Dietary Intake of Rosmarinic Acid Increases Serum Inhibitory Activity in Amyloid A Aggregation and Suppresses Deposition in the Organs of Mice

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
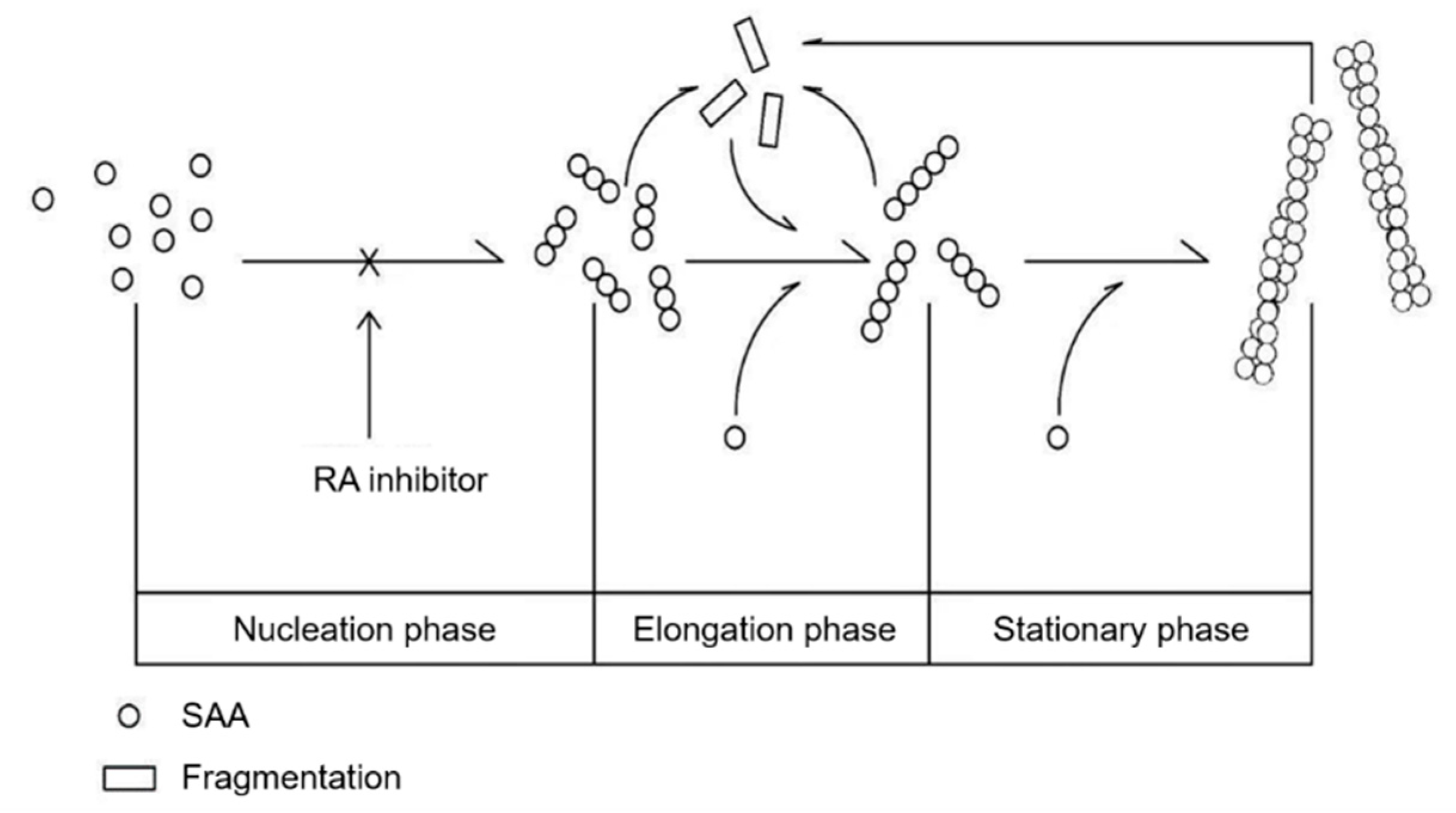
Abstract
:1. Introduction
2. Results
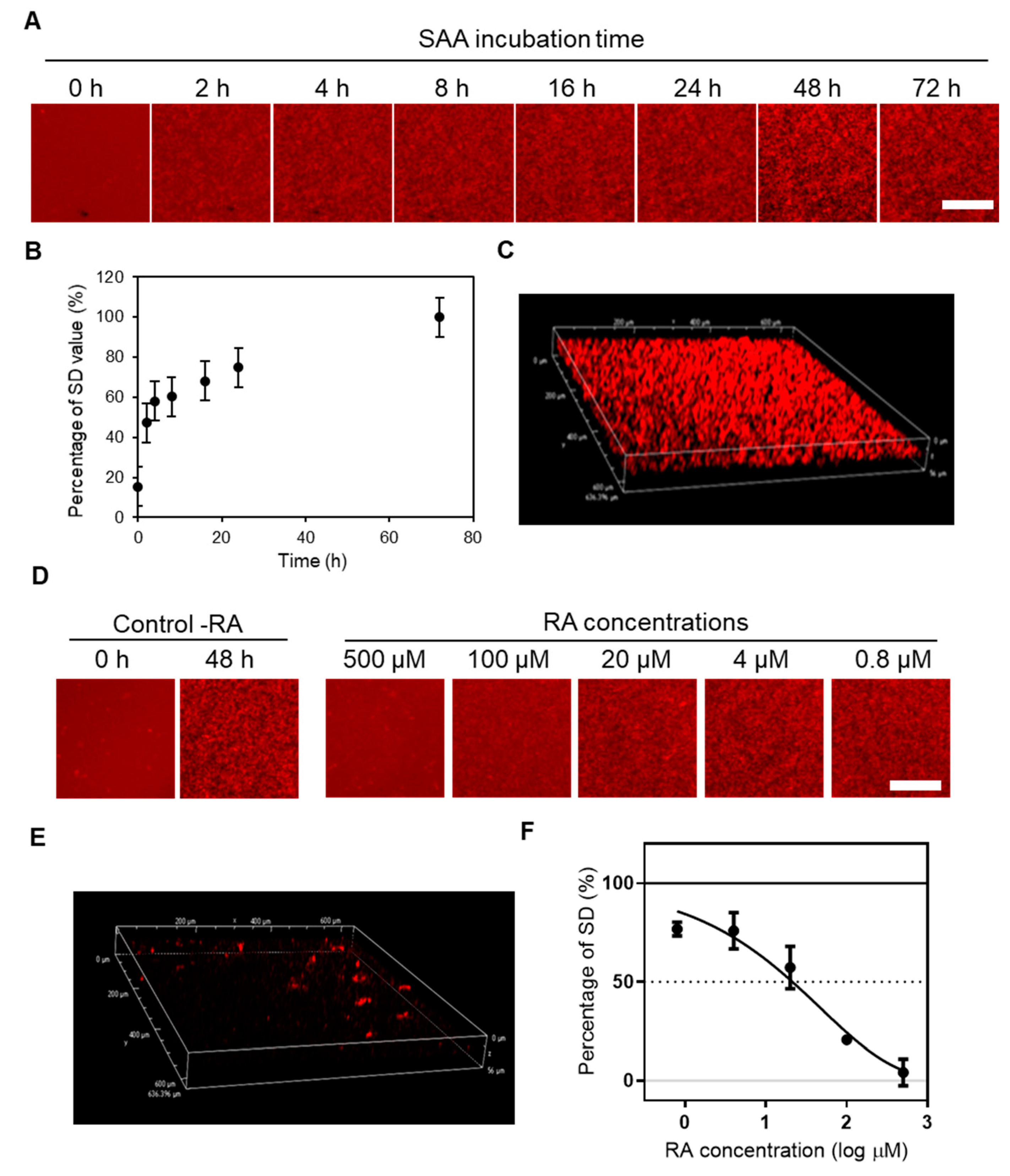
2.1. Real-Time Imaging of SAA Aggregation Using QDs and Evaluation of the Aggregation Inhibitory Activity of RA by the MSHTS System
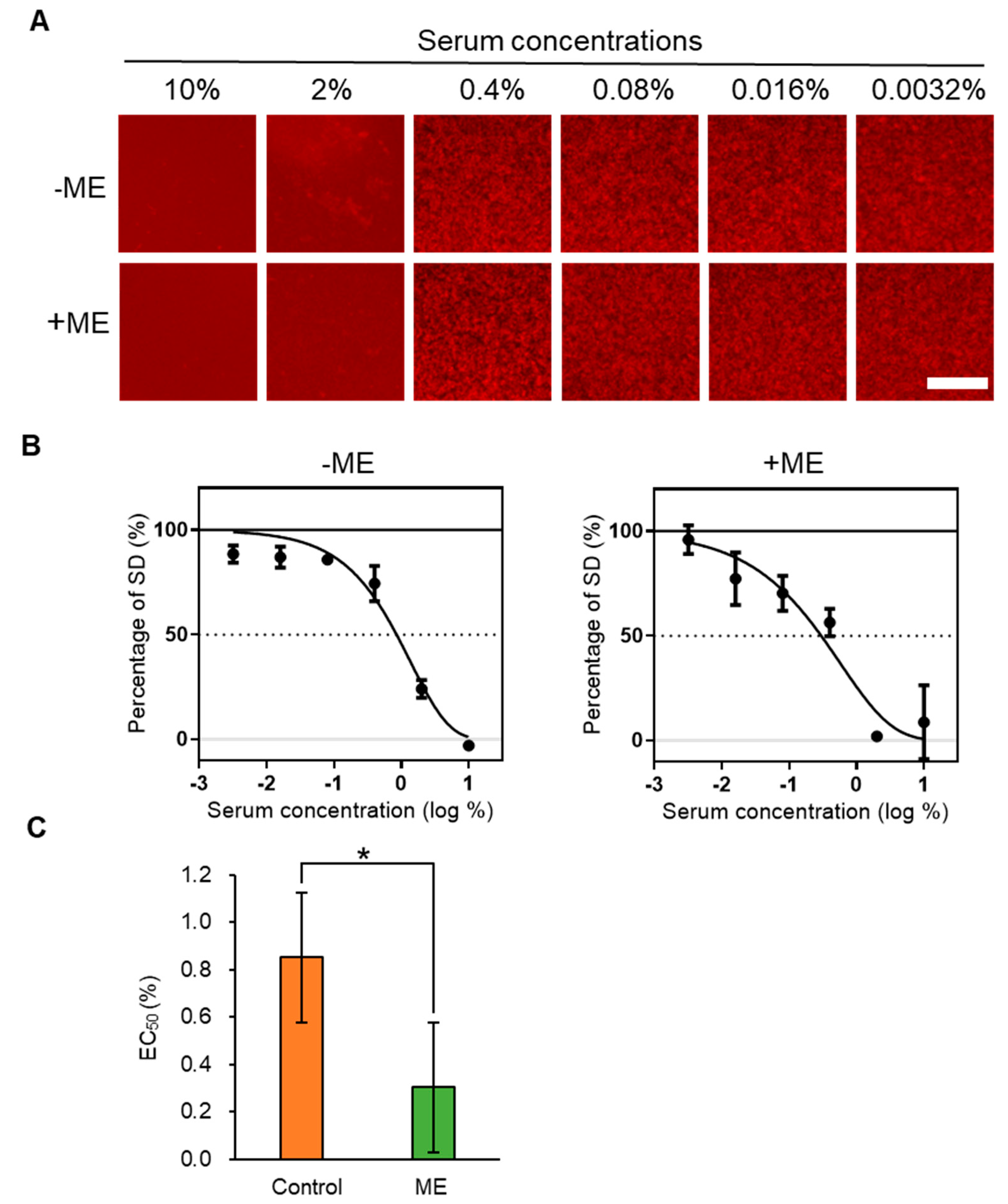
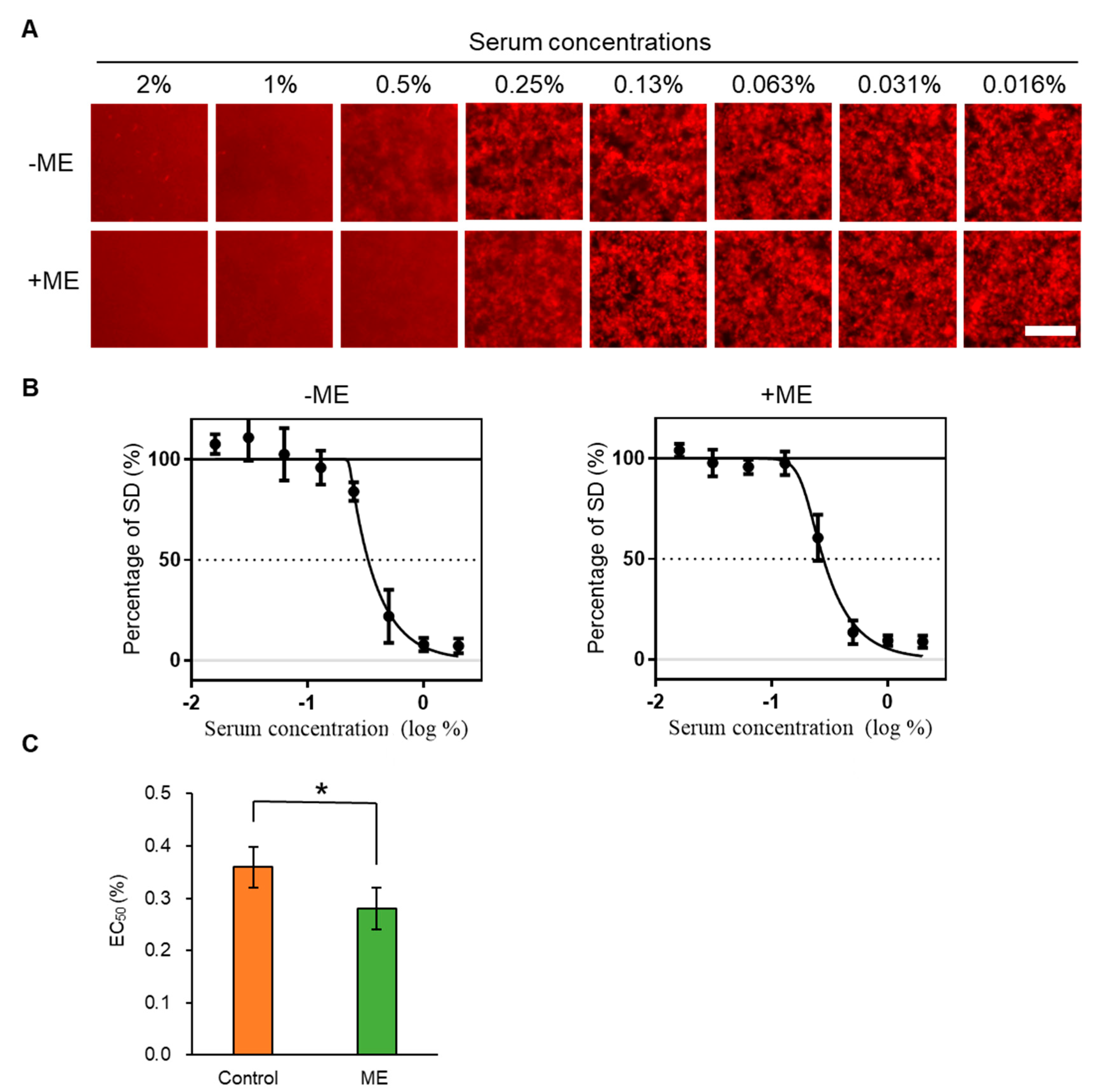
2.2. Inhibitory Activity of ME-Fed Mice Serum for Aggregation of mSAA and Aβ
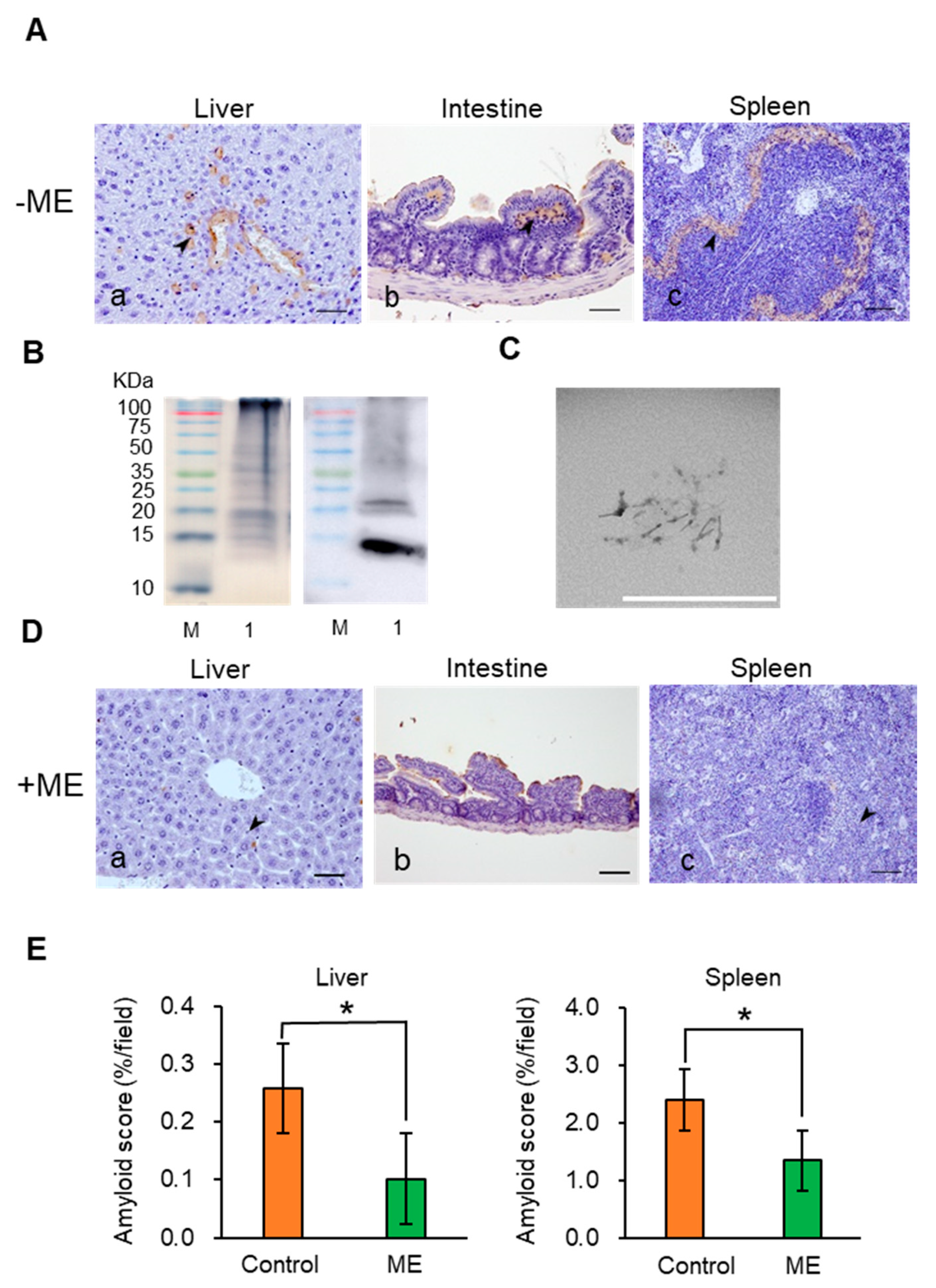
2.3. Suppression of AA Amyloidosis by ME Feeding
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Preparation of Mouse SAA Protein, AEF and Amyloid Extracts
4.3. Induction of AA Amyloidosis Using AEF
4.4. Histopathology
4.5. Image Analysis of Amyloid Deposition
4.6. Western Blot Analysis of Total Amyloid Extracts
4.7. Transmission Electron Microscopy Observations of Amyloid Fibrils
4.8. Imaging of mSAA Aggregation
4.9. Measurement of Amyloid Aggregation Inhibitory Activity of RA or Sera Using MSHTS System
4.10. RA Post-Aggregation Inhibition Experiment
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ogawa, S.; Murakami, T.; Inoshima, Y.; Ishiguro, N. Effect of heating on the stability of amyloid A (AA) fibrils and the intra- and cross-species transmission of AA amyloidosis. Amyloid 2015, 22, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Sipe, J.D.; Benson, M.D.; Buxbaum, J.N.; Ikeda, S.I.; Merlini, G.; Saraiva, M.J.M.; Westermark, P. Amyloid fibril protein nomenclature: 2012 recommendations from the Nomenclature Committee of the International Society of Amyloidosis. Amyloid 2012, 19, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Kluve-Beckerman, B.; Liepnieks, J.J.; Benson, M.D.; Lai, X.; Qi, G.; Wang, M. Carbamylation of the amino-terminal residue (Gly1) of mouse serum amyloid A promotes amyloid formation in a cell culture model. FEBS Lett. 2016, 590, 4296–4307. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Shiroo, M.; Migita, S. Diverse gene expression for isotypes of murine serum amyloid A protein during acute phase reaction. Science 1986, 232, 227–229. [Google Scholar] [CrossRef] [PubMed]
- Albisser, A.M.; McAdam, K.P.W.J.; Perlman, K.; Carson, S.; Bahoric, A.; Williamson, J.R. Unanticipated amyloidosis in dogs infused with insulin. Diabetes 1983, 32, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Lundmark, K.; Westermark, G.T.; Olsén, A.; Westermark, P. Protein fibrils in nature can enhance amyloid protein A amyloidosis in mice: Cross-seeding as a disease mechanism. Proc. Natl. Acad. Sci. USA 2005, 102, 6098–6102. [Google Scholar] [CrossRef] [Green Version]
- Brownlee, M.; Vlassara, H.; Cerami, A.; Martin, T.R.; Li, J.J.; McAdam, K.P. Association of insulin pump therapy with raised serum amyloid A in type I diabetes mellitus. Lancet 1984, 1, 411–413. [Google Scholar] [CrossRef]
- Anderberg, R.J.; Meek, R.L.; Hudkins, K.L.; Cooney, S.K.; Alpers, C.E.; LeBoeuf, R.C.; Tuttle, K.R. Serum amyloid A and inflammation in diabetic kidney disease and podocytes. Lab. Investig. 2015, 95, 250–262. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Ye, R.D. Serum amyloid A1: Structure, function and gene polymorphism. Gene 2016, 583, 48–57. [Google Scholar] [CrossRef] [Green Version]
- Iadanza, M.G.; Jackson, M.P.; Hewitt, E.W.; Ranson, N.A.; Radford, S.E. A new era for understanding amyloid structures and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 755–773. [Google Scholar] [CrossRef]
- Benditt, E.P.; Eriksen, N. Amyloid protein SAA is associated with high density lipoprotein from human serum. Proc. Natl. Acad. Sci. USA 1977, 74, 4025–4028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Z.; Cai, L.; Jiang, J.; Chang, K.-S.; van der Westhuyzen, D.R.; Luo, G. Human serum amyloid A protein inhibits hepatitis C virus entry into cells. J. Virol. 2007, 81, 6128–6133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsun, J.G.S.; Shiu, S.W.M.; Wong, Y.; Yung, S.; Chan, T.M.; Tan, K.C.B. Impact of serum amyloid A on cellular cholesterol efflux to serum intype 2 diabetes mellitus. Atherosclerosis 2013, 231, 405–410. [Google Scholar] [CrossRef]
- Lu, J.; Yu, Y.; Zhu, I.; Cheng, Y.; Sun, P.D. Structural mechanism of serum amyloid A-mediated inflammatory amyloidosis. Proc. Natl. Acad. Sci. USA 2014, 111, 5189–5194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couderc, E.; Morel, F.; Levillain, P.; Buffière-Morgado, A.; Camus, M.; Paquier, C.; Bodet, C.; Jégou, J.-F.; Pohin, M.; Favot, L.; et al. Interleukin-17A-induced production of acute serum amyloid A by keratinocytes contributes to psoriasis pathogenesis. PLoS ONE 2017, 12, e0181486. [Google Scholar] [CrossRef]
- Jayaraman, S.; Gantz, D.L.; Haupt, C.; Gursky, O. Serum amyloid A forms stable oligomers that disrupt vesicles at lysosomal pH and contribute to the pathogenesis of reactive amyloidosis. Proc. Natl. Acad. Sci. USA 2017, 114, E6507–E6515. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.Z.; Lee, M.J.; Hu, H.; Pollin, T.I.; Ryan, A.S.; Nicklas, B.J.; Snitker, S.; Horenstein, R.B.; Hull, K.; Goldberg, N.H.; et al. Acute-phase serum amyloid A: An inflammatory adipokine and potential link between obesity and its metabolic complications. PLoS Med. 2006, 3, e287. [Google Scholar] [CrossRef]
- Pettersson, T.; Konttinen, Y.T.; Maury, C.P.J. Treatment strategies for amyloid A amyloidosis. Expert Opin. Pharmacother. 2008, 9, 2117–2128. [Google Scholar] [CrossRef]
- McCubbin, W.D.; Kay, C.M.; Kisilevsky, R. Circular-dichroism studies on two murine serum amyloid A proteins. Biochem. J. 1988, 256, 775–783. [Google Scholar] [CrossRef] [Green Version]
- Westermark, G.T.; Engström, U.; Westermark, P. The N-terminal segment of protein AA determines its fibrillogenic property. Biochem. Biophys. Res. Commun. 1992, 182, 27–33. [Google Scholar] [CrossRef]
- Urieli-Shoval, S.; Linke, R.P.; Matzner, Y. Expression and function of serum amyloid A, a major acute-phase protein, in normal and disease states. Curr. Opin. Hematol. 2000, 7, 64–69. [Google Scholar] [CrossRef]
- Hoffman, J.S.; Benditt, E.P. Changes in high density lipoprotein content following endotoxin administration in the mouse. Formation of serum amyloid protein-rich subfractions. J. Biol. Chem. 1982, 257, 10510–10517. [Google Scholar] [PubMed]
- Zhang, B.; Une, Y.; Fu, X.; Yan, J.; Ge, F.; Yao, J.; Sawashita, J.; Mori, M.; Tomozawa, H.; Kametani, F.; et al. Fecal transmission of AA amyloidosis in the cheetah contributes to high incidence of disease. Proc. Natl. Acad. Sci. USA 2008, 105, 7263–7268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radamaker, L.; Lin, Y.-H.; Annamalai, K.; Huhn, S.; Hegenbart, U.; Schönland, S.O.; Fritz, G.; Schmidt, M.; Fändrich, M. Cryo-EM structure of a light chain-derived amyloid fibril from a patient with systemic AL amyloidosis. Nat. Commun. 2019, 10, 1103–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, M.; Wiese, S.; Adak, V.; Engler, J.; Agarwal, S.; Fritz, G.; Westermark, P.; Zacharias, M.; Fändrich, M. Cryo-EM structure of a transthyretin-derived amyloid fibril from a patient with hereditary ATTR amyloidosis. Nat. Commun. 2019, 10, 5008–5017. [Google Scholar] [CrossRef] [Green Version]
- Liberta, F.; Loerch, S.; Rennegarbe, M.; Schierhorn, A.; Westermark, P.; Westermark, G.T.; Hazenberg, B.P.C.; Grigorieff, N.; Fändrich, M.; Schmidt, M. Cryo-EM fibril structures from systemic AA amyloidosis reveal the species complementarity of pathological amyloids. Nat. Commun. 2019, 10, 1104–1124. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, R.; Tainaka, R.; Ando, Y.; Hashi, Y.; Deepak, H.V.; Suga, Y.; Murai, Y.; Anetai, M.; Monde, K.; Ohta, K.; et al. An automated microliter-scale high-throughput screening system (MSHTS) for real-time monitoring of protein aggregation using quantum-dot nanoprobes. Sci. Rep. 2019, 9, 2587–2596. [Google Scholar] [CrossRef]
- Ishigaki, Y.; Tanaka, H.; Akama, H.; Ogara, T.; Uwai, K.; Tokuraku, K. A microliter-scale high-throughput screening system with quantum-dot nanoprobes for amyloid-β aggregation inhibitors. PLoS ONE 2013, 8, e72992. [Google Scholar] [CrossRef] [Green Version]
- Tokuraku, K.; Marquardt, M.; Ikezu, T. Real-time imaging and quantification of amyloid-beta peptide aggregates by novel quantum-dot nanoprobes. PLoS ONE 2009, 4, e8492. [Google Scholar] [CrossRef]
- Ogara, T.; Takahashi, T.; Yasui, H.; Uwai, K.; Tokuraku, K. Evaluation of the effects of amyloid β aggregation from seaweed extracts by a microliter-scale high-throughput screening system with a quantum dot nanoprobe. J. Biosci. Bioeng. 2015, 120, 45–50. [Google Scholar] [CrossRef]
- Kuragano, M.; Yamashita, R.; Chikai, Y.; Kitamura, R.; Tokuraku, K. Three-dimensional real time imaging of amyloid β aggregation on living cells. Sci. Rep. 2020, 10, 9742–9754. [Google Scholar] [CrossRef]
- Ono, K.; Hasegawa, K.; Naiki, H.; Yamada, M. Curcumin has potent anti-amyloidogenic effects for Alzheimer’s beta-amyloid fibrils in vitro. J. Neurosci. Res. 2004, 75, 742–750. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, T.; Ono, K.; Murase, A.; Yamada, M. Phenolic compounds prevent Alzheimer’s pathology through different effects on the amyloid-β aggregation pathway. Am. J. Pathol. 2009, 175, 2557–2565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taguchi, R.; Hatayama, K.; Takahashi, T.; Hayashi, T.; Sato, Y.; Sato, D.; Ohta, K.; Nakano, H.; Seki, C.; Endo, Y.; et al. Structure–activity relations of rosmarinic acid derivatives for the amyloid β aggregation inhibition and antioxidant properties. Eur. J. Med. Chem. 2017, 138, 1066–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bu, X.L.; Rao, P.P.N.; Wang, Y.J. Anti-amyloid aggregation activity of natural compounds: Implications for Alzheimer’s drug discovery. Mol. Neurobiol. 2016, 53, 3565–3575. [Google Scholar] [CrossRef]
- Lin, X.; Galaqin, N.; Tainaka, R.; Shimamori, K.; Kuragano, M.; Noguchi, T.Q.P.; Tokuraku, K. Real-time 3D imaging and inhibition analysis of various amyloid aggregations using quantum dots. Int. J. Mol. Sci. 2020, 21, 1978. [Google Scholar] [CrossRef] [Green Version]
- Kuragano, M.; Yoshinari, W.; Lin, X.; Shimamori, K.; Uwai, K.; Tokuraku, K. Evaluation of Amyloid β42 Aggregation Inhibitory Activity of Commercial Dressings by A Microliter-Scale High-Throughput Screening System Using Quantum-Dot Nanoprobes. Foods 2020, 9, 825. [Google Scholar] [CrossRef]
- Watanabe, K.; Uchida, K.; Chambers, J.K.; Tei, M.; Shoji, A.; Ushio, N.; Nakayama, H. Experimental transmission of AA amyloidosis by injecting the AA amyloid protein into interleukin-1 receptor antagonist knockout (IL-1raKO) mice. Vet. Pathol. 2015, 52, 505–512. [Google Scholar] [CrossRef] [Green Version]
- Tei, M.; Uchida, K.; Chambers, J.K.; Watanabe, K.I.; Tamamoto, T.; Ohno, K.; Nakayama, H. Variation of amino acid sequences of serum amyloid a (SAA) and immunohistochemical analysis of amyloid a (AA) in Japanese domestic cats. J. Vet. Med. Sci. 2018, 80, 164–172. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, K.; Uchida, K.; Chambers, J.K.; Ushio, N.; Nakayama, H. Deposition, clearance, and reinduction of amyloid A amyloid in interleukin 1 receptor antagonist knockout mice. Vet. Pathol. 2017, 54, 99–110. [Google Scholar] [CrossRef]
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meisl, G.; Kirkegaard, J.B.; Arosio, P.; Michaels, T.C.T.; Vendruscolo, M.; Dobson, C.M.; Linse, S.; Knowles, T.P.J. Molecular mechanisms of protein aggregation from global fitting of kinetic models. Nat. Protoc. 2016, 11, 252–272. [Google Scholar] [CrossRef]
- Cohen, S.I.A.; Linse, S.; Luheshi, L.M.; Hellstrand, E.; White, D.A.; Rajah, L.; Otzen, D.E.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P.J. Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl. Acad. Sci. USA 2013, 110, 9758–9763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glabe, C.G.; Kayed, R. Common structure and toxic function of amyloid oligomers implies a common mechanism of pathogenesis. Neurology 2006, 66, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Glabe, C.G. Common mechanisms of amyloid oligomer pathogenesis in degenerative disease. Neurobiol. Aging 2006, 27, 570–575. [Google Scholar] [CrossRef]
- Soto, C.; Estrada, L.; Castilla, J. Amyloids, prions and the inherent infectious nature of misfolded protein aggregates. Trends Biochem. Sci. 2006, 31, 150–155. [Google Scholar] [CrossRef]
- Simoneau, S.; Rezaei, H.; Salès, N.; Kaiser-Schulz, G.; Lefebvre-Roque, M.; Vidal, C.; Fournier, J.G.; Comte, J.; Wopfner, F.; Grosclaude, J.; et al. In vitro and in vivo neurotoxicity of prion protein oligomers. PLoS Pathog. 2007, 3, e125. [Google Scholar] [CrossRef]
- Reixach, N.; Deechongkit, S.; Jiang, X.; Kelly, J.W.; Buxbaum, J.N. Tissue damage in the amyloidoses: Transthyretin monomers and nonnative oligomers are the major cytotoxic species in tissue culture. Proc. Natl. Acad. Sci. USA 2004, 101, 2817–2822. [Google Scholar] [CrossRef] [Green Version]
- Bucciantini, M. Inherent cytotoxicity of aggregates implies a common origin for protein misfolding diseases. Nature 2002, 416, 507–511. [Google Scholar] [CrossRef]
- Hase, T.; Shishido, S.; Yamamoto, S.; Yamashita, R.; Nukima, H.; Taira, S.; Toyoda, T.; Abe, K.; Hamaguchi, T.; Ono, K.; et al. Rosmarinic acid suppresses Alzheimer’s disease development by reducing amyloid β aggregation by increasing monoamine secretion. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef]
- Nuvolone, M.; Merlini, G. Systemic amyloidosis: Novel therapies and role of biomarkers. Nephrol. Dial. Transplant. 2017, 32, 770–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodin, K.; Ellmerich, S.; Kahan, M.C.; Tennent, G.A.; Loesch, A.; Gilbertson, J.A.; Hutchinson, W.L.; Mangione, P.P.; Gallimore, J.R.; Millar, D.J.; et al. Antibodies to human serum amyloid P component eliminate visceral amyloid deposits. Nature 2010, 468, 93–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Levites, Y.; Smithson, L.A.; Price, R.W.; Dakin, R.S.; Yuan, B.; Sierks, M.R.; Kim, J.; McGowan, E.; Reed, D.K.; Rosenberry, T.L.; et al. Insights into the mechanisms of action of anti-Abeta antibodies in Alzheimer’s disease mouse models. FASEB J. 2006, 20, 2576–2578. [Google Scholar] [CrossRef] [PubMed]
- Polanco, J.C.; Li, C.; Bodea, L.G.; Martinez-Marmol, R.; Meunier, F.A.; Götz, J. Amyloid-β and tau complexity-Towards improved biomarkers and targeted therapies. Nat. Rev. Neurol. 2018, 14, 22–40. [Google Scholar] [CrossRef] [PubMed]
- Kam, T.-I.; Mao, X.; Park, H.; Chou, S.-C.; Karuppagounder, S.S.; Umanah, G.E.; Yun, S.P.; Brahmachari, S.; Panicker, N.; Chen, R.; et al. Poly(ADP-ribose) drives pathologic α-synuclein neurodegeneration in Parkinson’s disease. Science 2018, 362, eaat8407. [Google Scholar] [CrossRef] [Green Version]
- Selkoe, D.J.; Hardy, J.; Sciences, B.; Hu, N.-W.; Nicoll, A.J.; Zhang, D.; Mably, A.J.; O’Malley, T.; Purro, S.A.; Terry, C.; et al. Amyloid β-Protein Dimers Isolated Directly from Alzheimer Brains Impair Synaptic Plasticity and Memory. Nat. Med. 2016, 7, 3374–3389. [Google Scholar]
- Lesné, S.; Ming, T.K.; Kotilinek, L.; Kayed, R.; Glabe, C.G.; Yang, A.; Gallagher, M.; Ashe, K.H. A specific amyloid-β protein assembly in the brain impairs memory. Nature 2006, 440, 352–357. [Google Scholar] [CrossRef]
- Gertz, M.A.; Dispenzieri, A.; Sher, T. Pathophysiology and treatment of cardiac amyloidosis. Nat. Rev. Cardiol. 2015, 12, 91–102. [Google Scholar] [CrossRef]
- Mikhael, J.R.; Schuster, S.R.; Jimenez-Zepeda, V.H.; Bello, N.; Spong, J.; Reeder, C.B.; Stewart, A.K.; Bergsagel, P.L.; Fonseca, R. Cyclophosphamide-bortezomib-dexamethasone (CyBorD) produces rapid and complete hematologic response in patients with AL amyloidosis. Blood 2012, 119, 4391–4394. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.; Das, P.; Switzer, R.C.; Golde, T.E.; Jankowsky, J.L. Robust amyloid clearance in a mouse model of Alzheimer’s disease provides novel insights into the mechanism of amyloid-β immunotherapy. J. Neurosci. 2011, 31, 4124–4136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horai, R.; Saijo, S.; Tanioka, H.; Nakae, S.; Sudo, K.; Okahara, A.; Ikuse, T.; Asano, M.; Iwakura, Y. Development of chronic inflammatory arthropathy resembling rheumatoid arthritis in interleukin I receptor antagonist-deficient mice. J. Exp. Med. 2000, 191, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Pras, M.; Schubert, M.; Zucker-Franklin, D.; Rimon, A.; Franklin, E.C. The characterization of soluble amyloid prepared in water. J. Clin. Investig. 1968, 47, 924–933. [Google Scholar] [PubMed] [Green Version]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, X.; Watanabe, K.; Kuragano, M.; Kurotaki, Y.; Nakanishi, U.; Tokuraku, K. Dietary Intake of Rosmarinic Acid Increases Serum Inhibitory Activity in Amyloid A Aggregation and Suppresses Deposition in the Organs of Mice. Int. J. Mol. Sci. 2020, 21, 6031. https://doi.org/10.3390/ijms21176031
Lin X, Watanabe K, Kuragano M, Kurotaki Y, Nakanishi U, Tokuraku K. Dietary Intake of Rosmarinic Acid Increases Serum Inhibitory Activity in Amyloid A Aggregation and Suppresses Deposition in the Organs of Mice. International Journal of Molecular Sciences. 2020; 21(17):6031. https://doi.org/10.3390/ijms21176031
Chicago/Turabian StyleLin, Xuguang, Kenichi Watanabe, Masahiro Kuragano, Yukina Kurotaki, Ushio Nakanishi, and Kiyotaka Tokuraku. 2020. "Dietary Intake of Rosmarinic Acid Increases Serum Inhibitory Activity in Amyloid A Aggregation and Suppresses Deposition in the Organs of Mice" International Journal of Molecular Sciences 21, no. 17: 6031. https://doi.org/10.3390/ijms21176031
APA StyleLin, X., Watanabe, K., Kuragano, M., Kurotaki, Y., Nakanishi, U., & Tokuraku, K. (2020). Dietary Intake of Rosmarinic Acid Increases Serum Inhibitory Activity in Amyloid A Aggregation and Suppresses Deposition in the Organs of Mice. International Journal of Molecular Sciences, 21(17), 6031. https://doi.org/10.3390/ijms21176031