R-Ras GTPases Signaling Role in Myelin Neurodegenerative Diseases



Abstract
:1. Introduction
1.1. Myelination and Oligodendrocytes
1.2. Myelin Neurodegenerative Diseases
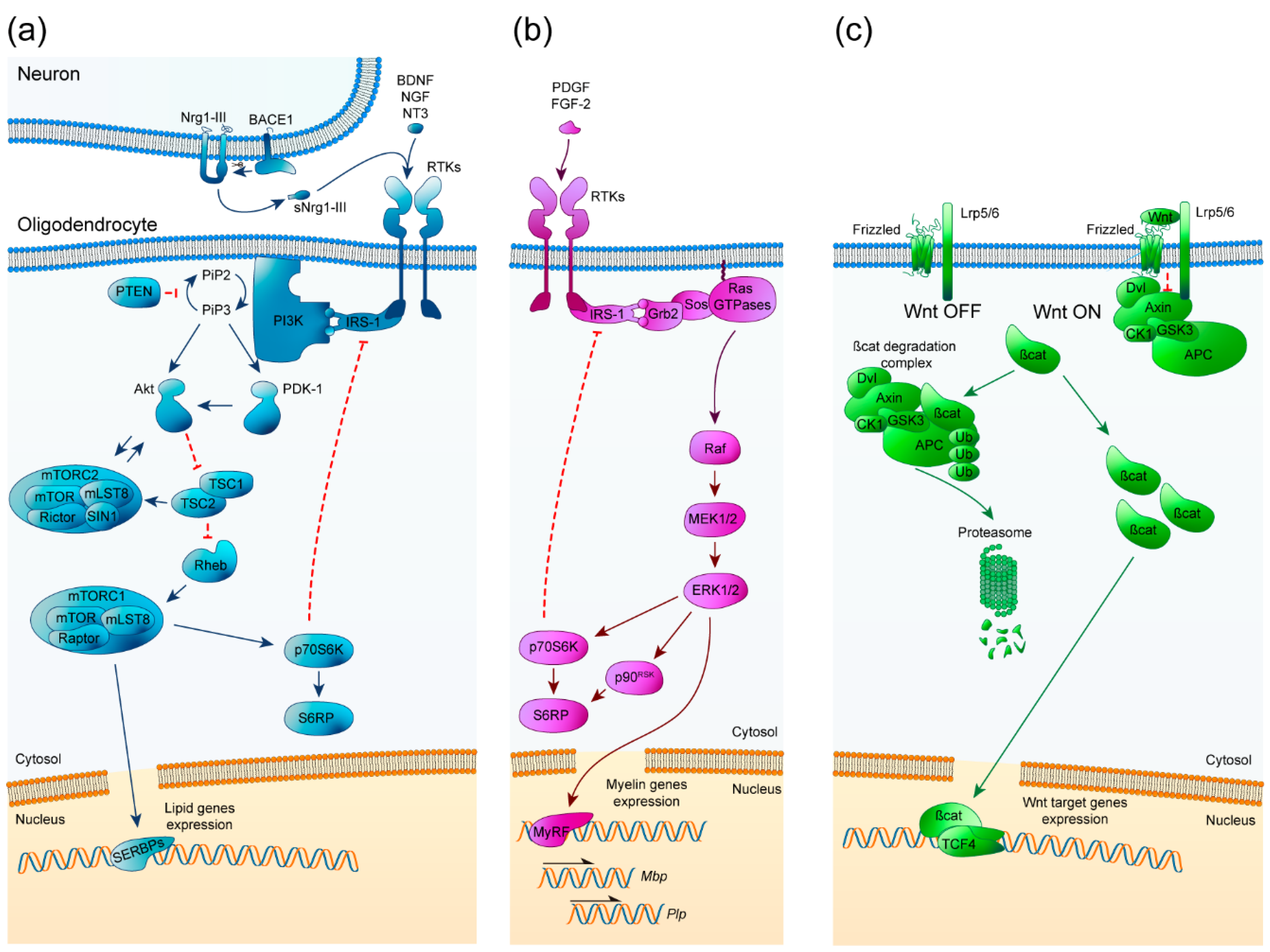
2. Cell Signaling Pathways Involved in Myelination
2.1. PI3K/Akt/mTOR
2.2. Erk1/2-MAPK
2.3. Wnt/β-Catenin
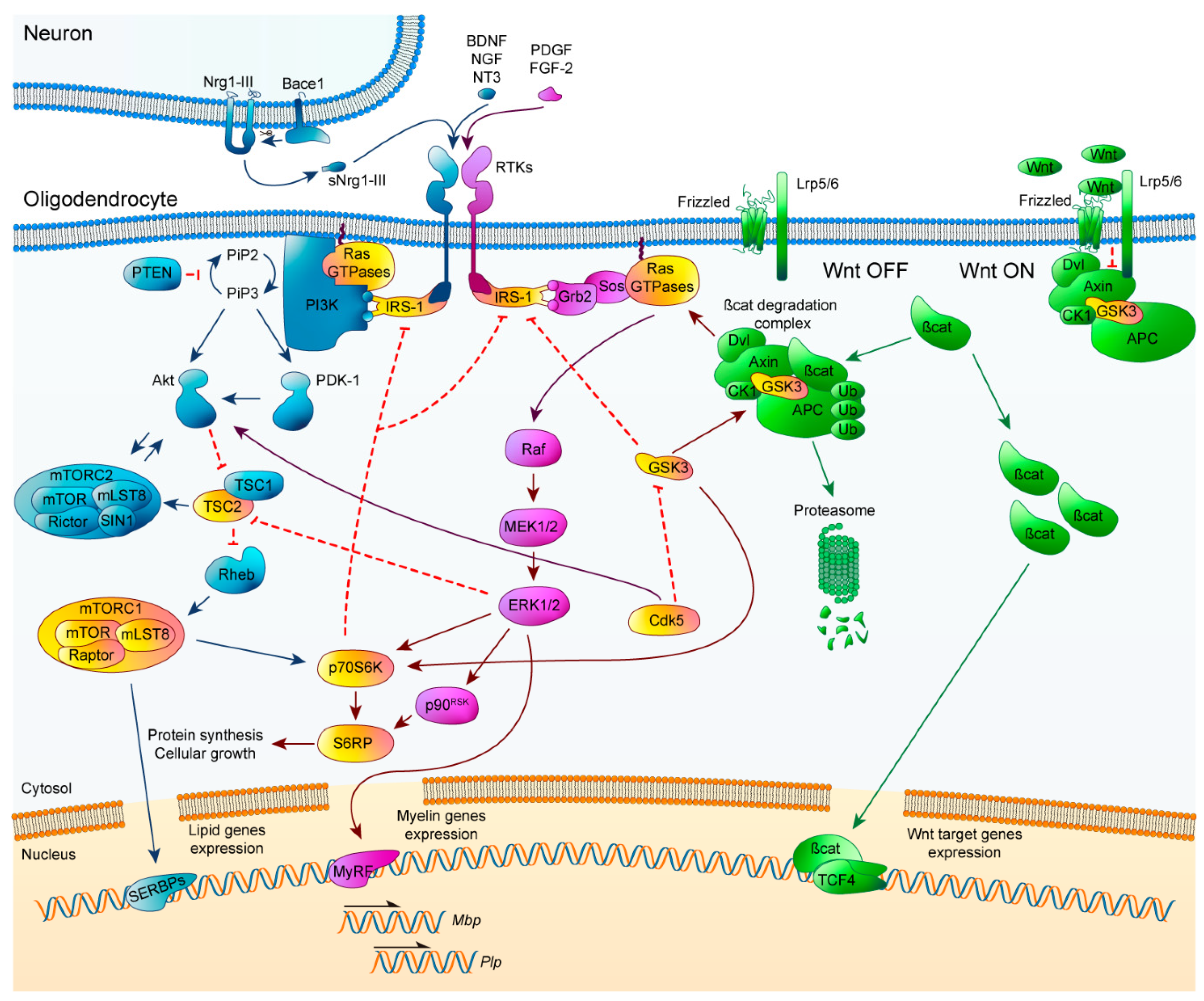
2.4. Crosstalk Signaling
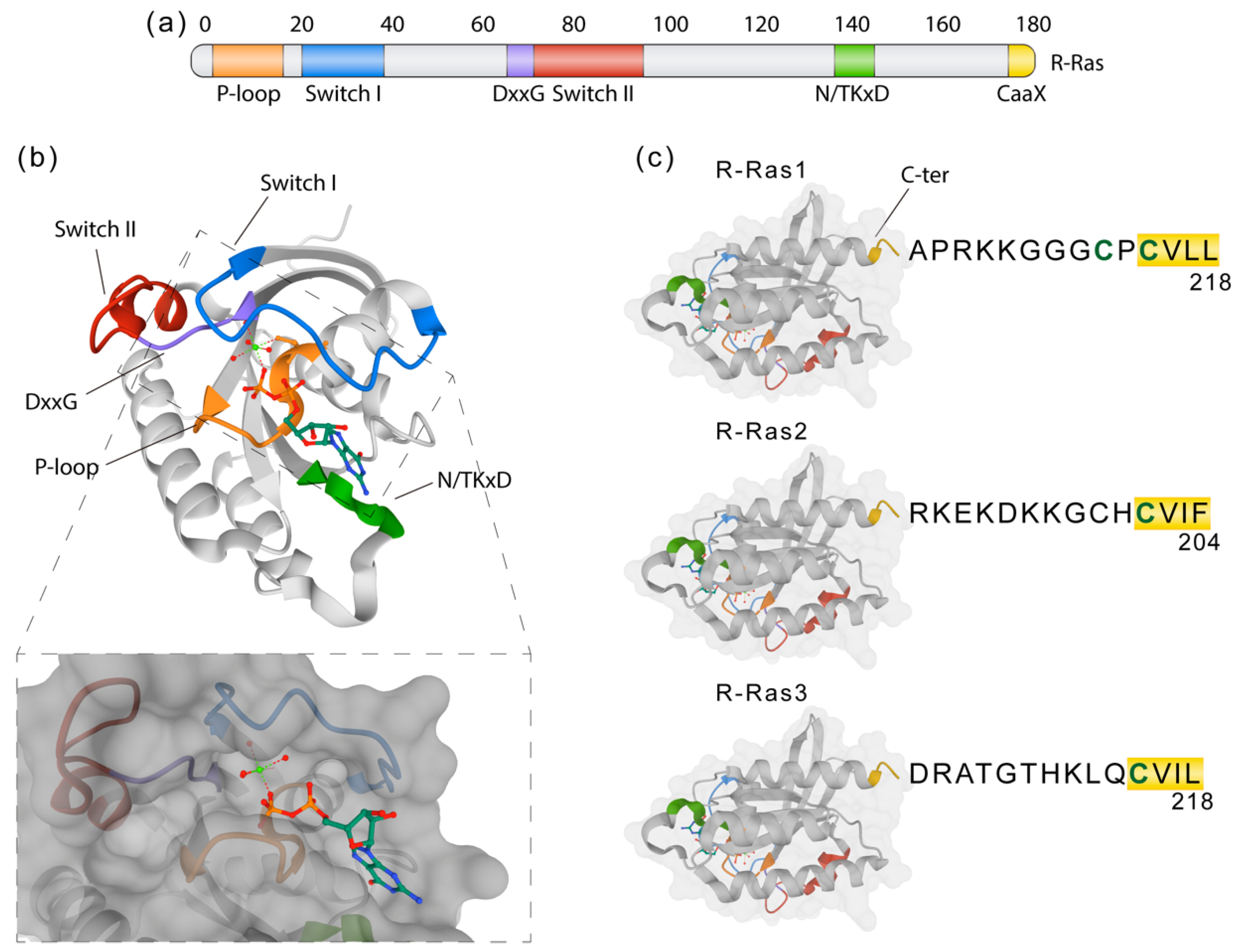
2.5. The Ras Family of GTPases and Their Role in Neurodegenerative Myelin Diseases
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bercury, K.K.; Macklin, W.B. Dynamics and mechanisms of CNS myelination. Dev. Cell 2015, 32, 447–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philips, T.; Rothstein, J.D.; Philips, T.; Rothstein, J.D. Oligodendroglia: Metabolic supporters of neurons. J. Clin. Investig. 2017, 127, 3271–3280. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, K.A.; Sheng, Z.-H. Mechanisms for the maintenance and regulation of axonal energy supply. J. Neurosci. Res. 2019, 97, 897–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, A.; Sherman, D.L.; Brophy, P.J. The Axonal Cytoskeleton and the Assembly of Nodes of Ranvier. Neuroscientist 2017, 24, 104–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lubetzki, C.; Sol-Foulon, N.; Desmazières, A. Nodes of Ranvier during development and repair in the CNS. Nat. Rev. Neurol. 2020, 1–14. [Google Scholar] [CrossRef]
- Nave, K.-A.; Werner, H.B. Myelination of the Nervous System: Mechanisms and Functions. Annu. Rev. Cell Dev. Boil. 2014, 30, 503–533. [Google Scholar] [CrossRef]
- Salzer, J.L.; Zalc, B. Myelination. Curr. Boil. 2016, 26, R971–R975. [Google Scholar] [CrossRef] [Green Version]
- Forbes, T.A.; Gallo, V. All Wrapped Up: Environmental Effects on Myelination. Trends Neurosci. 2017, 40, 572–587. [Google Scholar] [CrossRef]
- Goldman, S.A.; Kuypers, N.J. How to make an oligodendrocyte. Development 2015, 142, 3983–3995. [Google Scholar] [CrossRef] [Green Version]
- Foerster, S.; Hill, M.F.E.; Franklin, R.J.M. Diversity in the oligodendrocyte lineage: Plasticity or heterogeneity? Glia 2019, 67, 1797–1805. [Google Scholar] [CrossRef]
- E Bergles, D.; Richardson, W.D. Oligodendrocyte Development and Plasticity. Cold Spring Harb. Perspect. Boil. 2015, 8, a020453. [Google Scholar] [CrossRef] [PubMed]
- Marques, S.; Zeisel, A.; Codeluppi, S.; Van Bruggen, D.; Falcao, A.M.; Xiao, L.; Li, H.; Häring, M.; Hochgerner, H.; Romanov, R.A.; et al. Oligodendrocyte heterogeneity in the mouse juvenile and adult central nervous system. Science 2016, 352, 1326–1329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elbaz, B.; Popko, B. Molecular Control of Oligodendrocyte Development. Trends Neurosci. 2019, 42, 263–277. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Parpura, V.; Rodriguez-Arellano, J.J.; Zorec, R. Astroglia in Alzheimer’s Disease. Adv. Exp. Med. Biol. 2019, 1175, 273–324. [Google Scholar] [CrossRef] [PubMed]
- Marques, S.; Van Bruggen, D.; Vanichkina, D.P.; Floriddia, E.M.; Munguba, H.; Väremo, L.; Giacomello, S.; Falcao, A.M.; Meijer, M.; Björklund, Å.K.; et al. Transcriptional Convergence of Oligodendrocyte Lineage Progenitors during Development. Dev. Cell 2018, 46, 504–517.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barateiro, A.; Brites, D.; Fernandes, A. Oligodendrocyte Development and Myelination in Neurodevelopment: Molecular Mechanisms in Health and Disease. Curr. Pharm. Des. 2016, 22, 656–679. [Google Scholar] [CrossRef]
- Barateiro, A.; Fernandes, A. Temporal oligodendrocyte lineage progression: In vitro models of proliferation, differentiation and myelination. Biochim. Biophys. Acta (BBA) Bioenerg. 2014, 1843, 1917–1929. [Google Scholar] [CrossRef] [Green Version]
- Kirby, B.B.; Takada, N.; Latimer, A.J.; Shin, J.; Carney, T.; Kelsh, R.; Appel, B. In vivo time-lapse imaging shows dynamic oligodendrocyte progenitor behavior during zebrafish development. Nat. Neurosci. 2006, 9, 1506–1511. [Google Scholar] [CrossRef]
- Thornton, M.A.; Hughes, E.G. Neuron-oligodendroglia interactions: Activity-dependent regulation of cellular signaling. Neurosci. Lett. 2020, 727, 134916. [Google Scholar] [CrossRef]
- Snaidero, N.; Möbius, W.; Czopka, T.; Hekking, L.H.; Mathisen, C.; Verkleij, D.; Goebbels, S.; Edgar, J.M.; Merkler, D.; Lyons, D.A.; et al. Myelin membrane wrapping of CNS axons by PI(3,4,5)P3-dependent polarized growth at the inner tongue. Cell 2014, 156, 277–290. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.J.; Morrison, B.M.; Li, Y.; Lengacher, S.; Farah, M.H.; Hoffman, P.N.; Liu, Y.; Tsingalia, A.; Jin, L.; Zhang, P.-W.; et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 2012, 487, 443–448. [Google Scholar] [CrossRef]
- Saab, A.S.; Tzvetanova, I.D.; Nave, K.-A. The role of myelin and oligodendrocytes in axonal energy metabolism. Curr. Opin. Neurobiol. 2013, 23, 1065–1072. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Nave, K.-A. Oligodendrocytes: Myelination and Axonal Support. Cold Spring Harb. Perspect. Boil. 2015, 8, a020479. [Google Scholar] [CrossRef] [PubMed]
- Saab, A.S.; Tzvetavona, I.D.; Trevisiol, A.; Baltan, S.; Dibaj, P.; Kusch, K.; Möbius, W.; Goetze, B.; Jahn, H.M.; Huang, W.; et al. Oligodendroglial NMDA Receptors Regulate Glucose Import and Axonal Energy Metabolism. Neuron 2016, 91, 119–132. [Google Scholar] [CrossRef] [Green Version]
- Gibson, E.M.; Geraghty, A.C.; Monje, M. Bad wrap: Myelin and myelin plasticity in health and disease. Dev. Neurobiol. 2017, 78, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.; Reingold, S.C.; Cohen, J.A. International Panel on Diagnosis of Multiple Sclerosis Applying the 2017 McDonald diagnostic criteria for multiple sclerosis—Authors’ reply. Lancet Neurol. 2018, 17, 499–500. [Google Scholar] [CrossRef]
- Dobson, R.; Giovannoni, G. Multiple sclerosis—A review. Eur. J. Neurol. 2019, 26, 27–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghasemi, N.; Razavi, S.; Nikzad, E. Multiple Sclerosis: Pathogenesis, Symptoms, Diagnoses and Cell-Based Therapy Citation: Ghasemi N, Razavi Sh, Nikzad E. Multiple sclerosis: Pathogenesis, symptoms, diagnoses and cell-based therapy. Cell J. 2017, 19, 1–10. [Google Scholar] [PubMed]
- Nelson, M.J.; Miller, S.L.; McLain, L.W.; Gold, L.H.A. Multiple Sclerosis. J. Comput. Assist. Tomogr. 1981, 5, 892–894. [Google Scholar] [CrossRef]
- Faissner, S.; Plemel, J.R.; Gold, R.; Yong, V.W. Progressive multiple sclerosis: From pathophysiology to therapeutic strategies. Nat. Rev. Drug Discov. 2019, 18, 905–922. [Google Scholar] [CrossRef]
- Jasiak-Zatonska, M.; Kalinowska-Lyszczarz, A.; Michalak, S.; Kozubski, W. The Immunology of Neuromyelitis Optica—Current Knowledge, Clinical Implications, Controversies and Future Perspectives. Int. J. Mol. Sci. 2016, 17, 273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houzen, H.; Kondo, K.; Niino, M.; Horiuchi, K.; Takahashi, T.; Nakashima, I.; Tanaka, K. Prevalence and clinical features of neuromyelitis optica spectrum disorders in northern Japan. Neuroimaging Clin. 2017, 89, 1995–2001. [Google Scholar] [CrossRef] [PubMed]
- Stadelmann, C.; Timmler, S.; Barrantes-Freer, A.; Simons, M. Myelin in the Central Nervous System: Structure, Function, and Pathology. Physiol. Rev. 2019, 99, 1381–1431. [Google Scholar] [CrossRef] [PubMed]
- Morena, J.; Gupta, A.; Hoyle, J.C. Charcot-Marie-Tooth: From Molecules to Therapy. Int. J. Mol. Sci. 2019, 20, 3419. [Google Scholar] [CrossRef] [Green Version]
- Ishii, A.; Furusho, M.; Macklin, W.; Bansal, R. Independent and cooperative roles of the Mek/ERK1/2-MAPK and PI3K/Akt/mTOR pathways during developmental myelination and in adulthood. Glia 2019, 67, 1277–1295. [Google Scholar] [CrossRef]
- Gaesser, J.M.; Fyffe-Maricich, S.L. Intracellular signaling pathway regulation of myelination and remyelination in the CNS. Exp. Neurol. 2016, 283, 501–511. [Google Scholar] [CrossRef] [Green Version]
- Figlia, G.; Gerber, D.; Suter, U. Myelination and mTOR. Glia 2017, 66, 693–707. [Google Scholar] [CrossRef] [Green Version]
- Birchmeier, C.; Bennett, D.L. Neuregulin/ErbB Signaling in Developmental Myelin Formation and Nerve Repair. Curr. Topics Dev. Biol. 2016, 116, 45–64. [Google Scholar]
- Hu, X.; Hicks, C.W.; He, W.; Wong, P.; Macklin, W.B.; Trapp, B.D.; Yan, R. Bace1 modulates myelination in the central and peripheral nervous system. Nat. Neurosci. 2006, 9, 1520–1525. [Google Scholar] [CrossRef]
- Kataria, H.; Alizadeh, A.; Shahriary, G.M.; Rizi, S.S.; Henrie, R.; Santhosh, K.T.; Thliveris, J.A.; Karimi-Abdolrezaee, S. Neuregulin-1 promotes remyelination and fosters a pro-regenerative inflammatory response in focal demyelinating lesions of the spinal cord. Glia 2017, 66, 538–561. [Google Scholar] [CrossRef]
- Kataria, H.; Alizadeh, A.; Karimi-Abdolrezaee, S. Neuregulin-1/ErbB network: An emerging modulator of nervous system injury and repair. Prog. Neurobiol. 2019, 180, 101643. [Google Scholar] [CrossRef] [PubMed]
- Flores, A.I.; Narayanan, S.P.; Morse, E.N.; Shick, H.E.; Yin, X.; Kidd, G.; Avila, R.L.; Kirschner, D.; Macklin, W.B. Constitutively active Akt induces enhanced myelination in the CNS. J. Neurosci. 2008, 28, 7174–7183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goebbels, S.; Oltrogge, J.H.; Kemper, R.; Heilmann, I.; Bormuth, I.; Wolfer, S.; Wichert, S.P.; Möbius, W.; Liu, X.; Lappe-Siefke, C.; et al. Elevated Phosphatidylinositol 3,4,5-Trisphosphate in Glia Triggers Cell-Autonomous Membrane Wrapping and Myelination. J. Neurosci. 2010, 30, 8953–8964. [Google Scholar] [CrossRef] [PubMed]
- Ba, E.P.H.; Zhao, C.; Fancy, S.P.J.; Kaing, S.; Franklin, R.J.M.; Rowitch, D.H.; Dvm, S.P.J.F.; Franklin, R.J.M.; Rowitch, D.H. Oligodendrocyte PTEN is required for myelin and axonal integrity, not remyelination. Ann. Neurol. 2010, 68, 703–716. [Google Scholar] [CrossRef] [Green Version]
- Narayanan, S.P.; Flores, A.I.; Wang, F.; Macklin, W.B. Akt signals through the mammalian target of rapamycin pathway to regulate CNS myelination. J. Neurosci. 2009, 29, 6860–6870. [Google Scholar] [CrossRef]
- Bercury, K.K.; Dai, J.; Sachs, H.H.; Ahrendsen, J.T.; Wood, T.L.; Macklin, W.B. Conditional ablation of raptor or rictor has differential impact on oligodendrocyte differentiation and CNS myelination. J. Neurosci. 2014, 34, 4466–4480. [Google Scholar] [CrossRef] [Green Version]
- Lebrun-Julien, F.; Bachmann, L.; Norrmén, C.; Trötzmüller, M.; Köfeler, H.; Rüegg, M.A.; Hall, M.N.; Suter, U. Balanced mTORC1 Activity in Oligodendrocytes Is Required for Accurate CNS Myelination. J. Neurosci. 2014, 34, 8432–8448. [Google Scholar] [CrossRef] [Green Version]
- McLane, L.E.; Bourne, J.N.; Evangelou, A.V.; Khandker, L.; Macklin, W.B.; Wood, T.L. Loss of Tuberous Sclerosis Complex1 in Adult Oligodendrocyte Progenitor Cells Enhances Axon Remyelination and Increases Myelin Thickness after a Focal Demyelination. J. Neurosci. 2017, 37, 7534–7546. [Google Scholar] [CrossRef]
- Shi, Q.; Saifetiarova, J.; Taylor, A.M.; Bhat, M.A. mTORC1 Activation by Loss of Tsc1 in Myelinating Glia Causes Downregulation of Quaking and Neurofascin 155 Leading to Paranodal Domain Disorganization. Front. Cell. Neurosci. 2018, 12, 1–17. [Google Scholar] [CrossRef]
- Zou, Y.; Jiang, W.; Wang, J.; Li, Z.; Zhang, J.; Bu, J.; Zou, J.; Zhou, L.; Yu, S.; Cui, Y.; et al. Oligodendrocyte precursor cell-intrinsic effect of Rheb1 controls differentiation and mediates mTORC1-dependent myelination in brain. J. Neurosci. 2014, 34, 15764–15778. [Google Scholar] [CrossRef] [Green Version]
- Musah, A.S.; Brown, T.L.; Jeffries, M.A.; Shang, Q.; Hashimoto, H.; Evangelou, A.V.; Kowalski, A.; Batish, M.; Macklin, W.B.; Wood, T.L. Mechanistic Target of Rapamycin Regulates the Oligodendrocyte Cytoskeleton during Myelination. J. Neurosci. 2020, 40, 2993–3007. [Google Scholar] [CrossRef] [PubMed]
- Gonsalvez, D.; Ferner, A.H.; Peckham, H.; Murray, S.S.; Xiao, J. The roles of extracellular related-kinases 1 and 2 signaling in CNS myelination. Neuropharmacology 2016, 110, 586–593. [Google Scholar] [CrossRef] [PubMed]
- Ishii, A.; Fyffe-Maricich, S.L.; Furusho, M.; Miller, R.H.; Bansal, R. ERK1/ERK2 MAPK signaling is required to increase myelin thickness independent of oligodendrocyte differentiation and initiation of myelination. J. Neurosci. 2012, 32, 8855–8864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, A.; Furusho, M.; Dupree, J.L.; Bansal, R. Role of ERK1/2 MAPK signaling in the maintenance of myelin and axonal integrity in the adult CNS. J. Neurosci. 2014, 34, 16031–16045. [Google Scholar] [CrossRef] [PubMed]
- Fyffe-Maricich, S.L.; Karlo, J.C.; Landreth, G.E.; Miller, R.H. The ERK2 mitogen-activated protein kinase regulates the timing of oligodendrocyte differentiation. J. Neurosci. 2011, 31, 843–850. [Google Scholar] [CrossRef] [Green Version]
- Fyffe-Maricich, S.L.; Schott, A.; Karl, M.; Krasno, J.; Miller, R.H. Signaling through ERK1/2 controls myelin thickness during myelin repair in the adult central nervous system. J. Neurosci. 2013, 33, 18402–18408. [Google Scholar] [CrossRef] [Green Version]
- Ishii, A.; Furusho, M.; Bansal, R. Sustained activation of ERK1/2 MAPK in oligodendrocytes and schwann cells enhances myelin growth and stimulates oligodendrocyte progenitor expansion. J. Neurosci. 2013, 33, 175–186. [Google Scholar] [CrossRef] [Green Version]
- Furusho, M.; DuPree, J.L.; Nave, K.-A.; Bansal, R. Fibroblast growth factor receptor signaling in oligodendrocytes regulates myelin sheath thickness. J. Neurosci. 2012, 32, 6631–6641. [Google Scholar] [CrossRef] [Green Version]
- Guardiola-Diaz, H.M.; Ishii, A.; Bansal, R. Erk1/2 MAPK and mTOR signaling sequentially regulates progression through distinct stages of oligodendrocyte differentiation. Glia 2012, 60, 476–486. [Google Scholar] [CrossRef] [Green Version]
- Wong, A.W.; Xiao, J.; Kemper, D.; Kilpatrick, T.J.; Murray, S.S. Oligodendroglial Expression of TrkB Independently Regulates Myelination and Progenitor Cell Proliferation. J. Neurosci. 2013, 33, 4947–4957. [Google Scholar] [CrossRef] [Green Version]
- Ishii, A.; Furusho, M.; Dupree, J.L.; Bansal, R. Strength of ERK1/2 MAPK activation determines its effect on myelin and axonal integrity in the adult CNS. J. Neurosci. 2016, 36, 6471–6487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suo, N.; Guo, Y.-E.; He, B.; Gu, H.; Xie, X. Inhibition of MAPK/ERK pathway promotes oligodendrocytes generation and recovery of demyelinating diseases. Glia 2019, 67, 1320–1332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, F.; Lang, J.; Sohn, J.; Hammond, E.; Chang, M.; Pleasure, D.E. Canonical Wnt signaling in the oligodendroglial lineage-puzzles remain. Glia 2015, 63, 1671–1693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fancy, S.P.; Baranzini, S.E.; Zhao, C.; Yuk, D.-I.; Irvine, K.-A.; Kaing, S.; Sanai, N.; Franklin, R.J.M.; Rowitch, D.H. Dysregulation of the Wnt pathway inhibits timely myelination and remyelination in the mammalian CNS. Genes Dev. 2009, 23, 1571–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feigenson, K.; Reid, M.; See, J.; Crenshaw, E.B.; Grinspan, J.B.; Iii, E.B.C. Wnt signaling is sufficient to perturb oligodendrocyte maturation. Mol. Cell. Neurosci. 2009, 42, 255–265. [Google Scholar] [CrossRef]
- Azim, K.; Butt, A.M. GSK3? negatively regulates oligodendrocyte differentiation and myelination in vivo. Glia 2011, 59, 540–553. [Google Scholar] [CrossRef]
- Feigenson, K.; Reid, M.; See, J.; Crenshaw, E.B.; Grinspan, J.B.; Crenshaw, I.E.B.; Iii, E.B.C. Canonical Wnt Signalling Requires the BMP Pathway to Inhibit Oligodendrocyte Maturation. ASN Neuro 2011, 3, AN20110004. [Google Scholar] [CrossRef] [Green Version]
- Kalani, M.Y.S.; Cheshier, S.H.; Cord, B.J.; Bababeygy, S.R.; Vogel, H.; Weissman, I.L.; Palmer, T.D.; Nusse, R. Wnt-mediated self-renewal of neural stem/progenitor cells. Proc. Natl. Acad. Sci. USA 2008, 105, 16970–16975. [Google Scholar] [CrossRef] [Green Version]
- Ortega, F.; Gascón, S.; Masserdotti, G.; Deshpande, A.; Simon, C.; Fischer, J.; Dimou, L.; Lie, D.C.; Schroeder, T.; Berninger, B. Oligodendrogliogenic and neurogenic adult subependymal zone neural stem cells constitute distinct lineages and exhibit differential responsiveness to Wnt signalling. Nat. Cell Biol. 2013, 15, 602–613. [Google Scholar] [CrossRef] [Green Version]
- Tawk, M.; Makoukji, J.; Belle, M.; Fonte, C.; Trousson, A.; Hawkins, T.A.; Li, H.; Ghandour, S.; Schumacher, M.; Massaad, C. Wnt/β-Catenin Signaling Is an Essential and Direct Driver of Myelin Gene Expression and Myelinogenesis. J. Neurosci. 2011, 31, 3729–3742. [Google Scholar] [CrossRef]
- Lang, J.; Maeda, Y.; Bannerman, P.; Xu, J.; Horiuchi, M.; Pleasure, D.E.; Guo, F. Adenomatous polyposis coli regulates oligodendroglial development. J. Neurosci. 2013, 33, 3113–3130. [Google Scholar] [CrossRef] [PubMed]
- Fancy, S.P.; Harrington, E.P.; Yuen, T.J.; Silbereis, J.C.; Zhao, C.; Baranzini, S.E.; Bruce, C.C.; Otero, J.J.; Huang, E.J.; Nusse, R.; et al. Axin2 as regulatory and therapeutic target in newborn brain injury and remyelination. Nat. Neurosci. 2011, 14, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Cai, J.; Clevers, H.; Fast, E.; Gray, S.; Greenberg, R.; Jain, M.K.; Ma, Q.; Qiu, M.; Rowitch, D.H.; et al. A genome-wide screen for spatially restricted expression patterns identifies transcription factors that regulate glial development. J. Neurosci. 2009, 29, 11399–11408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, H.; Kesari, S.; Cai, J. Tcf7l2 is tightly controlled during myelin formation. Cell. Mol. Neurobiol. 2011, 32, 345–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lürbke, A.; Hagemeier, K.; Cui, Q.-L.; Metz, I.; Bruck, W.; Antel, J.; Kuhlmann, T. Limited TCF7L2 Expression in MS Lesions. PLoS ONE 2013, 8, e72822. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Chen, Y.; Hoang, T.; Montgomery, R.L.; Zhao, X.-H.; Bu, H.; Hu, T.; Taketo, M.M.; Van Es, J.H.; Clevers, H.; et al. HDAC1 and HDAC2 regulate oligodendrocyte differentiation by disrupting the β-catenin–TCF interaction. Nat. Neurosci. 2009, 12, 829–838. [Google Scholar] [CrossRef] [Green Version]
- Weng, C.; Ding, M.; Fan, S.; Cao, Q.; Lu, Z. Transcription factor 7 like 2 promotes oligodendrocyte differentiation and remyelination. Mol. Med. Rep. 2017, 16, 1864–1870. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Deng, Y.; Liu, L.; Yu, K.; Zhang, L.; Wang, H.; He, X.; Wang, J.; Lu, C.; Wu, L.N.; et al. Dual regulatory switch through interactions of Tcf7l2/Tcf4 with stage-specific partners propels oligodendroglial maturation. Nat. Commun. 2016, 7, 10883. [Google Scholar] [CrossRef] [Green Version]
- Xiao, J.; Ferner, A.H.; Wong, A.W.; Denham, M.; Kilpatrick, T.J.; Murray, S.S. Extracellular signal-regulated kinase 1/2 signaling promotes oligodendrocyte myelination in vitro. J. Neurochem. 2012, 122, 1167–1180. [Google Scholar] [CrossRef]
- Dai, J.; Bercury, K.K.; Macklin, W.B. Interaction of mTOR and Erk1/2 signaling to regulate oligodendrocyte differentiation. Glia 2014, 62, 2096–2109. [Google Scholar] [CrossRef]
- Ma, L.; Chen, Z.; Erdjument-Bromage, H.; Tempst, P.; Pandolfi, P.P. Phosphorylation and Functional Inactivation of TSC2 by Erk. Cell 2005, 121, 179–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michel, K.; Zhao, T.; Karl, M.; Lewis, K.; Fyffe-Maricich, S.L. Translational Control of Myelin Basic Protein Expression by ERK2 MAP Kinase Regulates Timely Remyelination in the Adult Brain. J. Neurosci. 2015, 35, 7850–7865. [Google Scholar] [CrossRef] [PubMed]
- Furusho, M.; Ishii, A.; Bansal, R. Signaling by FGF receptor 2, not FGF receptor 1, regulates myelin thickness through activation of ERK1/2–MAPK, which promotes mTORC1 activity in an Akt-independent manner. J. Neurosci. 2017, 37, 2931–2946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyler, W.A.; Gangoli, N.; Gokina, P.; Kim, H.A.; Covey, M.; Levison, S.W.; Wood, T.L. Activation of the Mammalian Target of Rapamycin (mTOR) is Essential for Oligodendrocyte Differentiation. J. Neurosci. 2009, 29, 6367–6378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, F.; Zhang, J.; Burke, K.; Miller, R.H.; Yang, Y. The Activators of Cyclin-Dependent Kinase 5 p35 and p39 Are Essential for Oligodendrocyte Maturation, Process Formation, and Myelination. J. Neurosci. 2016, 36, 3024–3037. [Google Scholar] [CrossRef] [PubMed]
- Luo, F.; Burke, K.; Kantor, C.; Miller, R.H.; Yang, Y. Cyclin-dependent kinase 5 mediates adult OPC maturation and myelin repair through modulation of Akt and GsK-3β signaling. J. Neurosci. 2014, 34, 10415–10429. [Google Scholar] [CrossRef]
- Luo, F.; Zhang, J.; Burke, K.; Romito-DiGiacomo, R.R.; Miller, R.H.; Yang, Y. Oligodendrocyte-specific loss of Cdk5 disrupts the architecture of nodes of Ranvier as well as learning and memory. Exp. Neurol. 2018, 306, 92–104. [Google Scholar] [CrossRef]
- Mccubrey, J.A.; Steelman, L.S.; Bertrand, F.E.; Davis, N.M.; Sokolosky, M.; Abrams, S.L. GSK-3 as potential target for therapeutic irvention in cancer. Oncotarget 2014, 5, 2881–2911. [Google Scholar] [CrossRef] [Green Version]
- Ding, Q.; Xia, W.; Liu, J.-C.; Yang, J.-Y.; Lee, D.-F.; Xia, J.; Bartholomeusz, G.; Li, Y.; Pan, Y.; Li, Z.; et al. Erk Associates with and Primes GSK-3β for Its Inactivation Resulting in Upregulation of β-Catenin. Mol. Cell 2005, 19, 159–170. [Google Scholar] [CrossRef]
- Shin, S.; Wolgamott, L.; Yu, Y.; Blenis, J.; Yoon, S.O. Glycogen synthase kinase (GSK)-3 promotes p70 ribosomal protein S6 kinase (p70S6K) activity and cell proliferation. Proc. Natl. Acad. Sci. USA 2011. [Google Scholar] [CrossRef] [Green Version]
- McCubrey, J.A.; Rakus, D.; Gizak, A.; Steelman, L.S.; Abrams, S.L.; Lertpiriyapong, K.; Fitzgerald, T.L.; Yang, L.V.; Montalto, G.; Cervello, M.; et al. Effects of mutations in Wnt/β-catenin, hedgehog, Notch and PI3K pathways on GSK-3 activity—Diverse effects on cell growth, metabolism and cancer. Biochim. Biophys. Acta (BBA) Bioenerg. 2016, 1863, 2942–2976. [Google Scholar] [CrossRef] [PubMed]
- Nagini, S.; Sophia, J.; Mishra, R. Glycogen synthase kinases: Moonlighting proteins with theranostic potential in cancer. Semin. Cancer Boil. 2019, 56, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Mo, S.P.; Coulson, J.M.; Prior, I.A. RAS variant signalling. Biochem. Soc. Trans. 2018, 46, 1325–1332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiner, D.J.; A Lundquist, E. Small GTPases. WormBook 2018, 2018, 1–65. [Google Scholar] [CrossRef]
- Goitre, L.; Trapani, E.; Trabalzini, L.; Retta, S.F. The Ras Superfamily of Small GTPases: The Unlocked Secrets. Adv. Struct. Saf. Stud. 2013, 1120, 1–18. [Google Scholar] [CrossRef]
- Liu, W.N.; Yan, M.; Chan, A.M. A thirty-year quest for a role of R-Ras in cancer: From an oncogene to a multitasking GTPase. Cancer Lett. 2017, 403, 59–65. [Google Scholar] [CrossRef]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef] [Green Version]
- Ohba, Y.; Mochizuki, N.; Yamashita, S.; Chan, A.M.; Schrader, J.W.; Hattori, S.; Nagashima, K.; Matsuda, M. Regulatory proteins of R-Ras, TC21/R-Ras2, and M-Ras/R-Ras3. J. Biol. Chem. 2000, 275, 20020–20026. [Google Scholar] [CrossRef] [Green Version]
- Colicelli, J. Human RAS Superfamily Proteins and Related GTPases. Sci. Signal. 2004, 2004, re13. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez-Erlandsson, S.; Vidal, P.M.H.; Fernández-Alfara, M.; Hernández-García, S.; Gonzalo-Flores, S.; Mudarra-Rubio, A.; Fresno, M.; Cubelos, B. R-RAS2 overexpression in tumors of the human central nervous system. Mol. Cancer 2013, 12, 127. [Google Scholar] [CrossRef] [Green Version]
- Drivas, G.T.; Shih, A.; Coutavas, E.; Rush, M.G.; D’Eustachio, P. Characterization of four novel ras-like genes expressed in a human teratocarcinoma cell line. Mol. Cell. Boil. 1990, 10, 1793–1798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, R.A.; Lu, J.; Carrasco, M.; Hunter, J.; Manandhar, A.; Gondi, S.; Westover, K.D.; Engen, J.R. Structural Dynamics in Ras and Related Proteins upon Nucleotide Switching. J. Mol. Boil. 2016, 428, 4723–4735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komatsu, M.; Ruoslahti, E. R-Ras is a global regulator of vascular regeneration that suppresses intimal hyperplasia and tumor angiogenesis. Nat. Med. 2005, 11, 1346–1350. [Google Scholar] [CrossRef] [PubMed]
- Larive, R.M.; Abad, A.; Cardaba, C.M.; Hernańdez, T.; Canãmero, M.; De Aĺava, E.; Santos, E.; Alarcoń, B.; Bustelo, X.R. The Ras-like protein R-Ras2/TC21 is important for proper mammary gland development. Mol. Biol. Cell 2012, 23, 2373–2387. [Google Scholar] [CrossRef] [Green Version]
- Kimmelman, A.C.; Rodriguez, N.N.; Chan, A.M.-L. R-Ras3/M-Ras Induces Neuronal Differentiation of PC12 Cells through Cell-Type-Specific Activation of the Mitogen-Activated Protein Kinase Cascade. Mol. Cell Biol. 2002, 22, 5946–5961. [Google Scholar] [CrossRef] [Green Version]
- Sun, P.; Watanabe, H.; Takano, K.; Yokoyama, T.; Fujisawa, J.-I.; Endo, T. Sustained activation of M-Ras induced by nerve growth factor is essential for neuronal differentiation of PC12 cells. Genes Cells 2006, 11, 1097–1113. [Google Scholar] [CrossRef]
- Olsen, I.M.; Ffrench-Constant, C. Dynamic regulation of integrin activation by intracellular and extracellular signals controls oligodendrocyte morphology. BMC Boil. 2005, 3, 25. [Google Scholar] [CrossRef] [Green Version]
- Rey, I.; Taylor-Harris, P.; Van Erp, H.; Hall, A. R-ras interacts with rasGAP, neurofibromin and c-raf but does not regulate cell growth or differentiation. Oncogene 1994, 9, 685–692. [Google Scholar]
- Huff, S.Y.; A Quilliam, L.; Cox, A.D.; Der, C.J. R-Ras is regulated by activators and effectors distinct from those that control Ras function. Oncogene 1997, 14, 133–143. [Google Scholar] [CrossRef]
- Marte, B.M.; Rodriguez-Viciana, P.; Wennström, S.; Warne, P.H.; Downward, J. R-Ras can activate the phosphoinositide 3-kinase but not the MAP kinase arm of the Ras effector pathways. Curr. Boil. 1997, 7, 63–71. [Google Scholar] [CrossRef] [Green Version]
- Osada, M.; Tolkacheva, T.; Li, W.; Chan, T.O.; Tsichlis, P.N.; Saez, R.; Kimmelman, A.C.; Chan, A.M. Differential Roles of Akt, Rac, and Ral in R-Ras-Mediated Cellular Transformation, Adhesion, and Survival. Mol. Cell. Boil. 1999, 19, 6333–6344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosário, M.; Paterson, H.F.; Marshall, C.J. Activation of the Ral and Phosphatidylinositol 3′ Kinase Signaling Pathways by the Ras-Related Protein TC21. Mol. Cell Biol. 2001, 21, 3750–3762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Self, A.J.; Caron, E.; Paterson, H.F.; Hall, A. Analysis of R-Ras signalling pathways. J. Cell Sci. 2001, 114, 1357–1366. [Google Scholar] [PubMed]
- Sanz-Rodríguez, M.; Gruart, A.; Escudero-Ramirez, J.; De Castro, F.; Delgado-García, J.M.; Wandosell, F.; Cubelos, B. R-Ras1 and R-Ras2 Are Essential for Oligodendrocyte Differentiation and Survival for Correct Myelination in the Central Nervous System. J. Neurosci. 2018, 38, 5096–5110. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Experimental Approach | Myelin Thickness | Myelination Degree | Phosphorylation of Downstream Elements | Myelin Proteins Expression | Nº of Mature OLs | Nº of OPCs | OL Differentiation | References |
|---|---|---|---|---|---|---|---|---|
| Bace1−/− | - | Normal | - | - | Normal | Normal | Normal | [39] |
| Akt-DD 1 | + | + | + | + | Normal | Normal | Enhanced | [42] |
| Rapamycin 2 | - | - | - | - | [45] | |||
| Ptenfl/fl; Cnp1Cre/+ Ptenfl/fl; Plp1CreERT2/+ | + | + | Normal | Normal | [43] | |||
| Ptenfl/fl; Olig2Cre/+ | + | + | + | Normal | Normal | Normal | [44] | |
| CNPCre/+; Raptorfl/fl | - | - | - | - | - | + | Delayed | [46] |
| CNPCre/+; Rictorfl/fl | Normal | Slightly - | - | Normal | - | Increased | [46] | |
| CNPCre/+; Rptorfl/fl | - | - | - | - | Normal | Normal | [47] | |
| CNPCre/+; Rictorfl/fl | - | Delayed myelination | Normal | Normal | Normal | Normal | [47] | |
| CNPCre/+; Rptorfl/fl; Rictorfl/fl | - | - | - | - | - | + | Delayed | [47] |
| CNPCre/+; Tsc1fl/fl | - | + | - | [47] | ||||
| Olig1Cre/+; Rheb1fl/fl | Normal | - | - | - | - | + | Impaired | [50] |
| NG2Cre/+; Tsc1fl/fl | + (later is normal) | Enhanced remyelination | + | Normal | Normal | Normal | [48] | |
| Plp1Cre/+; Tsc1fl/fl | - (later is normal) | Delayed remyelination | Normal | Normal | [48] |
| Experimental Approach | Myelin Thickness | Myelination Degree/Start of Myelination | Phosphorylation of Downstream Elements | Myelin Proteins Expression | Cellular Proliferation | Nº of Mature OLs | Nº of OPCs | OL Differentiation | References |
|---|---|---|---|---|---|---|---|---|---|
| MbpCre/+; TrkBfl/fl | - | -/Delayed | - | + | Normal | + | Normal | [60] | |
| CNPCre/+; Fgfr1/2fl/fl | - | -/Delayed | - | - | Normal | Normal | Normal | Normal | [58] |
| CNPCre/+; Erk1−/−; Erk2fl/fl | - | Normal/Normal | - | Normal | Normal | Normal | [53] | ||
| hGFAPCre/+; Erk2fl/fl NG2-Cre/+; Erk2fl/fl | Slightly -/Delayed | Normal | - | Normal | Delayed | [55] | |||
| CNPCre/+; MEK1DD+/− (1) | + | Normal/Enhanced remyelination | + | + | Normal | Normal | Normal | Normal | [53] |
| CNPCre/+; MEK1DD+/− (1) Olig1Cre/+; MEK1DD+/− (1) | + | +/Normal | + | + | Normal | + | [57] | ||
| Olig1Cre/+; Erk1−/−;Erk2fl/fl | - | - | - | [57] |
| Experimental Approach | Myelination Degree | Myelin Proteins Expression | Cellular Proliferation | Nº of Mature OLs | Nº of Immature OLs (OPCs) | OL Differentiation | References |
|---|---|---|---|---|---|---|---|
| Olig2CreERT2/+; APCfl/fl | - | - | - | - | - | Impaired | [71] |
| Axin2−/− | Delayed remyelination | Normal | - | Normal | Delayed | [72] | |
| Treatment with XAV939 1 | + | + | Normal | + | - | Enhanced | [72] |
| GSK-3Β inhibitors | + | + | + | + | + | Normal | [66] |
| Olig2Cre/+; DA-Cat 2 | - | PLP+ decreased | - | Normal | Delayed | [64] | |
| CnpCre/+; DA-Cat | - | - | Normal | - | Normal | Delayed | [65] |
| Tcf4−/− | - | - | - | Delayed | [73,74] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alcover-Sanchez, B.; Garcia-Martin, G.; Wandosell, F.; Cubelos, B. R-Ras GTPases Signaling Role in Myelin Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 5911. https://doi.org/10.3390/ijms21165911
Alcover-Sanchez B, Garcia-Martin G, Wandosell F, Cubelos B. R-Ras GTPases Signaling Role in Myelin Neurodegenerative Diseases. International Journal of Molecular Sciences. 2020; 21(16):5911. https://doi.org/10.3390/ijms21165911
Chicago/Turabian StyleAlcover-Sanchez, Berta, Gonzalo Garcia-Martin, Francisco Wandosell, and Beatriz Cubelos. 2020. "R-Ras GTPases Signaling Role in Myelin Neurodegenerative Diseases" International Journal of Molecular Sciences 21, no. 16: 5911. https://doi.org/10.3390/ijms21165911
APA StyleAlcover-Sanchez, B., Garcia-Martin, G., Wandosell, F., & Cubelos, B. (2020). R-Ras GTPases Signaling Role in Myelin Neurodegenerative Diseases. International Journal of Molecular Sciences, 21(16), 5911. https://doi.org/10.3390/ijms21165911