Anticancer Strategy Targeting Cell Death Regulators: Switching the Mechanism of Anticancer Floxuridine-Induced Cell Death from Necrosis to Apoptosis


Abstract
:
1. Introduction
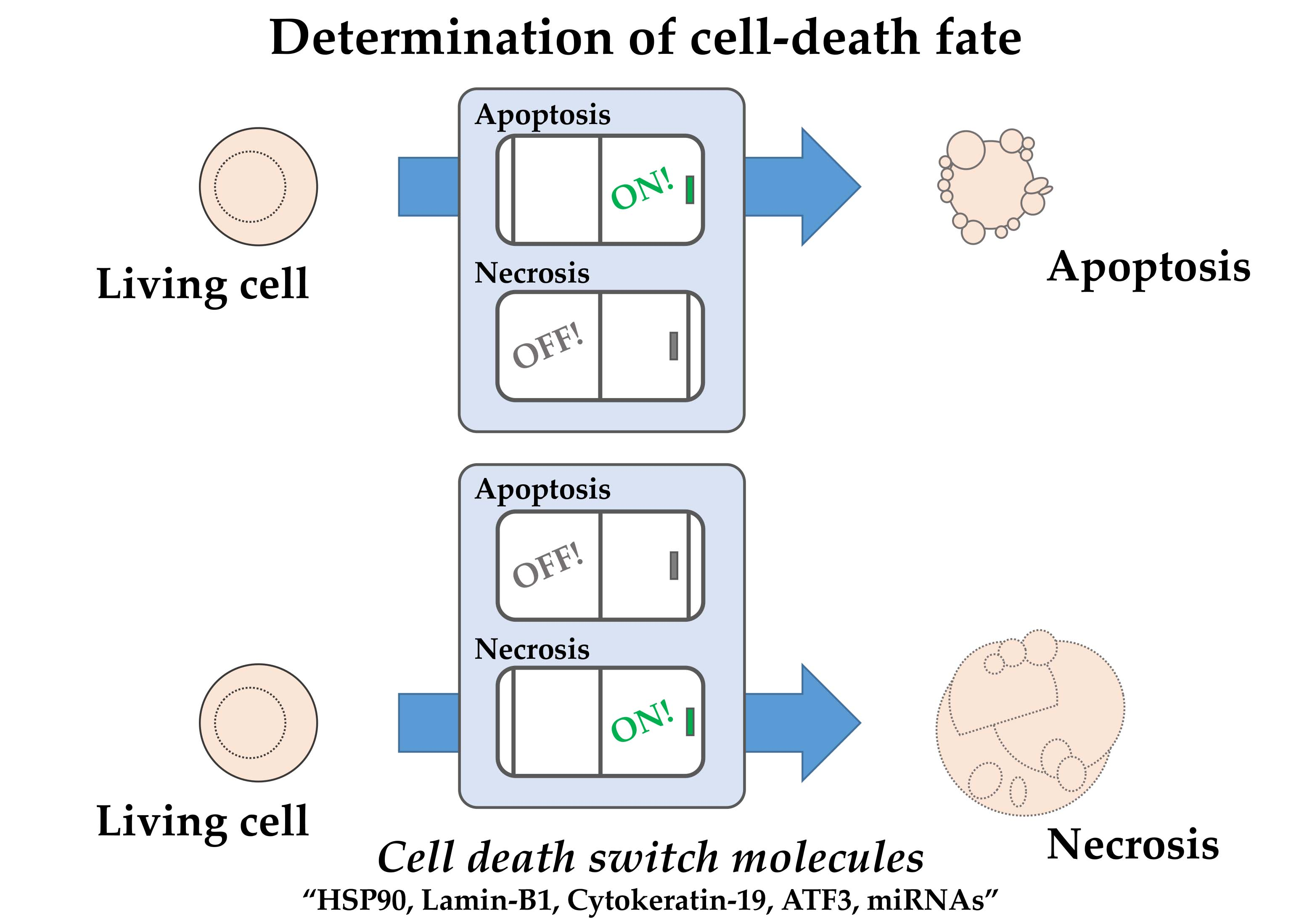
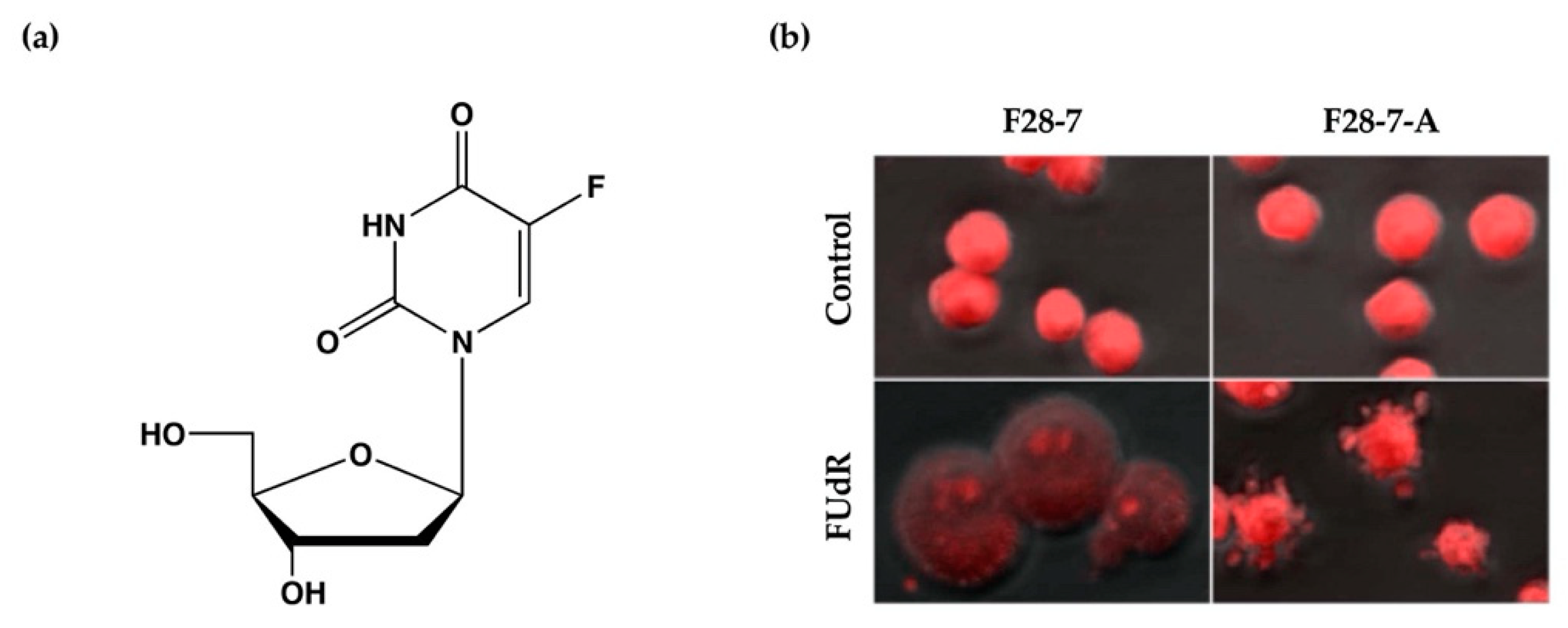
2. Characteristics of the Cell Death-Switching Model System
3. Functional Analysis of Candidate Molecular Switches Regulating Necrosis and Apoptosis
4. Anticancer Strategy Targeting Cell Death Regulators of Necrotic to Apoptotic Cell Death
5. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ATF3 | activating transcription factor 3 |
| FUdR | floxuridine |
| GA | geldanamycin |
| HSP | heat shock protein |
| miRNA | microRNA |
| miR | miRNA |
| siRNA | small interfering RNA |
| TNF | tumor necrosis factor |
References
- Nicotera, P.; Melino, G. Regulation of the apoptosis–necrosis switch. Oncogene 2004, 23, 2757–2765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2011, 19, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Pedro, J.M.B.-S.; Vitale, I.; A Aaronson, S.; Abrams, J.M.; Adam, D.; Alnemri, E.S.; Altucci, L.; Andrews, D.W.; Annicchiarico-Petruzzelli, M.; et al. Essential versus accessory aspects of cell death: Recommendations of the NCCD 2015. Cell Death Differ. 2014, 22, 58–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bredholt, G.; Mannelqvist, M.; Stefansson, I.M.; Birkeland, E.; Bø, T.H.; Øyan, A.M.; Trovik, J.; Kalland, K.-H.; Jonassen, I.; Salvesen, H.B.; et al. Tumor necrosis is an important hallmark of aggressive endometrial cancer and associates with hypoxia, angiogenesis and inflammation responses. Oncotarget 2015, 6, 39676–39691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vellayappan, B.; Tan, C.L.; Yong, C.; Khor, L.K.; Koh, W.Y.; Yeo, T.T.; Detsky, J.; Lo, S.; Sahgal, A. Diagnosis and Management of Radiation Necrosis in Patients With Brain Metastases. Front. Oncol. 2018, 8. [Google Scholar] [CrossRef]
- Karsch-Bluman, A.; Feiglin, A.; Arbib, E.; Stern, T.; Shoval, H.; Schwob, O.; Berger, M.; Benny, O. Tissue necrosis and its role in cancer progression. Oncogene 2018, 38, 1920–1935. [Google Scholar] [CrossRef]
- Kakutani, T.; Ebara, Y.; Kanja, K.; Hidaka, M.; Matsumoto, Y.; Nagano, A.; Wataya, Y. Different Modes of Cell Death Induced by 5-Fluoro-2′-deoxyuridine in Two Clones of the Mouse Mammary Tumor FM3A Cell Line. Biochem. Biophys. Res. Commun. 1998, 247, 773–779. [Google Scholar] [CrossRef]
- Santi, D.V.; Peña, V.A.; Lam, S.S. On the structure of the cofactor in the complex formed with thymidylate synthetase, 5,10-methylenetetrahydrofolate and 5-fluoro-2′-deoxyuridylate. Biochim. Biophys. Acta (BBA) Enzym. 1976, 438, 324–331. [Google Scholar] [CrossRef]
- Yoshioka, A.; Tanaka, S.; Hiraoka, O.; Koyama, Y.; Hirota, Y.; Ayusawa, D.; Seno, T.; Garrett, C.; Wataya, Y. Deoxyribonucleoside triphosphate imbalance. 5-Fluorodeoxyuridine-induced DNA double strand breaks in mouse FM3A cells and the mechanism of cell death. J. Boil. Chem. 1987, 262, 8235–8241. [Google Scholar]
- Kakutani, T.; Ebara, Y.; Kanja, K.; Takahashi, K.; Wataya, Y. Activation of c-junand c-fosGenes in dNTP Imbalance Cell Death Induced With 5-Fluoro-2′-Deoxyuridine in Mouse Mammary Tumor FM3A Cell Line. Nucleosides Nucleotides 1998, 17, 1299–1308. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Hiramoto, A.; Uchikubo, Y.; Miyazaki, E.; Satake, A.; Naito, T.; Hiraoka, O.; Miyake, T.; Kim, H.-S.; Wataya, Y. Gene expression profiles of necrosis and apoptosis induced by 5-fluoro-2′-deoxyuridine. Genomics 2008, 92, 9–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, A.; Nakama, K.; Watanabe, H.; Satake, A.; Yamamoto, A.; Omi, T.; Hiramoto, A.; Masutani, M.; Wataya, Y.; Kim, H.-S. Role of activating transcription factor 3 protein ATF3 in necrosis and apoptosis induced by 5-fluoro-2?-deoxyuridine. FEBS J. 2014, 281, 1892–1900. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Satake, A.; Hiramoto, A.; Wataya, Y.; Kim, H.-S. Protein Expression Profiles of Necrosis and Apoptosis Induced by 5-Fluoro-2′-deoxyuridine in Mouse Cancer Cells. J. Proteome Res. 2010, 9, 2329–2338. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Hiramoto, A.; Satake, A.; Miyazaki, E.; Naito, T.; Wataya, Y.; Kim, H.-S. Association of Nuclear Membrane Protein Lamin B1 with Necrosis and Apoptosis in Cell Death Induced by 5-Fluoro-2′-Deoxyuridine. Nucleosides Nucleotides Nucleic Acids 2008, 27, 433–438. [Google Scholar] [CrossRef]
- Hai, T.; Hartman, M.G. The molecular biology and nomenclature of the activating transcription factor/cAMP responsive element binding family of transcription factors: Activating transcription factor proteins and homeostasis. Gene 2001, 273, 1–11. [Google Scholar] [CrossRef]
- Thompson, M.R.; Xu, D.; Williams, B.R. ATF3 transcription factor and its emerging roles in immunity and cancer. J. Mol. Med. 2009, 87, 1053–1060. [Google Scholar] [CrossRef] [Green Version]
- Cohen, M.; Gruenbaum, Y.; Lee, K.K.; Wilson, K.L. Transcriptional repression, apoptosis, human disease and the functional evolution of the nuclear lamina. Trends Biochem. Sci. 2001, 26, 41–47. [Google Scholar] [CrossRef]
- Hutchison, C.J. Lamins: Building blocks or regulators of gene expression? Nat. Rev. Mol. Cell Boil. 2002, 3, 848–858. [Google Scholar] [CrossRef]
- Freund, A.; Laberge, R.-M.; DeMaria, M.; Campisi, J. Lamin B1 loss is a senescence-associated biomarker. Mol. Boil. Cell 2012, 23, 2066–2075. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-J.; Rheinwald, J.G. A new small (40 kd) keratin filament protein made by some cultured human squamous cell carcinomas. Cell 1981, 25, 627–635. [Google Scholar] [CrossRef]
- Moll, R.; Von Bassewitz, D.B.; Schulz, U.; Franke, W.W. An Unusual Type of Cytokeratin Filament in Cells of a Human Cloacogenic Carcinoma Derived from the Anorectal Transition Zone. Differentiation 1982, 22, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Ku, N.-O.; Omary, M.B. Effect of Mutation and Phosphorylation of Type I Keratins on Their Caspase-mediated Degradation. J. Boil. Chem. 2001, 276, 26792–26798. [Google Scholar] [CrossRef] [Green Version]
- Oshima, R.G. Apoptosis and keratin intermediate filaments. Cell Death Differ. 2002, 9, 486–492. [Google Scholar] [CrossRef] [Green Version]
- Kawahara, A.; Enari, M.; Talanian, R.V.; Wong, W.W.; Nagata, S. Fas-induced DNA fragmentation and proteolysis of nuclear proteins. Genes Cells 1998, 3, 297–306. [Google Scholar] [CrossRef]
- Panaretou, B.; Siligardi, G.; Meyer, P.; Maloney, A.; Sullivan, J.K.; Singh, S.; Millson, S.H.; Clarke, P.A.; Naaby-Hansen, S.; Stein, R.; et al. Activation of the ATPase Activity of Hsp90 by the Stress-Regulated Cochaperone Aha1. Mol. Cell 2002, 10, 1307–1318. [Google Scholar] [CrossRef] [Green Version]
- Lotz, G.P.; Lin, H.; Harst, A.; Obermann, W.M.J. Aha1 Binds to the Middle Domain of Hsp90, Contributes to Client Protein Activation, and Stimulates the ATPase Activity of the Molecular Chaperone. J. Boil. Chem. 2003, 278, 17228–17235. [Google Scholar] [CrossRef] [Green Version]
- Meyer, P. Structural basis for recruitment of the ATPase activator Aha1 to the Hsp90 chaperone machinery. EMBO J. 2004, 23, 1402–1410. [Google Scholar] [CrossRef] [Green Version]
- Siligardi, G.; Hu, B.; Panaretou, B.; Piper, P.W.; Pearl, L.H.; Prodromou, C. Co-chaperone Regulation of Conformational Switching in the Hsp90 ATPase Cycle. J. Boil. Chem. 2004, 279, 51989–51998. [Google Scholar] [CrossRef] [Green Version]
- Scheibel, T.; Buchner, J. The Hsp90 complex—A super-chaperone machine as a novel drug target. Biochem. Pharmacol. 1998, 56, 675–682. [Google Scholar] [CrossRef]
- Rutherford, S.L.; Lindquist, S. Hsp90 as a capacitor for morphological evolution. Nature 1998, 396, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.; Devin, A.; Miller, A.; Lin, Y.; Rodriguez, Y.; Neckers, L.; Liu, Z.-G. Disruption of Hsp90 Function Results in Degradation of the Death Domain Kinase, Receptor-interacting Protein (RIP), and Blockage of Tumor Necrosis Factor-induced Nuclear Factor-κB Activation. J. Boil. Chem. 2000, 275, 10519–10526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powers, M.V.; Workman, P. Targeting of multiple signalling pathways by heat shock protein 90 molecular chaperone inhibitors. Endocr.-Relat. Cancer 2006, 13, S125–S135. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Wang, E. Heat shock protein 90 suppresses tumor necrosis factor alpha induced apoptosis by preventing the cleavage of Bid in NIH3T3 fibroblasts. Cell. Signal. 2004, 16, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Berghe, T.V.; Van Loo, G.; Saelens, X.; Van Gurp, M.; Brouckaert, G.; Kalai, M.; Declercq, W.; Vandenabeele, P. Differential Signaling to Apoptotic and Necrotic Cell Death by Fas-associated Death Domain Protein FADD. J. Boil. Chem. 2003, 279, 7925–7933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoter, A.; El-Sabban, M.; Naim, H.Y. The HSP90 Family: Structure, Regulation, Function, and Implications in Health and Disease. Int. J. Mol. Sci. 2018, 19, 2560. [Google Scholar] [CrossRef] [Green Version]
- Grenert, J.P.; Johnson, B.D.; Toft, D.O. The Importance of ATP Binding and Hydrolysis by Hsp90 in Formation and Function of Protein Heterocomplexes. J. Boil. Chem. 1999, 274, 17525–17533. [Google Scholar] [CrossRef] [Green Version]
- Obermann, W.M.; Sondermann, H.; Russo, A.A.; Pavletich, N.P.; Hartl, F.U. In Vivo Function of Hsp90 Is Dependent on ATP Binding and ATP Hydrolysis. J. Cell Boil. 1998, 143, 901–910. [Google Scholar] [CrossRef] [Green Version]
- Panaretou, B.; Prodromou, C.; Roe, S.M.; O’Brien, R.; Ladbury, J.E.; Piper, P.W.; Pearl, L.H. ATP binding and hydrolysis are essential to the function of the Hsp90 molecular chaperone in vivo. EMBO J. 1998, 17, 4829–4836. [Google Scholar] [CrossRef] [Green Version]
- Connell, P.; Ballinger, C.A.; Jiang, J.; Wu, Y.; Thompson, L.J.; Höhfeld, J.; Patterson, C. The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nature 2000, 3, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Pearl, L.H.; Prodromou, C. Structure and Mechanism of the Hsp90 Molecular Chaperone Machinery. Annu. Rev. Biochem. 2006, 75, 271–294. [Google Scholar] [CrossRef] [PubMed]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Bartel, B. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Bartel, B. MicroRNAs: Target Recognition and Regulatory Functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [Green Version]
- Xu, P.; Guo, M.; Hay, B.A. MicroRNAs and the regulation of cell death. Trends Genet. 2004, 20, 617–624. [Google Scholar] [CrossRef]
- Lytle, J.R.; Yario, T.A.; Steitz, J.A. Target mRNAs are repressed as efficiently by microRNA-binding sites in the 5’ UTR as in the 3’ UTR. Proc. Natl. Acad. Sci. USA 2007, 104, 9667–9672. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.; Ajay, S.S.; Yook, J.I.; Kim, H.S.; Hong, S.H.; Kim, N.H.; Han, S.; Chinnaiyan, A.M.; Athey, B. New class of microRNA targets containing simultaneous 5’-UTR and 3’-UTR interaction sites. Genome Res. 2009, 19, 1175–1183. [Google Scholar] [CrossRef] [Green Version]
- Sato, A.; Omi, T.; Yamamoto, A.; Satake, A.; Hiramoto, A.; Masutani, M.; Tanuma, S.-I.; Wataya, Y.; Kim, H.-S. MicroRNA-351 Regulates Two-Types of Cell Death, Necrosis and Apoptosis, Induced by 5-fluoro-2’-deoxyuridine. PLoS ONE 2016, 11, e0153130. [Google Scholar] [CrossRef] [Green Version]
- Scaffidi, P.; Misteli, T.; Bianchi, M. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef]
- Sato, A.; Ogino, Y.; Shimotsuma, A.; Hiramoto, A.; Kim, H.-S.; Wataya, Y. Direct interaction analysis of microRNA-351-5p and nuclear scaffold lamin B1 mRNA by the cell-free in vitro mRNA/miRNA binding evaluation system. Nucleosides Nucleotides Nucleic Acids 2020, 39, 799–805. [Google Scholar] [CrossRef] [PubMed]
- Neckers, L.M.; Workman, P. Hsp90 molecular chaperone inhibitors: Are we there yet? Clin. Cancer Res. 2012, 18, 64–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatokoro, M.; Koga, F.; Yoshida, S.; Kihara, K. Heat shock protein 90 targeting therapy: State of the art and future perspective. EXCLI J. 2015, 14, 48–58. [Google Scholar] [PubMed]
- Shevtsov, M.; Multhoff, G.; Mikhaylova, E.R.; Shibata, A.; Ekimova, I.V.; Margulis, B.A. Combination of Anti-Cancer Drugs with Molecular Chaperone Inhibitors. Int. J. Mol. Sci. 2019, 20, 5284. [Google Scholar] [CrossRef] [Green Version]
- Roe, S.M.; Prodromou, C.; O’Brien, R.; Ladbury, J.E.; Piper, P.W.; Pearl, L.H. Structural Basis for Inhibition of the Hsp90 Molecular Chaperone by the Antitumor Antibiotics Radicicol and Geldanamycin. J. Med. Chem. 1999, 42, 260–266. [Google Scholar] [CrossRef]
- Kamal, A.; Thao, L.; Sensintaffar, J.; Zhang, L.; Boehm, M.F.; Fritz, L.C.; Burrows, F. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature 2003, 425, 407–410. [Google Scholar] [CrossRef]
- Berghe, T.V.; Kalai, M.; Van Loo, G.; Declercq, W.; Vandenabeele, P. Disruption of HSP90 Function Reverts Tumor Necrosis Factor-induced Necrosis to Apoptosis. J. Boil. Chem. 2002, 278, 5622–5629. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Xu, T.; Cao, Y.; Wang, H.; Li, L.; Chen, S.; Wang, X.; Shen, Z. A cytosolic heat shock protein 90 and cochaperone CDC37 complex is required for RIP3 activation during necroptosis. Proc. Natl. Acad. Sci. USA 2015, 112, 5017–5022. [Google Scholar] [CrossRef] [Green Version]
- Jacobsen, A.V.; Lowes, K.N.; Tanzer, M.C.; Lucet, I.S.; Hildebrand, J.M.; Petrie, E.J.; Van Delft, M.F.; Liu, Z.; A Conos, S.; Zhang, J.-G.; et al. HSP90 activity is required for MLKL oligomerisation and membrane translocation and the induction of necroptotic cell death. Cell Death Dis. 2016, 7, e2051. [Google Scholar] [CrossRef]
- Zhao, X.M.; Chen, Z.; Zhao, J.B.; Zhang, P.P.; Pu, Y.F.; Jiang, S.H.; Hou, J.J.; Cui, Y.M.; Jia, X.L.; Zhang, S.Q. Hsp90 modulates the stability of MLKL and is required for TNF-induced necroptosis. Cell Death Dis. 2016, 7, e2089. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | F28-7 Cells | F28-7-A Cells |
|---|---|---|
| FUdR (EC50, nM) | 1 | 1 |
| Cell death mode | Necrosis | Apoptosis |
| Cell death morphological features | Swelling (Cell and organelles) | Membrane blebbing, shrinking (Cell and organelles) |
| DNA fragmentation | Chromosome size (100–200 kbp) | Oligonucleosome size |
| Mitochondrial events | ||
| Membrane potential | Down | Down |
| Cytochrome c release | - | + |
| Cell death markers | ||
| Caspase-3 | Cleaved | Cleaved |
| PARP | Cleaved | Cleaved |
| Extracellular HMGB1 | + | − |
| Name | Expression | Cell Death | Experiments | Observation | Ref |
|---|---|---|---|---|---|
| HSP90 | NC | N > A | IH(GA) | MF, DL | [13] |
| Lamin-B1 | High F28-7 | N > A | KD(siR) | MF | [15,16] |
| Cytokeratin-19 | High F28-7 | N > A | KD(siR) | MF | [15] |
| ATF3 | High F28-7 | N > A | KD(siR) | MF | [14] |
| miR-351-5p | High F28-7-A | N > A/A > N | OE(miRm)/IH(miRi) | MF, HR | [49,51] |
| miR-743a-3p | High F28-7-A | N > A | OE(miRm) | MF | [49] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sato, A.; Hiramoto, A.; Kim, H.-S.; Wataya, Y. Anticancer Strategy Targeting Cell Death Regulators: Switching the Mechanism of Anticancer Floxuridine-Induced Cell Death from Necrosis to Apoptosis. Int. J. Mol. Sci. 2020, 21, 5876. https://doi.org/10.3390/ijms21165876
Sato A, Hiramoto A, Kim H-S, Wataya Y. Anticancer Strategy Targeting Cell Death Regulators: Switching the Mechanism of Anticancer Floxuridine-Induced Cell Death from Necrosis to Apoptosis. International Journal of Molecular Sciences. 2020; 21(16):5876. https://doi.org/10.3390/ijms21165876
Chicago/Turabian StyleSato, Akira, Akiko Hiramoto, Hye-Sook Kim, and Yusuke Wataya. 2020. "Anticancer Strategy Targeting Cell Death Regulators: Switching the Mechanism of Anticancer Floxuridine-Induced Cell Death from Necrosis to Apoptosis" International Journal of Molecular Sciences 21, no. 16: 5876. https://doi.org/10.3390/ijms21165876
APA StyleSato, A., Hiramoto, A., Kim, H. -S., & Wataya, Y. (2020). Anticancer Strategy Targeting Cell Death Regulators: Switching the Mechanism of Anticancer Floxuridine-Induced Cell Death from Necrosis to Apoptosis. International Journal of Molecular Sciences, 21(16), 5876. https://doi.org/10.3390/ijms21165876