Nanoparticle-Based Technology Approaches to the Management of Neurological Disorders

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Neurological Disorders
2.1. Neurodegenerative Diseases
2.1.1. Alzheimer’s Disease (AD)
2.1.2. Parkinson’s Disease (PD)
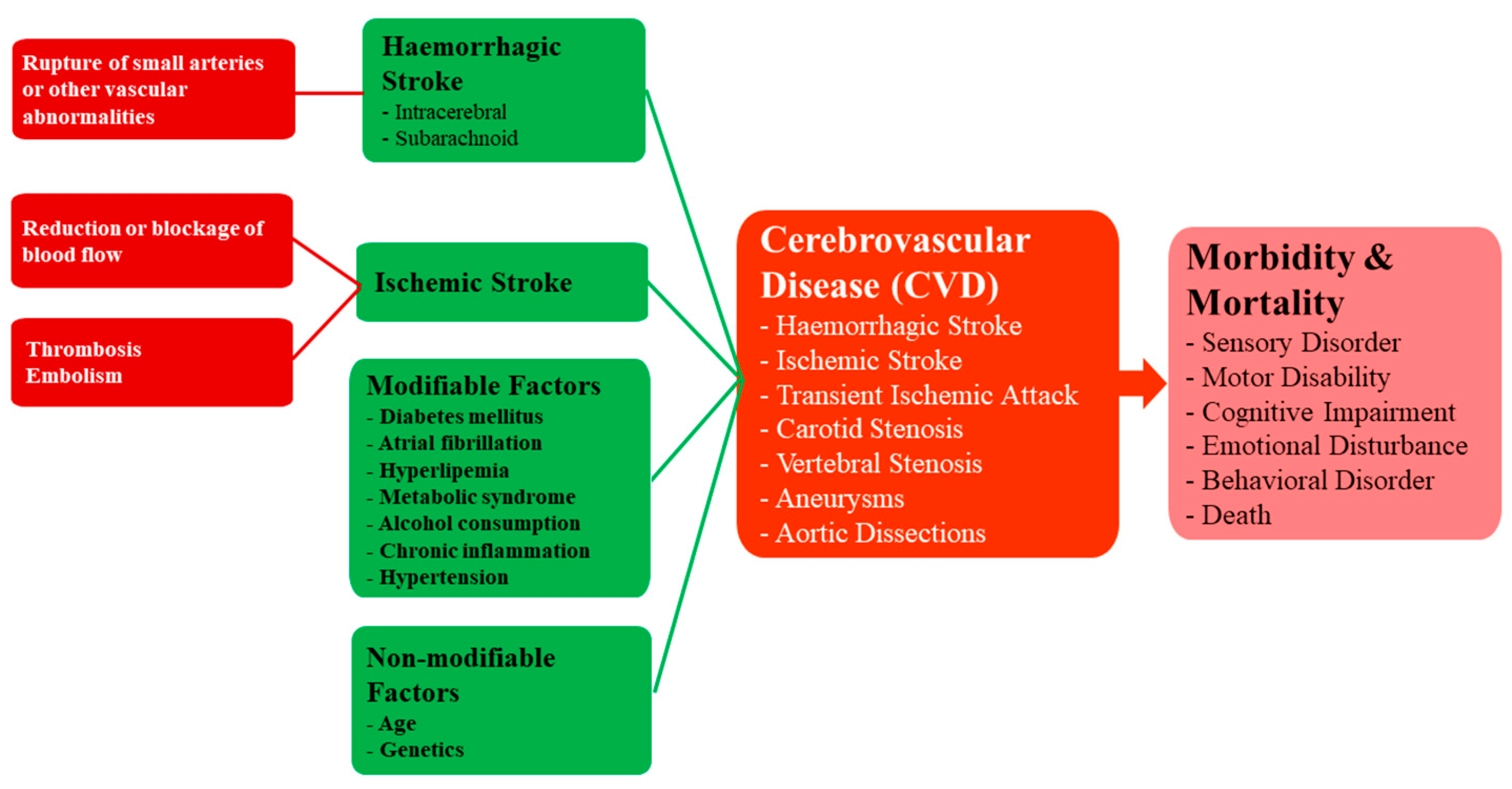
2.2. Cerebrovascular Diseases (CVDs)
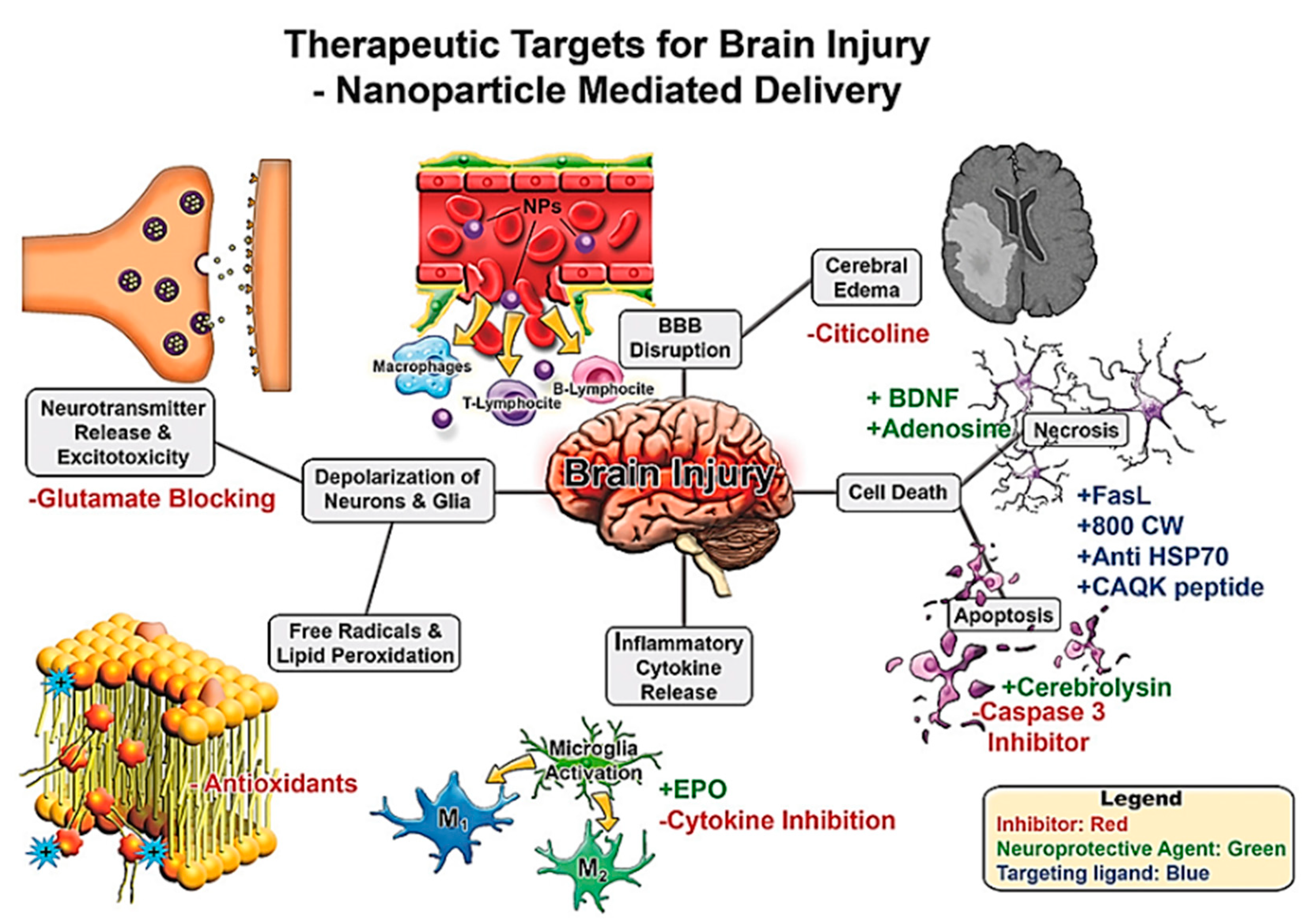
2.3. Traumatic Brain Injury (TBI)
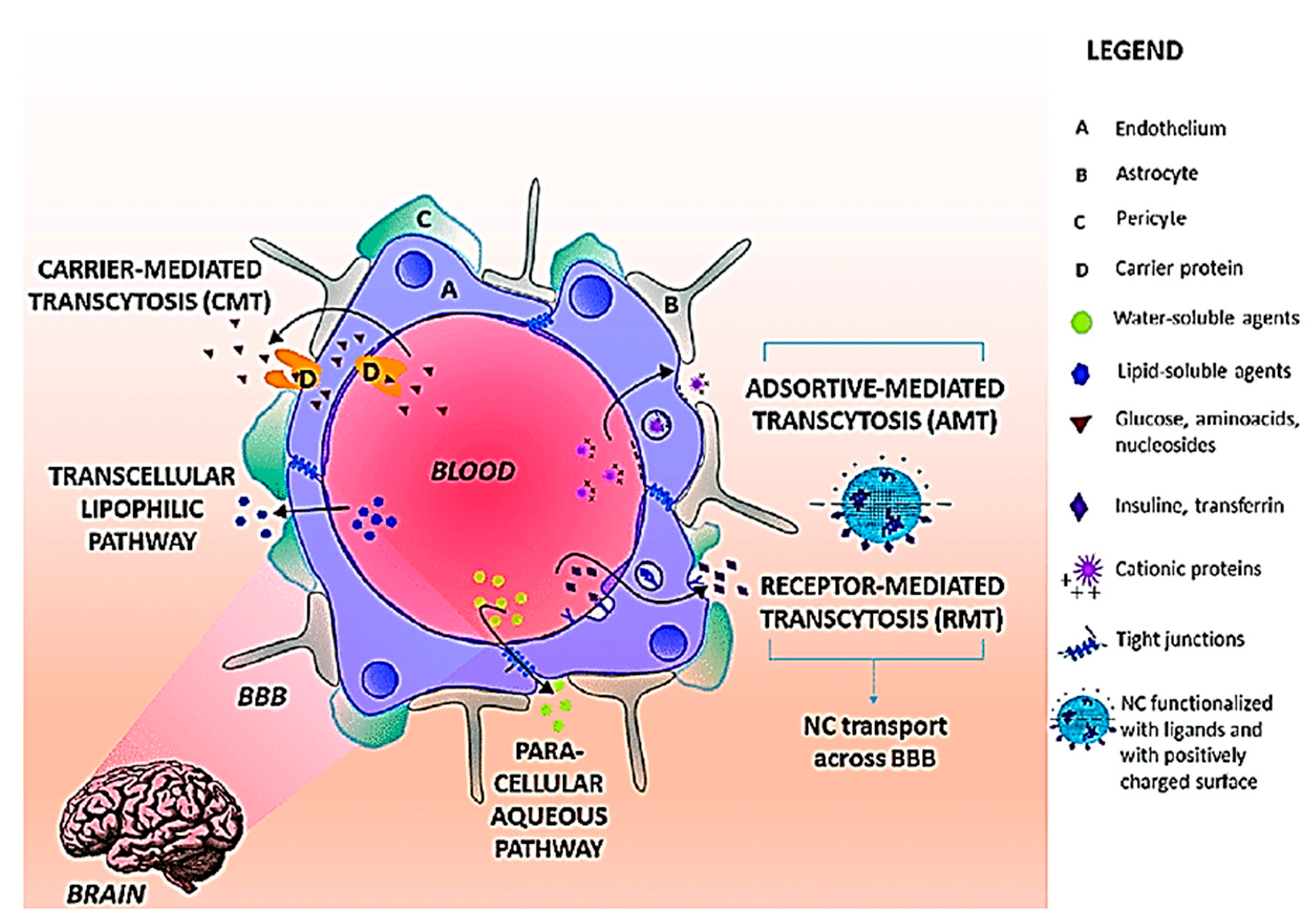
3. Blood Brain Barrier (BBB)—A Challenge for CNS Drug Delivery
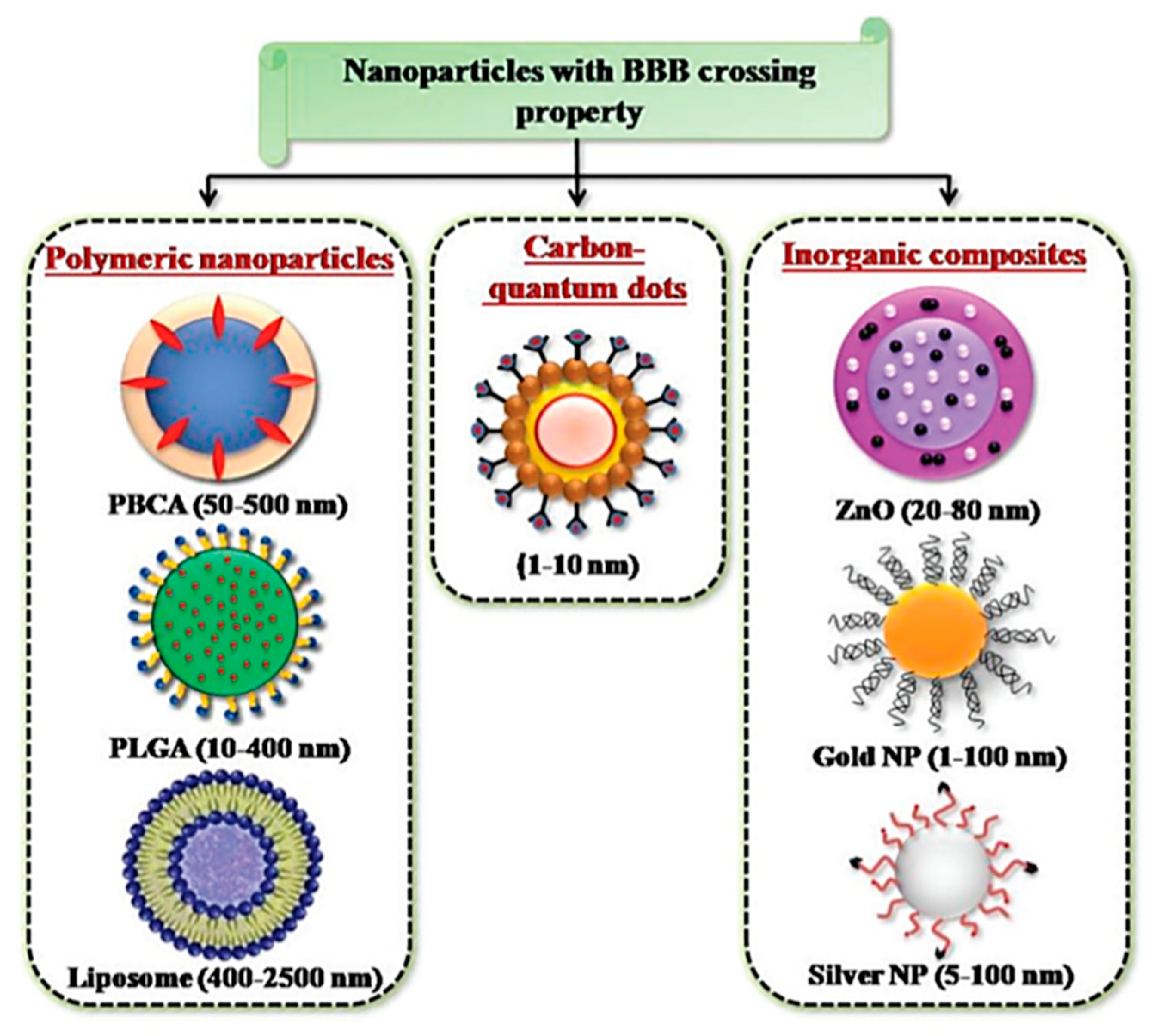
4. Nanoparticle-Based CNS Theranostics
4.1. Nanoparticle Applications in the Treatment and Diagnosis of AD
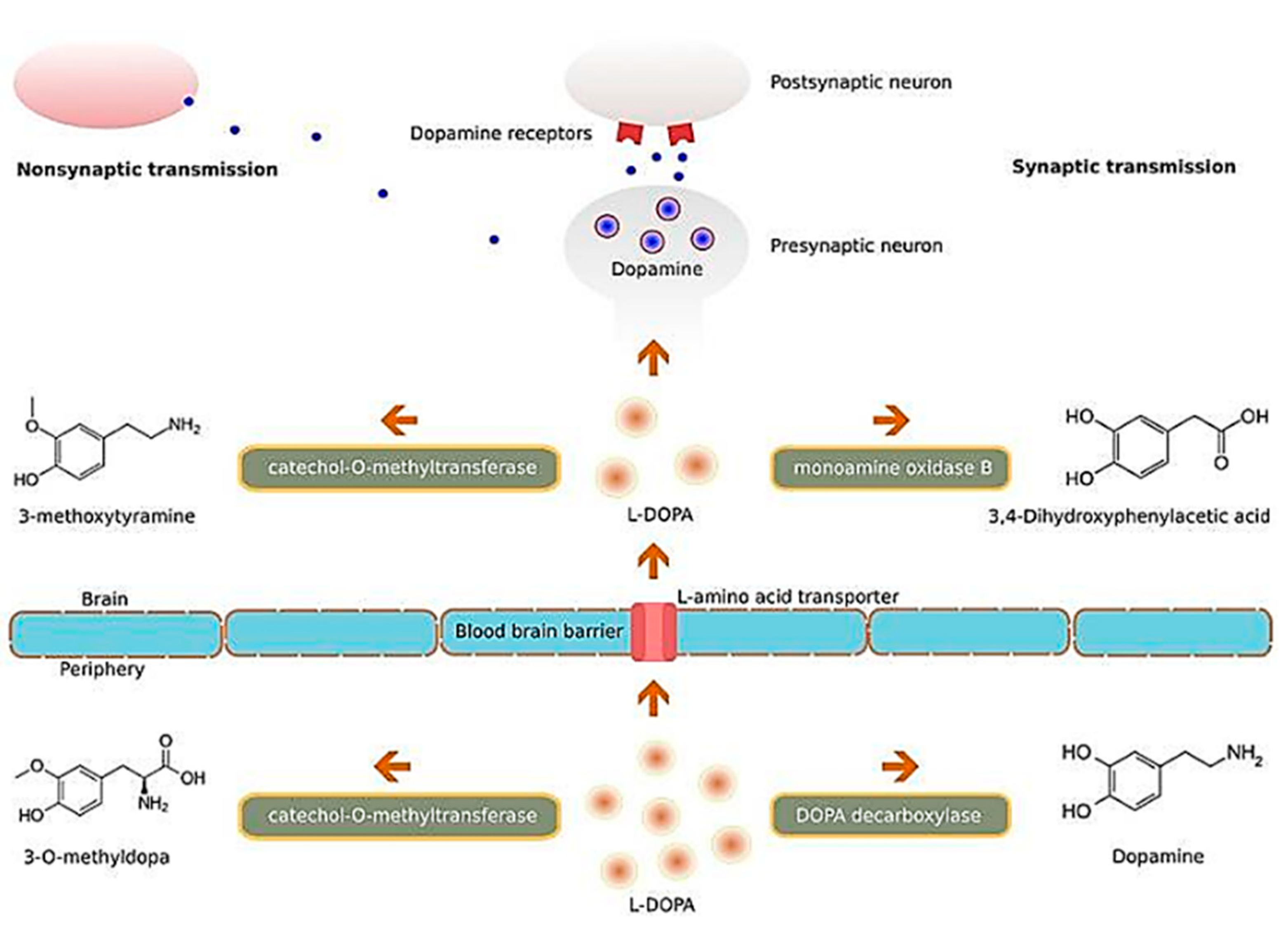
4.2. Nanoparticle Applications in the Treatment and Diagnosis of PD
4.3. Nanoparticle Applications in the Treatment and Diagnosis of CVDs
4.4. Nanoparticle Applications in the Treatment and Diagnosis of TBI
5. Clinical Status of NPs and Future Perspectives
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Aβ | Amyloid beta |
| ABC | ATP-binding cassette |
| ABCB1 | ATP-binding cassette sub-family B member 1 |
| Ach | Acetylcholine |
| AChE | Acetylcholinesterase |
| AD | Alzheimer’s disease |
| Ag/MoS2 | Silver-molybdenum disulfide |
| AMT | Adsorptive-mediated transcytosis |
| ApoE | Apolipoprotein E |
| APP | Amyloid precursor protein |
| AP-1 | Activator protein 1 |
| AP9 | CD147 antagonist peptide-9 |
| ATX | Astaxanthin |
| BBB | Blood-brain barrier |
| BDNF | Brain-derived neurotrophic factor |
| βCas | β casein |
| CAP | Cellulose acetate phthalate |
| CeO2 | Cerium oxide |
| CIs | Cholinesterase inhibitors |
| CNS | Central nervous system |
| CVDs | Cerebrovascular diseases |
| DALYs | Disability-adjusted life-years |
| ELISA | Enzyme-linked immunosorbent assay |
| EMA | European Medicines Agency |
| FDA | Food and Drug Administration |
| Gd | Gadolinium |
| GDNF | Glial cell-derived neurotrophic factor |
| HDL | High-density lipoprotein |
| ICH | Intracerebral haemorrhage |
| IL-1 | Interleukin-1 |
| IL-6 | Interleukin-6 |
| KP | Kynurenine pathway |
| MDCKII-MDR1 | Madin Darby canine kidney II - multi drug resistance gene-1 |
| MEM | 2-(methylthio)ethyl methacrylate |
| MION | Monocrystalline iron oxide nanoparticles |
| MNHS | Methacrylic acid N-hydroxysuccinimide ester |
| MRI | Magnetic resonance imaging |
| NGF | Nerve growth factor |
| NF-κB | Nuclear factor kappa B |
| NIR | Near infrared |
| NLCs | Nanostructured lipid carriers |
| NMDA | N-methyl-D-aspartate |
| NO | Nitric oxide |
| NP | Nanoparticle |
| NPS | Nanostructured plasmonic substrate |
| NSAIDs | Non-steroidal anti-inflammatory drugs |
| NSE | Neuron-specific enolase |
| ORP | Oxygen reactive polymer |
| PBCA | Poly (butyl cyanoacrylate) |
| PD | Parkinson’s disease |
| PEG-PLGA | Polyethylene glycol-poly lactic acid-co-glycolic acid |
| PFC | Perfluorocarbon |
| PGE2 | Prostaglandin E2 |
| PLGA | Poly (lactic-co-glycolic acid) |
| QUIN | Quinolinic acid |
| RES | Reticuloendothelial system |
| RGD | Arginylglycylaspartic acid |
| RMT | Receptor-mediated transcytosis |
| RNPs | redox-active nitroxide-containing nanoparticles |
| ROS | Reactive oxygen species |
| RP | Ropinirole |
| SLNs | Solid lipid nanoparticles |
| SNCA | Synuclein alpha gene |
| SNc | Substantia nigra pars compacta |
| TBI | Traumatic brain injury |
| ThT | Thioflavin-T |
| TNF-α | Tumor necrosis factor alpha |
| tPA | Tissue-type plasminogen activator |
| US | United States |
| VEcad | VE cadherin |
| VLDL | Very-low density lipoprotein |
| WGA | Wheat germ agglutinin |
| WHO | World Health Organization |
References
- Bartels, S.J.; Naslund, J.A. The underside of the silver tsunami--older adults and mental health care. N. Engl. J. Med. 2013, 368, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Blasi, P.; Giovagnoli, S.; Schoubben, A.; Ricci, M.; Rossi, C. Solid lipid nanoparticles for targeted brain drug delivery. Adv. Drug Deliv. Rev. 2007, 59, 454–477. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Neurological disorders; public health challenges. Available online: https://www.who.int/mental_health/publications/neurological_disorders_ph_challenges/en/ (accessed on 18 July 2020).
- Feigin, V.L.; Nichols, E.; Alam, T.; Bannick, M.S.; Beghi, E.; Blake, N.; Culpepper, W.J.; Dorsey, E.R.; Elbaz, A.; Ellenbogen, R.G.; et al. Global, regional, and national burden of neurological disorders, 1990-2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 459–480. [Google Scholar] [CrossRef] [Green Version]
- Wohlfart, S.; Gelperina, S.; Kreuter, J. Transport of drugs across the blood-brain barrier by nanoparticles. J. Control Release 2012, 161, 264–273. [Google Scholar] [CrossRef]
- Kanwar, J.R.; Sriramoju, B.; Kanwar, R.K. Neurological disorders and therapeutics targeted to surmount the blood-brain barrier. Int. J. Nanomed. 2012, 7, 3259–3278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pringsheim, T.; Fiest, K.; Jette, N. The international incidence and prevalence of neurologic conditions: How common are they? Neurology 2014, 83, 1661–1664. [Google Scholar] [CrossRef] [Green Version]
- Neuwelt, E.; Abbott, N.J.; Abrey, L.; Banks, W.A.; Blakley, B.; Davis, T.; Engelhardt, B.; Grammas, P.; Nedergaard, M.; Nutt, J.; et al. Strategies to advance translational research into brain barriers. Lancet Neurol. 2008, 7, 84–96. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, L. Modern methods for delivery of drugs across the blood-brain barrier. Adv. Drug Deliv. Rev. 2012, 64, 640–665. [Google Scholar] [CrossRef]
- Lee, G.; Dallas, S.; Hong, M.; Bendayan, R. Drug transporters in the central nervous system: Brain barriers and brain parenchyma considerations. Pharmacol. Rev. 2001, 53, 569–596. [Google Scholar] [CrossRef]
- Pardridge, W.M. Drug targeting to the brain. Pharm. Res. 2007, 24, 1733–1744. [Google Scholar] [CrossRef]
- Holmes, D. The next big things are tiny. Lancet Neurol. 2013, 12, 31–32. [Google Scholar] [CrossRef]
- Youns, M.; Hoheisel, J.D.; Efferth, T. Therapeutic and diagnostic applications of nanoparticles. Curr. Drug Targets 2011, 12, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Shakeri, S.; Ashrafizadeh, M.; Zarrabi, A.; Roghanian, R.; Afshar, E.G.; Pardakhty, A.; Mohammadinejad, R.; Kumar, A.; Thakur, V.K. Multifunctional Polymeric Nanoplatforms for Brain Diseases Diagnosis, Therapy and Theranostics. Biomedicines 2020, 8, 13. [Google Scholar] [CrossRef] [Green Version]
- Xie, A.; Gao, J.; Xu, L.; Meng, D. Shared mechanisms of neurodegeneration in Alzheimer’s disease and Parkinson’s disease. Biomed. Res. Int. 2014, 2014, 648740. [Google Scholar] [CrossRef] [PubMed]
- Van Deerlin, V.M. The genetics and neuropathology of neurodegenerative disorders: Perspectives and implications for research and clinical practice. Acta Neuropathol. 2012, 124, 297–303. [Google Scholar] [CrossRef]
- Garcia, J.C.; Bustos, R.H. The Genetic Diagnosis of Neurodegenerative Diseases and Therapeutic Perspectives. Brain Sci. 2018, 8, 222. [Google Scholar] [CrossRef] [Green Version]
- Zlokovic, B.V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 2011, 12, 723–738. [Google Scholar] [CrossRef]
- Banks, W.A. Drug delivery to the brain in Alzheimer’s disease: Consideration of the blood-brain barrier. Adv. Drug Deliv. Rev. 2012, 64, 629–639. [Google Scholar] [CrossRef] [Green Version]
- Crous-Bou, M.; Minguillon, C.; Gramunt, N.; Molinuevo, J.L. Alzheimer’s disease prevention: From risk factors to early intervention. Alzheimers Res. Ther. 2017, 9, 71. [Google Scholar] [CrossRef]
- Guerreiro, R.; Bras, J. The age factor in Alzheimer’s disease. Genome Med. 2015, 7, 106. [Google Scholar] [CrossRef] [Green Version]
- Weintraub, S.; Wicklund, A.H.; Salmon, D.P. The neuropsychological profile of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006171. [Google Scholar] [CrossRef]
- Schultz, C.; Tredici, K.D.; Braak, H. Neuropathology of Alzheimer’s Disease. In Alzheimer’s Disease. Current Clinical Neurology; Richter, R.W., Richter, B.Z.e., Eds.; Humana Press: Totowa, NJ, USA, 2004. [Google Scholar]
- Perl, D.P. Neuropathology of Alzheimer’s disease. Mt. Sinai J. Med. 2010, 77, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yilmazer-Hanke, D.M.; Hanke, J. Progression of Alzheimer-related neuritic plaque pathology in the entorhinal region, perirhinal cortex and hippocampal formation. Dement. Geriatr. Cogn. Disord. 1999, 10, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Flores-Martinez, E.; Pena-Ortega, F. Amyloid beta Peptide-Induced Changes in Prefrontal Cortex Activity and Its Response to Hippocampal Input. Int. J. Pept. 2017, 2017, 7386809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selkoe, D.J. The molecular pathology of Alzheimer’s disease. Neuron 1991, 6, 487–498. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Pan, M.H.; Lai, C.S.; Ho, C.T. Anti-inflammatory activity of natural dietary flavonoids. Food Funct. 2010, 1, 15–31. [Google Scholar] [CrossRef]
- Price, J.L.; Davis, P.B.; Morris, J.C.; White, D.L. The distribution of tangles, plaques and related immunohistochemical markers in healthy aging and Alzheimer’s disease. Neurobiol. Aging. 1991, 12, 295–312. [Google Scholar] [CrossRef]
- Ricciarelli, R.; Fedele, E. The amyloid cascade hypothesis in Alzheimer’s disease: It’s time to change our mind. Curr. Neuropharmacol. 2017, 15, 926–935. [Google Scholar] [CrossRef] [Green Version]
- Whitehouse, P.J.; Price, D.L.; Clark, A.W.; Coyle, J.T.; DeLong, M.R. Alzheimer disease: Evidence for selective loss of cholinergic neurons in the nucleus basalis. Ann. Neurol. 1981, 10, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s disease: Targeting the Cholinergic System. Curr. Neuropharmacol 2016, 14, 101–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farlow, M. A clinical overview of cholinesterase inhibitors in Alzheimer’s disease. Int. Psychogeriatr. 2002, 14 (Suppl. 1), 93–126. [Google Scholar] [CrossRef] [PubMed]
- Pope, C.; Karanth, S.; Liu, J. Pharmacology and toxicology of cholinesterase inhibitors: Uses and misuses of a common mechanism of action. Environ. Toxicol. Pharmacol. 2005, 19, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Ogura, H.; Kosasa, T.; Kuriya, Y.; Yamanishi, Y. Comparison of inhibitory activities of donepezil and other cholinesterase inhibitors on acetylcholinesterase and butyrylcholinesterase in vitro. Methods Find. Exp. Clin. Pharmacol. 2000, 22, 609–613. [Google Scholar] [CrossRef]
- Folch, J.; Busquets, O.; Ettcheto, M.; Sanchez-Lopez, E.; Castro-Torres, R.D.; Verdaguer, E.; Garcia, M.L.; Olloquequi, J.; Casadesus, G.; Beas-Zarate, C.; et al. Memantine for the Treatment of Dementia: A Review on its Current and Future Applications. J. Alzheimers Dis. 2018, 62, 1223–1240. [Google Scholar] [CrossRef] [Green Version]
- Kutzing, M.K.; Luo, V.; Firestein, B.L. Protection from glutamate-induced excitotoxicity by memantine. Ann. Biomed. Eng. 2012, 40, 1170–1181. [Google Scholar] [CrossRef] [Green Version]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and future treatments for Alzheimer’s disease. Ther. Adv. Neurol. Disord. 2013, 6, 19–33. [Google Scholar] [CrossRef] [Green Version]
- Imbimbo, B.P.; Giardina, G.A. Gamma-secretase inhibitors and modulators for the treatment of Alzheimer’s disease: Disappointments and hopes. Curr. Top. Med. Chem. 2011, 11, 1555–1570. [Google Scholar] [CrossRef]
- Green, R.C.; Schneider, L.S.; Amato, D.A.; Beelen, A.P.; Wilcock, G.; Swabb, E.A.; Zavitz, K.H. Tarenflurbil Phase 3 Study, G., Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild Alzheimer disease: A randomized controlled trial. JAMA 2009, 302, 2557–2564. [Google Scholar] [CrossRef] [Green Version]
- Von Campenhausen, S.; Bornschein, B.; Wick, R.; Botzel, K.; Sampaio, C.; Poewe, W.; Oertel, W.; Siebert, U.; Berger, K.; Dodel, R. Prevalence and incidence of Parkinson’s disease in Europe. Eur. Neuropsychopharmacol. 2005, 15, 473–490. [Google Scholar] [CrossRef] [PubMed]
- De Rijk, M.C.; Tzourio, C.; Breteler, M.M.; Dartigues, J.F.; Amaducci, L.; Lopez-Pousa, S.; Manubens-Bertran, J.M.; Alperovitch, A.; Rocca, W.A. Prevalence of parkinsonism and Parkinson’s disease in Europe: The EUROPARKINSON Collaborative Study. European Community Concerted Action on the Epidemiology of Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 1997, 62, 10–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizek, P.; Kumar, N.; Jog, M.S. An update on the diagnosis and treatment of Parkinson disease. CMAJ 2016, 188, 1157–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lima, M.M.; Reksidler, A.B.; Vital, M.A. The neurobiology of the substantia nigra pars compacta: From motor to sleep regulation. J. Neural. Transm. Suppl. 2009, 73, 135–145. [Google Scholar]
- Shults, C.W. Lewy bodies. Proc. Natl. Acad. Sci. USA 2006, 103, 1661–1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [Green Version]
- Sherer, T.B.; Betarbet, R.; Greenamyre, J.T. Pathogenesis of Parkinson’s disease. Curr. Opin. Investig. Drugs 2001, 2, 657–662. [Google Scholar]
- Behari, M.; Bhattacharyya, K.B.; Borgohain, R.; Das, S.K.; Ghosh, B.; Kishore, A.; Krishnan, S.; Mridula, K.R.; Muthane, U.; Pal, P.K.; et al. Parkinson’s disease. Ann. Indian Acad Neurol. 2011, 14, 2–6. [Google Scholar] [CrossRef]
- Jankovic, J.; Sherer, T. The future of research in Parkinson disease. JAMA Neurol. 2014, 71, 1351–1352. [Google Scholar] [CrossRef]
- Lang, A.E. The progression of Parkinson disease: A hypothesis. Neurology 2007, 68, 948–952. [Google Scholar] [CrossRef]
- Feng, J. Microtubule: A common target for parkin and Parkinson’s disease toxins. Neuroscientist 2006, 12, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Davis, R.L.; Sue, C.M. Mitochondrial Dysfunction in Parkinson’s Disease: New Mechanistic Insights and Therapeutic Perspectives. Curr. Neurol. Neurosci. Rep. 2018, 18, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maragakis, N.J.; Rothstein, J.D. Mechanisms of Disease: Astrocytes in neurodegenerative disease. Nat. Clin. Pract. Neurol. 2006, 2, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Moffett, J.R.; Namboodiri, M.A. Tryptophan and the immune response. Immunol. Cell Biol. 2003, 81, 247–265. [Google Scholar] [CrossRef]
- Zinger, A.; Barcia, C.; Herrero, M.T.; Guillemin, G.J. The involvement of neuroinflammation and kynurenine pathway in Parkinson’s disease. Parkinsons Dis. 2011, 2011, 716859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, C.K.; Fernandez-Gomez, F.J.; Braidy, N.; Estrada, C.; Costa, C.; Costa, S.; Bessede, A.; Fernandez-Villalba, E.; Zinger, A.; Herrero, M.T.; et al. Involvement of the kynurenine pathway in the pathogenesis of Parkinson’s disease. Prog. Neurobiol. 2017, 155, 76–95. [Google Scholar] [CrossRef]
- Pirtosek, Z.; Bajenaru, O.; Kovacs, N.; Milanov, I.; Relja, M.; Skorvanek, M. Update on the Management of Parkinson’s Disease for General Neurologists. Parkinsons Dis. 2020, 2020, 9131474. [Google Scholar] [CrossRef]
- Jankovic, J. Levodopa strengths and weaknesses. Neurology 2002, 58 (Suppl. 1), S19–S32. [Google Scholar] [CrossRef]
- Celesia, G.G.; Wanamaker, W.M. L-dopa-carbidopa: Combined therapy for the treatment of Parkinson’s disease. Dis. Nerv. Syst. 1976, 37, 123–125. [Google Scholar]
- Jankovic, J. Motor fluctuations and dyskinesias in Parkinson’s disease: Clinical manifestations. Mov. Disord. 2005, 20 (Suppl. 11), S11–S16. [Google Scholar] [CrossRef]
- Stoker, T.B.; Torsney, K.M.; Barker, R.A. Emerging Treatment Approaches for Parkinson’s Disease. Front Neurosci 2018, 12, 693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- A classification and outline of cerebrovascular diseases. II. Stroke 1975, 6, 564–616. [CrossRef] [PubMed] [Green Version]
- Chandra, A.; Stone, C.R.; Du, X.; Li, W.A.; Huber, M.; Bremer, R.; Geng, X.; Ding, Y. The cerebral circulation and cerebrovascular disease III: Stroke. Brain Circ. 2017, 3, 66–77. [Google Scholar] [PubMed]
- Rosamond, W.; Flegal, K.; Furie, K.; Go, A.; Greenlund, K.; Haase, N.; Hailpern, S.M.; Ho, M.; Howard, V.; Kissela, B.; et al. Heart Disease and Stroke Statistics—2008 Update. Circulation 2008, 117, e25–e146. [Google Scholar]
- Sacco, R.L.; Kasner, S.E.; Broderick, J.P.; Caplan, L.R.; Connors, J.J.; Culebras, A.; Elkind, M.S.; George, M.G.; Hamdan, A.D.; Higashida, R.T.; et al. An updated definition of stroke for the 21st century: A statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2013, 44, 2064–2089. [Google Scholar] [CrossRef] [Green Version]
- Qureshi, A.I.; Mendelow, A.D.; Hanley, D.F. Intracerebral haemorrhage. Lancet 2009, 373, 1632–1644. [Google Scholar] [CrossRef] [Green Version]
- Boehme, A.K.; Esenwa, C.; Elkind, M.S. Stroke Risk Factors, Genetics, and Prevention. Circ. Res. 2017, 120, 472–495. [Google Scholar] [CrossRef]
- Mestre, H.; Cohen-Minian, Y.; Zajarias-Fainsod, D.; Ibarra, A. Pharmacological treatment of acute ischemic stroke. In Neurodegenerative Diseases; Kishore, U., Ed.; IntechOpen: London, UK, 2013. [Google Scholar]
- Barreto, A.D. Intravenous thrombolytics for ischemic stroke. Neurotherapeutics 2011, 8, 388–399. [Google Scholar] [CrossRef] [Green Version]
- Coull, B.M.; Williams, L.S.; Goldstein, L.B.; Meschia, J.F.; Heitzman, D.; Chaturvedi, S.; Johnston, K.C.; Starkman, S.; Morgenstern, L.B.; Wilterdink, J.L.; et al. Anticoagulants and antiplatelet agents in acute ischemic stroke: Report of the Joint Stroke Guideline Development Committee of the American Academy of Neurology and the American Stroke Association (a division of the American Heart Association). Stroke 2002, 33, 1934–1942. [Google Scholar] [CrossRef]
- Hemphill, J.C., 3rd; Greenberg, S.M.; Anderson, C.S.; Becker, K.; Bendok, B.R.; Cushman, M.; Fung, G.L.; Goldstein, J.N.; Macdonald, R.L.; Mitchell, P.H.; et al. Guidelines for the Management of Spontaneous Intracerebral Hemorrhage: A Guideline for Healthcare Professionals from the American Heart Association/American Stroke Association. Stroke 2015, 46, 2032–2060. [Google Scholar] [CrossRef] [Green Version]
- Saver, J.L. Time is brain--quantified. Stroke 2006, 37, 263–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewan, M.C.; Rattani, A.; Gupta, S.; Baticulon, R.E.; Hung, Y.C.; Punchak, M.; Agrawal, A.; Adeleye, A.O.; Shrime, M.G.; Rubiano, A.M.; et al. Estimating the global incidence of traumatic brain injury. J. Neurosurg. 2019, 130, 1080–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faul, M.; Xu, L.; Wald, M.M.; Coronado, V.G. Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations and Deaths 2002–2006. Available online: https://www.cdc.gov/traumaticbraininjury/pdf/blue_book.pdf (accessed on 9 August 2020).
- Taylor, C.A.; Bell, J.M.; Breiding, M.J.; Xu, L. Traumatic brain injury—Related Emergency Department visits, hospitalizations, and deaths—United States, 2007 and 2013. MMWR Surveill Summ. 2017, 66, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Galgano, M.; Toshkezi, G.; Qiu, X.; Russell, T.; Chin, L.; Zhao, L.R. Traumatic Brain Injury: Current Treatment Strategies and Future Endeavors. Cell Transpl. 2017, 26, 1118–1130. [Google Scholar] [CrossRef] [Green Version]
- Rutland-Brown, W.; Langlois, J.A.; Thomas, K.E.; Xi, Y.L. Incidence of traumatic brain injury in the United States, 2003. J. Head Trauma Rehabil. 2006, 21, 544–548. [Google Scholar] [CrossRef]
- Hawthorne, G.; Gruen, R.L.; Kaye, A.H. Traumatic brain injury and long-term quality of life: Findings from an Australian study. J. Neurotrauma 2009, 26, 1623–1633. [Google Scholar] [CrossRef] [PubMed]
- Walker, W.C.; Pickett, T.C. Motor impairment after severe traumatic brain injury: A longitudinal multicenter study. J. Rehabil Res. Dev. 2007, 44, 975–982. [Google Scholar] [CrossRef]
- Pagulayan, K.F.; Temkin, N.R.; Machamer, J.E.; Dikmen, S.S. The measurement and magnitude of awareness difficulties after traumatic brain injury: A longitudinal study. J. Int. Neuropsychol. Soc. 2007, 13, 561–570. [Google Scholar] [CrossRef]
- Prins, M.; Greco, T.; Alexander, D.; Giza, C.C. The pathophysiology of traumatic brain injury at a glance. Dis. Model. Mech. 2013, 6, 1307–1315. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, K. Traumatic brain injury: Pathophysiology for neurocritical care. J. Intensive. Care 2016, 4, 29. [Google Scholar] [CrossRef] [Green Version]
- Gentile, N.T.; McIntosh, T.K. Antagonists of excitatory amino acids and endogenous opioid peptides in the treatment of experimental central nervous system injury. Ann. Emerg. Med. 1993, 22, 1028–1034. [Google Scholar] [CrossRef]
- Morganti-Kossmann, M.C.; Rancan, M.; Stahel, P.F.; Kossmann, T. Inflammatory response in acute traumatic brain injury: A double-edged sword. Curr. Opin. Crit. Care 2002, 8, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Yakovlev, A.G.; Knoblach, S.M.; Fan, L.; Fox, G.B.; Goodnight, R.; Faden, A.I. Activation of CPP32-like caspases contributes to neuronal apoptosis and neurological dysfunction after traumatic brain injury. J. Neurosci. 1997, 17, 7415–7424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, Y.; Gu, Q.; Peterson, P.L.; Muizelaar, J.P.; Lee, C.P. Mitochondrial dysfunction and calcium perturbation induced by traumatic brain injury. J. Neurotrauma 1997, 14, 23–34. [Google Scholar] [CrossRef]
- Gupta, R.; Sen, N. Traumatic brain injury: A risk factor for neurodegenerative diseases. Rev. Neurosci. 2016, 27, 93–100. [Google Scholar] [CrossRef]
- Xiong, Y.; Mahmood, A.; Chopp, M. Emerging treatments for traumatic brain injury. Expert Opin. Emerg. Drugs 2009, 14, 67–84. [Google Scholar] [CrossRef]
- Helmy, A.; Vizcaychipi, M.; Gupta, A.K. Traumatic brain injury: Intensive care management. Br. J. Anaesth. 2007, 99, 32–42. [Google Scholar] [CrossRef] [Green Version]
- Maas, A.I.; Stocchetti, N.; Bullock, R. Moderate and severe traumatic brain injury in adults. Lancet Neurol. 2008, 7, 728–741. [Google Scholar] [CrossRef]
- Narayan, R.K.; Michel, M.E.; Ansell, B.; Baethmann, A.; Biegon, A.; Bracken, M.B.; Bullock, M.R.; Choi, S.C.; Clifton, G.L.; Contant, C.F.; et al. Clinical trials in head injury. J. Neurotrauma 2002, 19, 503–557. [Google Scholar] [CrossRef]
- Saunders, N.R.; Habgood, M.D.; Mollgard, K.; Dziegielewska, K.M. The biological significance of brain barrier mechanisms: Help or hindrance in drug delivery to the central nervous system? F1000 Res. 2016, 5, 313. [Google Scholar] [CrossRef]
- Gonzalez-Mariscal, L.; Betanzos, A.; Nava, P.; Jaramillo, B.E. Tight junction proteins. Prog. Biophys. Mol. Biol. 2003, 81, 1–44. [Google Scholar] [CrossRef]
- Rubin, L.L.; Staddon, J.M. The cell biology of the blood-brain barrier. Annu. Rev. Neurosci. 1999, 22, 11–28. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pulgar, V.M. Transcytosis to Cross the Blood Brain Barrier, New Advancements and Challenges. Front. Neurosci. 2018, 12, 1019. [Google Scholar] [CrossRef]
- Jones, A.R.; Shusta, E.V. Blood-brain barrier transport of therapeutics via receptor-mediation. Pharm. Res. 2007, 24, 1759–1771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffy, K.R.; Pardridge, W.M. Blood-brain barrier transcytosis of insulin in developing rabbits. Brain Res. 1987, 420, 32–38. [Google Scholar] [CrossRef]
- Jefferies, W.A.; Brandon, M.R.; Hunt, S.V.; Williams, A.F.; Gatter, K.C.; Mason, D.Y. Transferrin receptor on endothelium of brain capillaries. Nature 1984, 312, 162–163. [Google Scholar] [CrossRef]
- Golden, P.L.; Maccagnan, T.J.; Pardridge, W.M. Human blood-brain barrier leptin receptor. Binding and endocytosis in isolated human brain microvessels. J. Clin. Invest. 1997, 99, 14–18. [Google Scholar] [CrossRef] [Green Version]
- Bickel, U. Antibody delivery through the blood-brain barrier. Adv. Drug Deliv. Rev. 1995, 15, 53–72. [Google Scholar] [CrossRef]
- Kumagai, A.K.; Eisenberg, J.B.; Pardridge, W.M. Absorptive-mediated endocytosis of cationized albumin and a beta-endorphin-cationized albumin chimeric peptide by isolated brain capillaries. Model system of blood-brain barrier transport. J. Biol. Chem. 1987, 262, 15214–15219. [Google Scholar]
- Dean, M.; Rzhetsky, A.; Allikmets, R. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 2001, 11, 1156–1166. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, I.; Hamada, H.; Tsuruo, T.; Mori, S. Specialized localization of P-glycoprotein recognized by MRK 16 monoclonal antibody in endothelial cells of the brain and the spinal cord. Jpn. J. Cancer Res. 1990, 81, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Endicott, J.A.; Ling, V. The biochemistry of P-glycoprotein-mediated multidrug resistance. Annu. Rev. Biochem. 1989, 58, 137–171. [Google Scholar] [CrossRef] [PubMed]
- Schinkel, A.H. P-Glycoprotein, a gatekeeper in the blood-brain barrier. Adv. Drug Deliv. Rev. 1999, 36, 179–194. [Google Scholar] [CrossRef]
- Kemper, E.M.; van Zandbergen, A.E.; Cleypool, C.; Mos, H.A.; Boogerd, W.; Beijnen, J.H.; van Tellingen, O. Increased penetration of paclitaxel into the brain by inhibition of P-Glycoprotein. Clin. Cancer Res. 2003, 9, 2849–2855. [Google Scholar] [PubMed]
- Sanchez-Covarrubias, L.; Slosky, L.M.; Thompson, B.J.; Davis, T.P.; Ronaldson, P.T. Transporters at CNS barrier sites: Obstacles or opportunities for drug delivery? Curr. Pharm. Des. 2014, 20, 1422–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soares, T.B.; Loureiro, L.; Carvalho, A.; Oliveira, M.; Dias, A.; Sarmento, B.; Lucio, M. Lipid nanocarriers loaded with natural compounds: Potential new therapies for age related neurodegenerative diseases? Prog. Neurobiol. 2018, 168, 21–41. [Google Scholar] [CrossRef]
- Athira, S.S.; Prajitha, N.; Mohanan, P.V. Interaction of nanoparticles with central nervous system and its consequences. Am. J. Res. Med. Sci. 2018, 4, 12–32. [Google Scholar] [CrossRef]
- Buzea, C.; Pacheco, I.I.; Robbie, K. Nanomaterials and nanoparticles: Sources and toxicity. Biointerphases 2007, 2, MR17-71. [Google Scholar] [CrossRef] [Green Version]
- Friedman, A.D.; Claypool, S.E.; Liu, R. The smart targeting of nanoparticles. Curr. Pharm. Des. 2013, 19, 6315–6329. [Google Scholar] [CrossRef] [Green Version]
- Montet, X.; Funovics, M.; Montet-Abou, K.; Weissleder, R.; Josephson, L. Multivalent effects of RGD peptides obtained by nanoparticle display. J. Med. Chem. 2006, 49, 6087–6093. [Google Scholar] [CrossRef] [PubMed]
- De Matteis, V.; Rinaldi, R. Toxicity Assessment in the Nanoparticle Era. Adv. Exp. Med. Biol. 2018, 1048, 1–19. [Google Scholar] [PubMed]
- Petkar, K.C.; Chavhan, S.S.; Agatonovik-Kustrin, S.; Sawant, K.K. Nanostructured materials in drug and gene delivery: A review of the state of the art. Crit. Rev. Ther. Drug Carr. Syst. 2011, 28, 101–164. [Google Scholar] [CrossRef] [PubMed]
- Li, S.D.; Huang, L. Nanoparticles evading the reticuloendothelial system: Role of the supported bilayer. Biochim. Biophys. Acta 2009, 1788, 2259–2266. [Google Scholar] [CrossRef] [Green Version]
- Klibanov, A.L.; Maruyama, K.; Torchilin, V.P.; Huang, L. Amphipathic polyethyleneglycols effectively prolong the circulation time of liposomes. FEBS Lett. 1990, 268, 235–237. [Google Scholar] [CrossRef] [Green Version]
- Woodle, M.C.; Collins, L.R.; Sponsler, E.; Kossovsky, N.; Papahadjopoulos, D.; Martin, F.J. Sterically stabilized liposomes. Reduction in electrophoretic mobility but not electrostatic surface potential. Biophys J. 1992, 61, 902–910. [Google Scholar] [CrossRef] [Green Version]
- Teleanu, D.M.; Chircov, C.; Grumezescu, A.M.; Volceanov, A.; Teleanu, R.I. Blood-Brain Delivery Methods Using Nanotechnology. Pharmaceutics 2018, 10, 269. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Wang, C.; Sun, J.; Xue, Y. Neurotoxicity of silica nanoparticles: Brain localization and dopaminergic neurons damage pathways. ACS Nano. 2011, 5, 4476–4489. [Google Scholar] [CrossRef]
- Sousa, F.; Mandal, S.; Garrovo, C.; Astolfo, A.; Bonifacio, A.; Latawiec, D.; Menk, R.H.; Arfelli, F.; Huewel, S.; Legname, G.; et al. Functionalized gold nanoparticles: A detailed in vivo multimodal microscopic brain distribution study. Nanoscale 2010, 2, 2826–2834. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.; Bae, K.H.; Yang, H.; Lee, S.J.; Kim, H.; Kim, Y.; Joo, K.M.; Seo, S.W.; Park, T.G.; Nam, D.H. In vivo specific delivery of c-Met siRNA to glioblastoma using cationic solid lipid nanoparticles. Bioconjug. Chem. 2011, 22, 2568–2572. [Google Scholar] [CrossRef]
- Bu, G. Apolipoprotein E and its receptors in Alzheimer’s disease: Pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 2009, 10, 333–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laskowitz, D.T.; Thekdi, A.D.; Thekdi, S.D.; Han, S.K.; Myers, J.K.; Pizzo, S.V.; Bennett, E.R. Downregulation of microglial activation by apolipoprotein E and apoE-mimetic peptides. Exp. Neurol. 2001, 167, 74–85. [Google Scholar] [CrossRef]
- Moos, T.; Rosengren Nielsen, T.; Skjorringe, T.; Morgan, E.H. Iron trafficking inside the brain. J. Neurochem. 2007, 103, 1730–1740. [Google Scholar] [CrossRef] [PubMed]
- Mishra, V.; Mahor, S.; Rawat, A.; Gupta, P.N.; Dubey, P.; Khatri, K.; Vyas, S.P. Targeted brain delivery of AZT via transferrin anchored pegylated albumin nanoparticles. J. Drug Target. 2006, 14, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Ulbrich, K.; Hekmatara, T.; Herbert, E.; Kreuter, J. Transferrin- and transferrin-receptor-antibody-modified nanoparticles enable drug delivery across the blood-brain barrier (BBB). Eur. J. Pharm. Biopharm. 2009, 71, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.Z.; Ahmad, J.; Amin, S.; Rahman, M.; Anwar, M.; Mallick, N.; Ahmad, F.J.; Rahman, Z.; Kamal, M.A.; Akhter, S. Role of nanomedicines in delivery of anti-acetylcholinesterase compounds to the brain in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2014, 13, 1315–1324. [Google Scholar] [CrossRef]
- Wilson, B.; Samanta, M.K.; Santhi, K.; Kumar, K.P.; Paramakrishnan, N.; Suresh, B. Poly(n-butylcyanoacrylate) nanoparticles coated with polysorbate 80 for the targeted delivery of rivastigmine into the brain to treat Alzheimer’s disease. Brain Res. 2008, 1200, 159–168. [Google Scholar] [CrossRef]
- Borchard, G.; Audus, K.L.; Shi, F.; Kreuter, J. Uptake of surfactant-coated poly(methyl methacrylate)-nanoparticles by bovine brain microvessel endothelial cell monolayers. Int. J. Pharm. 1994, 110, 29–35. [Google Scholar] [CrossRef]
- Sun, W.; Xie, C.; Wang, H.; Hu, Y. Specific role of polysorbate 80 coating on the targeting of nanoparticles to the brain. Biomaterials 2004, 25, 3065–3071. [Google Scholar] [CrossRef]
- Markesbery, W.R. The role of oxidative stress in Alzheimer disease. Arch. Neurol. 1999, 56, 1449–1452. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Garrett, M.R.; Men, P.; Zhu, X.; Perry, G.; Smith, M.A. Nanoparticle and other metal chelation therapeutics in Alzheimer disease. Biochim. Biophys. Acta 2005, 1741, 246–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.; Hu, M.J.; Wang, Y.Q.; Cui, Y.L. Antioxidant Activities of Quercetin and Its Complexes for Medicinal Application. Molecules 2019, 24, 1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Park, B.S.; Lee, K.G.; Choi, C.Y.; Jang, S.S.; Kim, Y.H.; Lee, S.E. Effects of naturally occurring compounds on fibril formation and oxidative stress of beta-amyloid. J. Agric. Food Chem. 2005, 53, 8537–8541. [Google Scholar] [CrossRef] [PubMed]
- Nday, C.M.; Halevas, E.; Jackson, G.E.; Salifoglou, A. Quercetin encapsulation in modified silica nanoparticles: Potential use against Cu(II)-induced oxidative stress in neurodegeneration. J. Inorg. Biochem. 2015, 145, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, S.; Kapil, R.; Singh, B. Formulation development and systematic optimization of solid lipid nanoparticles of quercetin for improved brain delivery. J. Pharm. Pharmacol. 2011, 63, 342–351. [Google Scholar] [CrossRef]
- Thorn, D.C.; Heath, E.; Carver, J.A. The two-faced nature of milk casein proteins: Amyloid fibril formation and chaperone-like activity. Aust. J. Dairy Technol. 2009, 64, 34–40. [Google Scholar]
- Javed, I.; Peng, G.; Xing, Y.; Yu, T.; Zhao, M.; Kakinen, A.; Faridi, A.; Parish, C.L.; Ding, F.; Davis, T.P.; et al. Inhibition of amyloid beta toxicity in zebrafish with a chaperone-gold nanoparticle dual strategy. Nat. Commun. 2019, 10, 3780. [Google Scholar] [CrossRef]
- Brambilla, D.; Le Droumaguet, B.; Nicolas, J.; Hashemi, S.H.; Wu, L.P.; Moghimi, S.M.; Couvreur, P.; Andrieux, K. Nanotechnologies for Alzheimer’s disease: Diagnosis, therapy, and safety issues. Nanomedicine 2011, 7, 521–540. [Google Scholar] [CrossRef]
- Biancalana, M.; Koide, S. Molecular mechanism of Thioflavin-T binding to amyloid fibrils. Biochim. Biophys. Acta 2010, 1804, 1405–1412. [Google Scholar] [CrossRef] [Green Version]
- Klunk, W.E.; Wang, Y.; Huang, G.F.; Debnath, M.L.; Holt, D.P.; Mathis, C.A. Uncharged thioflavin-T derivatives bind to amyloid-beta protein with high affinity and readily enter the brain. Life Sci. 2001, 69, 1471–1484. [Google Scholar] [CrossRef]
- Schroeder, U.; Sommerfeld, P.; Ulrich, S.; Sabel, B.A. Nanoparticle technology for delivery of drugs across the blood-brain barrier. J. Pharm. Sci. 1998, 87, 1305–1307. [Google Scholar] [CrossRef] [PubMed]
- Hartig, W.; Paulke, B.R.; Varga, C.; Seeger, J.; Harkany, T.; Kacza, J. Electron microscopic analysis of nanoparticles delivering thioflavin-T after intrahippocampal injection in mouse: Implications for targeting beta-amyloid in Alzheimer’s disease. Neurosci. Lett. 2003, 338, 174–176. [Google Scholar] [CrossRef]
- Wadghiri, Y.Z.; Sigurdsson, E.M.; Sadowski, M.; Elliott, J.I.; Li, Y.; Scholtzova, H.; Tang, C.Y.; Aguinaldo, G.; Pappolla, M.; Duff, K.; et al. Detection of Alzheimer’s amyloid in transgenic mice using magnetic resonance microimaging. Magn. Reson. Med. 2003, 50, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Trapani, A.; De Giglio, E.; Cafagna, D.; Denora, N.; Agrimi, G.; Cassano, T.; Gaetani, S.; Cuomo, V.; Trapani, G. Characterization and evaluation of chitosan nanoparticles for dopamine brain delivery. Int J. Pharm. 2011, 419, 296–307. [Google Scholar] [CrossRef]
- Pillay, S.; Pillay, V.; Choonara, Y.E.; Naidoo, D.; Khan, R.A.; du Toit, L.C.; Ndesendo, V.M.; Modi, G.; Danckwerts, M.P.; Iyuke, S.E. Design, biometric simulation and optimization of a nano-enabled scaffold device for enhanced delivery of dopamine to the brain. Int. J. Pharm. 2009, 382, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Mao, S.; Sun, W.; Kissel, T. Chitosan-based formulations for delivery of DNA and siRNA. Adv. Drug Deliv. Rev. 2010, 62, 12–27. [Google Scholar] [CrossRef]
- Jost, W.H.; Buhmann, C.; Fuchs, G.; Greulich, W.; Hummel, S.; Korchounov, A.; Mungersdorf, M.; Schwarz, M.; Spiegel-Meixensberger, M. Initial experience with ropinirole PR (prolonged release). J. Neurol. 2008, 255 (Suppl. 5), 60–63. [Google Scholar] [CrossRef]
- Barcia, E.; Boeva, L.; Garcia-Garcia, L.; Slowing, K.; Fernandez-Carballido, A.; Casanova, Y.; Negro, S. Nanotechnology-based drug delivery of ropinirole for Parkinson’s disease. Drug Deliv. 2017, 24, 1112–1123. [Google Scholar] [CrossRef] [Green Version]
- Dudhipala, N.; Gorre, T. Neuroprotective Effect of Ropinirole Lipid Nanoparticles Enriched Hydrogel for Parkinson’s Disease: In Vitro, Ex Vivo, Pharmacokinetic and Pharmacodynamic Evaluation. Pharmaceutics 2020, 12, 448. [Google Scholar] [CrossRef]
- Tsai, M.J.; Huang, Y.B.; Wu, P.C.; Fu, Y.S.; Kao, Y.R.; Fang, J.Y.; Tsai, Y.H. Oral apomorphine delivery from solid lipid nanoparticles with different monostearate emulsifiers: Pharmacokinetic and behavioral evaluations. J. Pharm. Sci. 2011, 100, 547–557. [Google Scholar] [CrossRef]
- Lorigados Pedre, L.; Pavon Fuentes, N.; Alvarez Gonzalez, L.; McRae, A.; Serrano Sanchez, T.; Blanco Lescano, L.; Macias Gonzalez, R. Nerve growth factor levels in Parkinson disease and experimental parkinsonian rats. Brain Res. 2002, 952, 122–127. [Google Scholar] [CrossRef]
- Kurakhmaeva, K.B.; Djindjikhashvili, I.A.; Petrov, V.E.; Balabanyan, V.U.; Voronina, T.A.; Trofimov, S.S.; Kreuter, J.; Gelperina, S.; Begley, D.; Alyautdin, R.N. Brain targeting of nerve growth factor using poly(butyl cyanoacrylate) nanoparticles. J. Drug Target. 2009, 17, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Hernando, S.; Herran, E.; Figueiro-Silva, J.; Pedraz, J.L.; Igartua, M.; Carro, E.; Hernandez, R.M. Intranasal Administration of TAT-Conjugated Lipid Nanocarriers Loading GDNF for Parkinson’s Disease. Mol. Neurobiol. 2018, 55, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Ngwuluka, N.C.; Pillay, V.; Choonara, Y.E.; Modi, G.; Naidoo, D.; du Toit, L.C.; Kumar, P.; Ndesendo, V.M.; Khan, R.A. Fabrication, modeling and characterization of multi-crosslinked methacrylate copolymeric nanoparticles for oral drug delivery. Int. J. Mol. Sci. 2011, 12, 6194–6225. [Google Scholar] [CrossRef]
- Muthuraman, M.; Koirala, N.; Ciolac, D.; Pintea, B.; Glaser, M.; Groppa, S.; Tamas, G.; Groppa, S. Deep Brain Stimulation and L-DOPA Therapy: Concepts of Action and Clinical Applications in Parkinson’s Disease. Front. Neurol. 2018, 9, 711. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Tao, W.; Lu, W.; Zhang, Q.; Zhang, Y.; Jiang, X.; Fu, S. Lectin-conjugated PEG-PLA nanoparticles: Preparation and brain delivery after intranasal administration. Biomaterials 2006, 27, 3482–3490. [Google Scholar] [CrossRef]
- Arisoy, S.; Sayiner, O.; Comoglu, T.; Onal, D.; Atalay, O.; Pehlivanoglu, B. In vitro and in vivo evaluation of levodopa-loaded nanoparticles for nose to brain delivery. Pharm. Dev. Technol. 2020, 25, 735–747. [Google Scholar] [CrossRef]
- Shin, J.-W.; Yoon, J.; Shin, M.; Choi, J.-W. Electrochemical Dopamine Biosensor Composed of Silver Encapsulated MoS2 Hybrid Nanoparticle. Biotechnol. Bioprocess Eng. 2019, 24, 135–144. [Google Scholar] [CrossRef]
- Vazquez-Guardado, A.; Barkam, S.; Peppler, M.; Biswas, A.; Dennis, W.; Das, S.; Seal, S.; Chanda, D. Enzyme-Free Plasmonic Biosensor for Direct Detection of Neurotransmitter Dopamine from Whole Blood. Nano Lett. 2019, 19, 449–454. [Google Scholar] [CrossRef]
- You, X.; Gopinath, S.C.B.; Lakshmipriya, T.; Li, D. High-Affinity Detection of Alpha-Synuclein by Aptamer-Gold Conjugates on an Amine-Modified Dielectric Surface. J. Anal. Methods Chem. 2019, 2019, 6526850. [Google Scholar] [CrossRef] [Green Version]
- Powers, W.J.; Rabinstein, A.A.; Ackerson, T.; Adeoye, O.M.; Bambakidis, N.C.; Becker, K.; Biller, J.; Brown, M.; Demaerschalk, B.M.; Hoh, B.; et al. American Heart Association Stroke, C., 2018 Guidelines for the Early Management of Patients With Acute Ischemic Stroke: A Guideline for Healthcare Professionals From the American Heart Association/American Stroke Association. Stroke 2018, 49, e46–e110. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Li, C.; Zhou, D.; Ding, C.; Jin, Y.; Tian, Q.; Meng, X.; Pu, K.; Zhu, Y. Cyclic RGD functionalized liposomes encapsulating urokinase for thrombolysis. Acta Biomater. 2018, 70, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Juenet, M.; Aid-Launais, R.; Li, B.; Berger, A.; Aerts, J.; Ollivier, V.; Nicoletti, A.; Letourneur, D.; Chauvierre, C. Thrombolytic therapy based on fucoidan-functionalized polymer nanoparticles targeting P-selectin. Biomaterials 2018, 156, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Juenet, M.; Aid-Launais, R.; Maire, M.; Ollivier, V.; Letourneur, D.; Chauvierre, C. Development of Polymer Microcapsules Functionalized with Fucoidan to Target P-Selectin Overexpressed in Cardiovascular Diseases. Adv. Healthc. Mater. 2017, 6, 1601200. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Jin, R.; Wang, M.; Li, G. Nanoparticle Delivery of CD147 Antagonistic Peptide-9 Protects against Acute Ischemic Brain Injury and tPA-Induced Intracerebral Hemorrhage in Mice. ACS Appl. Bio. Mater. 2020, 3, 1976–1985. [Google Scholar] [CrossRef]
- Jeong, H.G.; Cha, B.G.; Kang, D.W.; Kim, D.Y.; Ki, S.K.; Kim, S.I.; Han, J.H.; Yang, W.; Kim, C.K.; Kim, J.; et al. Ceria nanoparticles synthesized with aminocaproic acid for the treatment of subarachnoid Hemorrhage. Stroke 2018, 49, 3030–3038. [Google Scholar] [CrossRef]
- You, Z.Q.; Wu, Q.; Zhou, X.M.; Zhang, X.S.; Yuan, B.; Wen, L.L.; Xu, W.D.; Cui, S.; Tang, X.L.; Zhang, X. Receptor-mediated delivery of Astaxanthin-loaded nanoparticles to neurons: An enhanced potential for subarachnoid hemorrhage treatment. Front. Neurosci. 2019, 13, 989. [Google Scholar] [CrossRef]
- Bertram, J.P.; Williams, C.A.; Robinson, R.; Segal, S.S.; Flynn, N.T.; Lavik, E.B. Intravenous hemostat: Nanotechnology to halt bleeding. Sci. Transl. Med. 2009, 1, 11ra22. [Google Scholar] [CrossRef] [Green Version]
- Anselmo, A.C.; Modery-Pawlowski, C.L.; Menegatti, S.; Kumar, S.; Vogus, D.R.; Tian, L.L.; Chen, M.; Squires, T.M.; Sen Gupta, A.; Mitragotri, S. Platelet-like nanoparticles: Mimicking shape, flexibility, and surface biology of platelets to target vascular injuries. ACS Nano 2014, 8, 11243–11253. [Google Scholar] [CrossRef]
- Gryshchuk, V.; Galagan, N. Silica Nanoparticles Effects on Blood Coagulation Proteins and Platelets. Biochem. Res. Int. 2016, 2016, 2959414. [Google Scholar] [CrossRef] [Green Version]
- Jin, A.Y.; Tuor, U.I.; Rushforth, D.; Filfil, R.; Kaur, J.; Ni, F.; Tomanek, B.; Barber, P.A. Magnetic resonance molecular imaging of post-stroke neuroinflammation with a P-selectin targeted iron oxide nanoparticle. Contrast Media Mol. Imaging 2009, 4, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Hubert, V.; Dumot, C.; Ong, E.; Amaz, C.; Canet-Soulas, E.; Chauveau, F.; Wiart, M. MRI coupled with clinically-applicable iron oxide nanoparticles reveals choroid plexus involvement in a murine model of neuroinflammation. Sci. Rep. 2019, 9, 10046. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.Y.; Kwong, G.A.; Warren, A.D.; Wood, D.K.; Bhatia, S.N. Nanoparticles that sense thrombin activity as synthetic urinary biomarkers of thrombosis. ACS Nano 2013, 7, 9001–9009. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, V.N.; Nguyen, D.T.; Kodibagkar, V.D.; Stabenfeldt, S.E. Nanoparticle-Based Therapeutics for Brain Injury. Adv. Healthc. Mater. 2018, 7, 1700668. [Google Scholar] [CrossRef]
- Xu, J.; Ypma, M.; Chiarelli, P.A.; Park, J.; Ellenbogen, R.G.; Stayton, P.S.; Mourad, P.D.; Lee, D.; Convertine, A.J.; Kievit, F.M. Theranostic Oxygen Reactive Polymers for Treatment of Traumatic Brain Injury. Adv. Funct. Mater. 2016, 26, 4124–4133. [Google Scholar] [CrossRef]
- Bailey, Z.S.; Nilson, E.; Bates, J.A.; Oyalowo, A.; Hockey, K.S.; Sajja, V.; Thorpe, C.; Rogers, H.; Dunn, B.; Frey, A.S.; et al. Cerium Oxide Nanoparticles Improve Outcome after In Vitro and In Vivo Mild Traumatic Brain Injury. J. Neurotrauma 2020, 37, 1452–1462. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Ifergan, I.; Kurz, J.E.; Linsenmeier, R.A.; Xu, D.; Cooper, J.G.; Miller, S.D.; Kessler, J.A. Intravenous Immunomodulatory Nanoparticle Treatment for Traumatic Brain Injury. Ann. Neurol. 2020, 87, 442–455. [Google Scholar] [CrossRef]
- Yoo, D.; Magsam, A.W.; Kelly, A.M.; Stayton, P.S.; Kievit, F.M.; Convertine, A.J. Core-Cross-Linked Nanoparticles Reduce Neuroinflammation and Improve Outcome in a Mouse Model of Traumatic Brain Injury. ACS Nano 2017, 11, 8600–8611. [Google Scholar] [CrossRef]
- Khalin, I.; Alyautdin, R.; Wong, T.W.; Gnanou, J.; Kocherga, G.; Kreuter, J. Brain-derived neurotrophic factor delivered to the brain using poly (lactide-co-glycolide) nanoparticles improves neurological and cognitive outcome in mice with traumatic brain injury. Drug Deliv. 2016, 23, 3520–3528. [Google Scholar] [CrossRef] [Green Version]
- Allen, S.J.; Watson, J.J.; Shoemark, D.K.; Barua, N.U.; Patel, N.K. GDNF, NGF and BDNF as therapeutic options for neurodegeneration. Pharmacol. Ther. 2013, 138, 155–175. [Google Scholar] [CrossRef]
- Clond, M.A.; Lee, B.S.; Yu, J.J.; Singer, M.B.; Amano, T.; Lamb, A.W.; Drazin, D.; Kateb, B.; Ley, E.J.; Yu, J.S. Reactive oxygen species-activated nanoprodrug of Ibuprofen for targeting traumatic brain injury in mice. PLoS ONE 2013, 8, e61819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, T.; Marushima, A.; Nagasaki, Y.; Hirayama, A.; Muroi, A.; Puentes, S.; Mujagic, A.; Ishikawa, E.; Matsumura, A. Novel neuroprotection using antioxidant nanoparticles in a mouse model of head trauma. J. Trauma Acute Care Surg. 2020, 88, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Hosoo, H.; Marushima, A.; Nagasaki, Y.; Hirayama, A.; Ito, H.; Puentes, S.; Mujagic, A.; Tsurushima, H.; Tsuruta, W.; Suzuki, K.; et al. Neurovascular unit protection from cerebral ischemia-reperfusion injury by radical-containing nanoparticles in mice. Stroke 2017, 48, 2238–2247. [Google Scholar] [CrossRef] [PubMed]
- Chonpathompikunlert, P.; Fan, C.H.; Ozaki, Y.; Yoshitomi, T.; Yeh, C.K.; Nagasaki, Y. Redox nanoparticle treatment protects against neurological deficit in focused ultrasound-induced intracerebral hemorrhage. Nanomedicine (Lond.) 2012, 7, 1029–1043. [Google Scholar] [CrossRef]
- Marushima, A.; Suzuki, K.; Nagasaki, Y.; Yoshitomi, T.; Toh, K.; Tsurushima, H.; Hirayama, A.; Matsumura, A. Newly synthesized radical-containing nanoparticles enhance neuroprotection after cerebral ischemia-reperfusion injury. Neurosurgery 2011, 68, 1418–1425. [Google Scholar] [CrossRef] [PubMed]
- Mei, T.; Kim, A.; Vong, L.B.; Marushima, A.; Puentes, S.; Matsumaru, Y.; Matsumura, A.; Nagasaki, Y. Encapsulation of tissue plasminogen activator in pH-sensitive self-assembled antioxidant nanoparticles for ischemic stroke treatment—Synergistic effect of thrombolysis and antioxidant. Biomaterials 2019, 215, 119209. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, P.; Mao, L.; Hou, Y.; Li, D. Determination of brain injury biomarkers by surface-enhanced Raman scattering using hollow gold nanospheres. RSC Adv. 2018, 8, 3143–3150. [Google Scholar] [CrossRef] [Green Version]
- Cruz, L.J.; Que, I.; Aswendt, M.; Chan, A.; Hoehn, M.; Löwik, C. Targeted nanoparticles for the non-invasive detection of traumatic brain injury by optical imaging and fluorine magnetic resonance imaging. Nano Res. 2016, 9, 1276–1289. [Google Scholar] [CrossRef]
- Anselmo, A.C.; Mitragotri, S. Nanoparticles in the clinic: An update. Bioeng. Transl. Med. 2019, 4, e10143. [Google Scholar] [CrossRef] [Green Version]
- Havel, H.; Finch, G.; Strode, P.; Wolfgang, M.; Zale, S.; Bobe, I.; Youssoufian, H.; Peterson, M.; Liu, M. Nanomedicines: From Bench to Bedside and Beyond. AAPS J. 2016, 18, 1373–1378. [Google Scholar] [CrossRef]
- Van der Worp, H.B.; Howells, D.W.; Sena, E.S.; Porritt, M.J.; Rewell, S.; O’Collins, V.; Macleod, M.R. Can animal models of disease reliably inform human studies? PLoS Med. 2010, 7, e1000245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunay, M.S.; Ozer, A.Y.; Chalon, S. Drug Delivery Systems for Imaging and Therapy of Parkinson’s Disease. Curr. Neuropharmacol. 2016, 14, 376–391. [Google Scholar] [CrossRef] [PubMed]
- Alam Bony, B.; Kievit, F.M. A Role for Nanoparticles in Treating Traumatic Brain Injury. Pharmaceutics 2019, 11, 473. [Google Scholar] [CrossRef] [PubMed] [Green Version]











© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sim, T.M.; Tarini, D.; Dheen, S.T.; Bay, B.H.; Srinivasan, D.K. Nanoparticle-Based Technology Approaches to the Management of Neurological Disorders. Int. J. Mol. Sci. 2020, 21, 6070. https://doi.org/10.3390/ijms21176070
Sim TM, Tarini D, Dheen ST, Bay BH, Srinivasan DK. Nanoparticle-Based Technology Approaches to the Management of Neurological Disorders. International Journal of Molecular Sciences. 2020; 21(17):6070. https://doi.org/10.3390/ijms21176070
Chicago/Turabian StyleSim, Tao Ming, Dinesh Tarini, S. Thameem Dheen, Boon Huat Bay, and Dinesh Kumar Srinivasan. 2020. "Nanoparticle-Based Technology Approaches to the Management of Neurological Disorders" International Journal of Molecular Sciences 21, no. 17: 6070. https://doi.org/10.3390/ijms21176070
APA StyleSim, T. M., Tarini, D., Dheen, S. T., Bay, B. H., & Srinivasan, D. K. (2020). Nanoparticle-Based Technology Approaches to the Management of Neurological Disorders. International Journal of Molecular Sciences, 21(17), 6070. https://doi.org/10.3390/ijms21176070