Delbue, S. The Use of Antimalarial Drugs against Viral Infection. Microorganisms 2020, 8, 85. [Google Scholar] [CrossRef] [Green Version]
- Devaux, C.A.; Rolain, J.M.; Colson, P.; Raoult, D. New insights on the antiviral effects of chloroquine against coronavirus: What to expect for COVID-19? Int. J. Antimicrob. Agents 2020, 55, 105938. [Google Scholar] [CrossRef]
- Kooi, C.; Cervin, M.; Anderson, R. Differentiation of acid-pH-dependent and -nondependent entry pathways for mouse hepatitis virus. Virology 1991, 180, 108–119. [Google Scholar] [CrossRef]
- Zeichhardt, H.; Wetz, K.; Willingmann, P.; Habermehl, K.O. Entry of poliovirus type 1 and Mouse Elberfeld (ME) virus into HEp-2 cells: Receptor-mediated endocytosis and endosomal or lysosomal uncoating. J. Gen. Virol. 1985, 66, 483–492. [Google Scholar] [CrossRef]
- Kronenberger, P.; Vrijsen, R.; Boeye, A. Chloroquine induces empty capsid formation during poliovirus eclipse. J. Virol. 1991, 65, 7008–7011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savarino, A.; Lucia, M.B.; Rastrelli, E.; Rutella, S.; Golotta, C.; Morra, E.; Tamburrini, E.; Perno, C.F.; Boelaert, J.R.; Sperber, K.; et al. Anti-HIV effects of chloroquine: Inhibition of viral particle glycosylation and synergism with protease inhibitors. J. Acquir. Immune Defic. Syndr. 2004, 35, 223–232. [Google Scholar] [CrossRef]
- Vincent, M.J.; Bergeron, E.; Benjannet, S.; Erickson, B.R.; Rollin, P.E.; Ksiazek, T.G.; Seidah, N.G.; Nichol, S.T. Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol. J. 2005, 2, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferri, C.; Sebastiani, M.; Antonelli, A.; Colaci, M.; Manfredi, A.; Giuggioli, D. Current treatment of hepatitis C-associated rheumatic diseases. Arthritis Res. 2012, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marti-Carvajal, A.; Ramon-Pardo, P.; Javelle, E.; Simon, F.; Aldighieri, S.; Horvath, H.; Rodriguez-Abreu, J.; Reveiz, L. Interventions for treating patients with chikungunya virus infection-related rheumatic and musculoskeletal disorders: A systematic review. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [Green Version]
- Savarino, A.; Gennero, L.; Sperber, K.; Boelaert, J.R. The anti-HIV-1 activity of chloroquine. J. Clin. Virol. 2001, 20, 131–135. [Google Scholar] [CrossRef]
- Romanelli, F.; Smith, K.M.; Hoven, A.D. Chloroquine and hydroxychloroquine as inhibitors of human immunodeficiency virus (HIV-1) activity. Curr. Pharm. Des. 2004, 10, 2643–2648. [Google Scholar] [CrossRef]
- Bhushan, P.; Aggarwal, A.; Baliyan, V. Complete Clearance of Cutaneous Warts with Hydroxychloroquine: Antiviral Action? Indian J. Derm. 2014, 59. [Google Scholar] [CrossRef]
- Andreani, J.; Le Bideau, M.; Duflot, I.; Jardot, P.; Rolland, C.; Boxberger, M.; Wurtz, N.; Rolain, J.M.; Colson, P.; La Scola, B.; et al. In vitro testing of combined hydroxychloroquine and azithromycin on SARS-CoV-2 shows synergistic effect. Microb. Pathog. 2020, 145, 104228. [Google Scholar] [CrossRef]
- Meo, S.A.; Klonoff, D.C.; Akram, J. Efficacy of chloroquine and hydroxychloroquine in the treatment of COVID-19. Eur. Rev. Med. Pharm. Sci. 2020, 24, 4539–4547. [Google Scholar]
- Sarma, P.; Kaur, H.; Kumar, H.; Mahendru, D.; Avti, P.; Bhattacharyya, A.; Prajapat, M.; Shekhar, N.; Kumar, S.; Singh, R.; et al. Virological and clinical cure in COVID-19 patients treated with hydroxychloroquine: A systematic review and meta-analysis. J. Med. Virol. 2020. [Google Scholar] [CrossRef]
- Gentile, I.; Maraolo, A.E.; Piscitelli, P.; Colao, A. COVID-19: Time for Post-Exposure Prophylaxis? Int. J. Environ. Res. Public. Health 2020, 17, 3997. [Google Scholar] [CrossRef] [PubMed]
- Fantini, J.; Chahinian, H.; Yahi, N. Synergistic antiviral effect of hydroxychloroquine and azithromycin in combination against SARS-CoV-2: What molecular dynamics studies of virus-host interactions reveal. Int. J. Antimicrob. Agents 2020, 106020. [Google Scholar] [CrossRef] [PubMed]
- Quiros Roldan, E.; Biasiotto, G.; Magro, P.; Zanella, I. The possible mechanisms of action of 4-aminoquinolines (chloroquine/hydroxychloroquine) against Sars-Cov-2 infection (COVID-19): A role for iron homeostasis? Pharm. Res. 2020, 158, 104904. [Google Scholar] [CrossRef] [PubMed]
- Ostaszewski, M.; Mazein, A.; Gillespie, M.E.; Kuperstein, I.; Niarakis, A.; Hermjakob, H.; Pico, A.R.; Willighagen, E.L.; Evelo, C.T.; Hasenauer, J.; et al. COVID-19 Disease Map, building a computational repository of SARS-CoV-2 virus-host interaction mechanisms. Sci Data 2020, 7, 136. [Google Scholar] [CrossRef] [PubMed]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Coutard, B.; Valle, C.; de Lamballerie, X.; Canard, B.; Seidah, N.G.; Decroly, E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antivir. Res. 2020, 176, 104742. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar]
- Ma, Y.; Wu, L.; Shaw, N.; Gao, Y.; Wang, J.; Sun, Y.; Lou, Z.; Yan, L.; Zhang, R.; Rao, Z. Structural basis and functional analysis of the SARS coronavirus nsp14-nsp10 complex. Proc. Natl. Acad. Sci. USA 2015, 112, 9436–9441. [Google Scholar] [CrossRef] [Green Version]
- Schoeman, D.; Fielding, B.C. Coronavirus envelope protein: Current knowledge. Virol. J. 2019, 16, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viswanathan, T.; Arya, S.; Chan, S.H.; Qi, S.; Dai, N.; Hromas, R.A.; Park, J.G.; Oladunni, F.; Martinez-Sobrido, L.; Gupta, Y.K. Structural Basis of RNA Cap Modification by SARS-CoV-2 Coronavirus. Nat. Commun. 2020, 11, 3718. [Google Scholar] [CrossRef] [PubMed]
- Verdia-Baguena, C.; Nieto-Torres, J.L.; Alcaraz, A.; Dediego, M.L.; Enjuanes, L.; Aguilella, V.M. Analysis of SARS-CoV E protein ion channel activity by tuning the protein and lipid charge. Biochim. Biophys. Acta 2013, 1828, 2026–2031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeDiego, M.L.; Nieto-Torres, J.L.; Jimenez-Guardeno, J.M.; Regla-Nava, J.A.; Castano-Rodriguez, C.; Fernandez-Delgado, R.; Usera, F.; Enjuanes, L. Coronavirus virulence genes with main focus on SARS-CoV envelope gene. Virus Res. 2014, 194, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.R.; Lin, L.D.; Machamer, C.E. Identification of a Golgi complex-targeting signal in the cytoplasmic tail of the severe acute respiratory syndrome coronavirus envelope protein. J. Virol. 2011, 85, 5794–5803. [Google Scholar] [CrossRef] [Green Version]
- EA, J.A.; Jones, I.M. Membrane binding proteins of coronaviruses. Future Virol. 2019, 14, 275–286. [Google Scholar]
- Ruch, T.R.; Machamer, C.E. The coronavirus E protein: Assembly and beyond. Viruses 2012, 4, 363–382. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.X.; Yuan, Q.; Liao, Y. Coronavirus envelope protein: A small membrane protein with multiple functions. Cell. Mol. Life Sci. 2007, 64, 2043–2048. [Google Scholar] [CrossRef] [Green Version]
- Fung, T.S.; Liu, D.X. Post-translational modifications of coronavirus proteins: Roles and function. Future Virol. 2018, 13, 405–430. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Q.; Liao, Y.; Torres, J.; Tam, J.P.; Liu, D.X. Biochemical evidence for the presence of mixed membrane topologies of the severe acute respiratory syndrome coronavirus envelope protein expressed in mammalian cells. Febs Lett. 2006, 580, 3192–3200. [Google Scholar] [CrossRef] [Green Version]
- Nieva, J.L.; Madan, V.; Carrasco, L. Viroporins: Structure and biological functions. Nat. Rev. Microbiol. 2012, 10, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Castano-Rodriguez, C.; Honrubia, J.M.; Gutierrez-Alvarez, J.; DeDiego, M.L.; Nieto-Torres, J.L.; Jimenez-Guardeno, J.M.; Regla-Nava, J.A.; Fernandez-Delgado, R.; Verdia-Baguena, C.; Queralt-Martin, M.; et al. Role of Severe Acute Respiratory Syndrome Coronavirus Viroporins E, 3a, and 8a in Replication and Pathogenesis. mBio 2018, 9, e02325-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Y.; Hogue, B.G. Role of the coronavirus E viroporin protein transmembrane domain in virus assembly. J. Virol. 2007, 81, 3597–3607. [Google Scholar] [CrossRef] [Green Version]
- Nieto-Torres, J.L.; Verdia-Baguena, C.; Jimenez-Guardeno, J.M.; Regla-Nava, J.A.; Castano-Rodriguez, C.; Fernandez-Delgado, R.; Torres, J.; Aguilella, V.M.; Enjuanes, L. Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome. Virology 2015, 485, 330–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, Y.H.; Huang, Y.W.; Wu, Y.H.; Tsai, C.S.; Lin, Y.C.; Mo, S.T.; Kuo, W.C.; Chuang, Y.T.; Jiang, S.T.; Shih, H.M.; et al. Selective inhibition of the NLRP3 inflammasome by targeting to promyelocytic leukemia protein in mouse and human. Blood 2013, 121, 3185–3194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeDiego, M.L.; Alvarez, E.; Almazan, F.; Rejas, M.T.; Lamirande, E.; Roberts, A.; Shieh, W.J.; Zaki, S.R.; Subbarao, K.; Enjuanes, L. A severe acute respiratory syndrome coronavirus that lacks the E gene is attenuated in vitro and in vivo. J. Virol. 2007, 81, 1701–1713. [Google Scholar] [CrossRef] [Green Version]
- Teoh, K.T.; Siu, Y.L.; Chan, W.L.; Schluter, M.A.; Liu, C.J.; Peiris, J.S.; Bruzzone, R.; Margolis, B.; Nal, B. The SARS coronavirus E protein interacts with PALS1 and alters tight junction formation and epithelial morphogenesis. Mol. Biol. Cell 2010, 21, 3838–3852. [Google Scholar] [CrossRef] [Green Version]
- DeDiego, M.L.; Nieto-Torres, J.L.; Jimenez-Guardeno, J.M.; Regla-Nava, J.A.; Alvarez, E.; Oliveros, J.C.; Zhao, J.; Fett, C.; Perlman, S.; Enjuanes, L. Severe acute respiratory syndrome coronavirus envelope protein regulates cell stress response and apoptosis. PLoS Pathog. 2011, 7, e1002315. [Google Scholar] [CrossRef] [Green Version]
- Scott, C.; Griffin, S. Viroporins: Structure, function and potential as antiviral targets. J. Gen. Virol. 2015, 96, 2000–2027. [Google Scholar] [CrossRef]
- Surya, W.; Li, Y.; Torres, J. Structural model of the SARS coronavirus E channel in LMPG micelles. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1309–1317. [Google Scholar] [CrossRef]
- Ogando, N.S.; Ferron, F.; Decroly, E.; Canard, B.; Posthuma, C.C.; Snijder, E.J. The Curious Case of the Nidovirus Exoribonuclease: Its Role in RNA Synthesis and Replication Fidelity. Front. Microbiol. 2019, 10, 1813. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Cai, H.; Pan, J.; Xiang, N.; Tien, P.; Ahola, T.; Guo, D. Functional screen reveals SARS coronavirus nonstructural protein nsp14 as a novel cap N7 methyltransferase. Proc. Natl. Acad. Sci. USA 2009, 106, 3484–3489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subissi, L.; Posthuma, C.C.; Collet, A.; Zevenhoven-Dobbe, J.C.; Gorbalenya, A.E.; Decroly, E.; Snijder, E.J.; Canard, B.; Imbert, I. One severe acute respiratory syndrome coronavirus protein complex integrates processive RNA polymerase and exonuclease activities. Proc. Natl. Acad. Sci. USA 2014, 111, E3900–E3909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckerle, L.D.; Lu, X.; Sperry, S.M.; Choi, L.; Denison, M.R. High fidelity of murine hepatitis virus replication is decreased in nsp14 exoribonuclease mutants. J. Virol. 2007, 81, 12135–12144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckerle, L.D.; Becker, M.M.; Halpin, R.A.; Li, K.; Venter, E.; Lu, X.; Scherbakova, S.; Graham, R.L.; Baric, R.S.; Stockwell, T.B.; et al. Infidelity of SARS-CoV Nsp14-exonuclease mutant virus replication is revealed by complete genome sequencing. PLoS Pathog. 2010, 6, e1000896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denison, M.R.; Graham, R.L.; Donaldson, E.F.; Eckerle, L.D.; Baric, R.S. Coronaviruses: An RNA proofreading machine regulates replication fidelity and diversity. Rna Biol. 2011, 8, 270–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, A.; Le, N.T.; Selisko, B.; Eydoux, C.; Alvarez, K.; Guillemot, J.C.; Decroly, E.; Peersen, O.; Ferron, F.; Canard, B. Remdesivir and SARS-CoV-2: Structural requirements at both nsp12 RdRp and nsp14 Exonuclease active-sites. Antivir. Res. 2020, 178, 104793. [Google Scholar] [CrossRef]
- Neuman, B.W.; Chamberlain, P.; Bowden, F.; Joseph, J. Atlas of coronavirus replicase structure. Virus Res. 2014, 194, 49–66. [Google Scholar] [CrossRef]
- Snijder, E.J.; Decroly, E.; Ziebuhr, J. The Nonstructural Proteins Directing Coronavirus RNA Synthesis and Processing. Adv. Virus Res. 2016, 96, 59–126. [Google Scholar]
- Bouvet, M.; Debarnot, C.; Imbert, I.; Selisko, B.; Snijder, E.J.; Canard, B.; Decroly, E. In vitro reconstitution of SARS-coronavirus mRNA cap methylation. PLoS Pathog. 2010, 6, e1000863. [Google Scholar] [CrossRef]
- Bouvet, M.; Imbert, I.; Subissi, L.; Gluais, L.; Canard, B.; Decroly, E. RNA 3’-end mismatch excision by the severe acute respiratory syndrome coronavirus nonstructural protein nsp10/nsp14 exoribonuclease complex. Proc. Natl. Acad. Sci. USA 2012, 109, 9372–9377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouvet, M.; Lugari, A.; Posthuma, C.C.; Zevenhoven, J.C.; Bernard, S.; Betzi, S.; Imbert, I.; Canard, B.; Guillemot, J.C.; Lecine, P.; et al. Coronavirus Nsp10, a critical co-factor for activation of multiple replicative enzymes. J. Biol. Chem. 2014, 289, 25783–25796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lugari, A.; Betzi, S.; Decroly, E.; Bonnaud, E.; Hermant, A.; Guillemot, J.C.; Debarnot, C.; Borg, J.P.; Bouvet, M.; Canard, B.; et al. Molecular mapping of the RNA Cap 2’-O-methyltransferase activation interface between severe acute respiratory syndrome coronavirus nsp10 and nsp16. J. Biol. Chem. 2010, 285, 33230–33241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ke, M.; Chen, Y.; Wu, A.; Sun, Y.; Su, C.; Wu, H.; Jin, X.; Tao, J.; Wang, Y.; Ma, X.; et al. Short peptides derived from the interaction domain of SARS coronavirus nonstructural protein nsp10 can suppress the 2’-O-methyltransferase activity of nsp10/nsp16 complex. Virus Res. 2012, 167, 322–328. [Google Scholar] [CrossRef]
- Marcotrigiano, J.; Gingras, A.C.; Sonenberg, N.; Burley, S.K. Cocrystal structure of the messenger RNA 5’ cap-binding protein (eIF4E) bound to 7-methyl-GDP. Cell 1997, 89, 951–961. [Google Scholar] [CrossRef] [Green Version]
- Nallagatla, S.R.; Toroney, R.; Bevilacqua, P.C. A brilliant disguise for self RNA: 5’-end and internal modifications of primary transcripts suppress elements of innate immunity. Rna Biol. 2008, 5, 140–144. [Google Scholar] [CrossRef] [Green Version]
- Decroly, E.; Ferron, F.; Lescar, J.; Canard, B. Conventional and unconventional mechanisms for capping viral mRNA. Nat. Rev. Microbiol. 2011, 10, 51–65. [Google Scholar] [CrossRef]
- Yoshimoto, F.K. The Proteins of Severe Acute Respiratory Syndrome Coronavirus-2 (SARS CoV-2 or n-COV19), the Cause of COVID-19. Protein J. 2020, 39, 198–216. [Google Scholar]
- Decroly, E.; Imbert, I.; Coutard, B.; Bouvet, M.; Selisko, B.; Alvarez, K.; Gorbalenya, A.E.; Snijder, E.J.; Canard, B. Coronavirus nonstructural protein 16 is a cap-0 binding enzyme possessing (nucleoside-2’O)-methyltransferase activity. J. Virol. 2008, 82, 8071–8084. [Google Scholar] [CrossRef] [Green Version]
- Encinar, J.A.; Menendez, J.A. Potential Drugs Targeting Early Innate Immune Evasion of SARS-Coronavirus 2 via 2’-O-Methylation of Viral RNA. Viruses 2020, 12, 525. [Google Scholar] [CrossRef]
- Menachery, V.D.; Yount, B.L., Jr.; Josset, L.; Gralinski, L.E.; Scobey, T.; Agnihothram, S.; Katze, M.G.; Baric, R.S. Attenuation and restoration of severe acute respiratory syndrome coronavirus mutant lacking 2’-o-methyltransferase activity. J. Virol. 2014, 88, 4251–4264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods IV: Extension of MNDO, AM1, and PM3 to more main group elements. J. Mol. Model. 2004, 10, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Koraimann, G.; Vriend, G. Increasing the precision of comparative models with YASARA NOVA--a self-parameterizing force field. Proteins 2002, 47, 393–402. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. YASARA View—Molecular graphics for all devices—From smartphones to workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef] [Green Version]
- Floresta, G.; Patamia, V.; Gentile, D.; Molteni, F.; Santamato, A.; Rescifina, A.; Vecchio, M. Repurposing of FDA-Approved Drugs for Treating Iatrogenic Botulism: A Paired 3D-QSAR/Docking Approach. ChemMedChem 2020, 15, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Gentile, D.; Patamia, V.; Scala, A.; Sciortino, M.T.; Piperno, A.; Rescifina, A. Putative Inhibitors of SARS-CoV-2 Main Protease from A Library of Marine Natural Products: A Virtual Screening and Molecular Modeling Study. Mar. Drugs 2020, 18, 225. [Google Scholar] [CrossRef] [Green Version]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef]
- Krieger, E.; Dunbrack, R.L., Jr.; Hooft, R.W.; Krieger, B. Assignment of protonation states in proteins and ligands: Combining pKa prediction with hydrogen bonding network optimization. Methods Mol. Biol. 2012, 819, 405–421. [Google Scholar]
- Krieger, E.; Nielsen, J.E.; Spronk, C.A.; Vriend, G. Fast empirical pKa prediction by Ewald summation. J. Mol. Graph. Model. 2006, 25, 481–486. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.M.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef] [PubMed]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple amber force fields and development of improved protein backbone parameters. Proteins-Struct. Funct. Bioinform. 2006, 65, 712–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Krieger, E.; Vriend, G. New Ways to Boost Molecular Dynamics Simulations. J. Comput. Chem. 2015, 36, 996–1007. [Google Scholar] [CrossRef]
- Hooft, R.W.W.; Vriend, G.; Sander, C.; Abola, E.E. Errors in protein structures. Nature 1996, 381, 272. [Google Scholar] [CrossRef]
- Liu, J.; Cao, R.; Xu, M.; Wang, X.; Zhang, H.; Hu, H.; Li, Y.; Hu, Z.; Zhong, W.; Wang, M. Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov. 2020, 6, 16. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | N7-MTase (kcal/mol) | ExoN (kcal/mol) |
|---|---|---|
| CQ | −6.2 | −6.0 |
| HCQ | −7.0 | −7.3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gentile, D.; Fuochi, V.; Rescifina, A.; Furneri, P.M. New Anti SARS-Cov-2 Targets for Quinoline Derivatives Chloroquine and Hydroxychloroquine. Int. J. Mol. Sci. 2020, 21, 5856. https://doi.org/10.3390/ijms21165856
Gentile D, Fuochi V, Rescifina A, Furneri PM. New Anti SARS-Cov-2 Targets for Quinoline Derivatives Chloroquine and Hydroxychloroquine. International Journal of Molecular Sciences. 2020; 21(16):5856. https://doi.org/10.3390/ijms21165856
Chicago/Turabian StyleGentile, Davide, Virginia Fuochi, Antonio Rescifina, and Pio Maria Furneri. 2020. "New Anti SARS-Cov-2 Targets for Quinoline Derivatives Chloroquine and Hydroxychloroquine" International Journal of Molecular Sciences 21, no. 16: 5856. https://doi.org/10.3390/ijms21165856
APA StyleGentile, D., Fuochi, V., Rescifina, A., & Furneri, P. M. (2020). New Anti SARS-Cov-2 Targets for Quinoline Derivatives Chloroquine and Hydroxychloroquine. International Journal of Molecular Sciences, 21(16), 5856. https://doi.org/10.3390/ijms21165856