Endoplasmic Reticulum Stress and Unfolded Protein Response in Neurodegenerative Diseases


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. ER Functions and Connections with Other Organelles
1.1. ER and Plasma Membrane
1.2. ER and Mitochondria
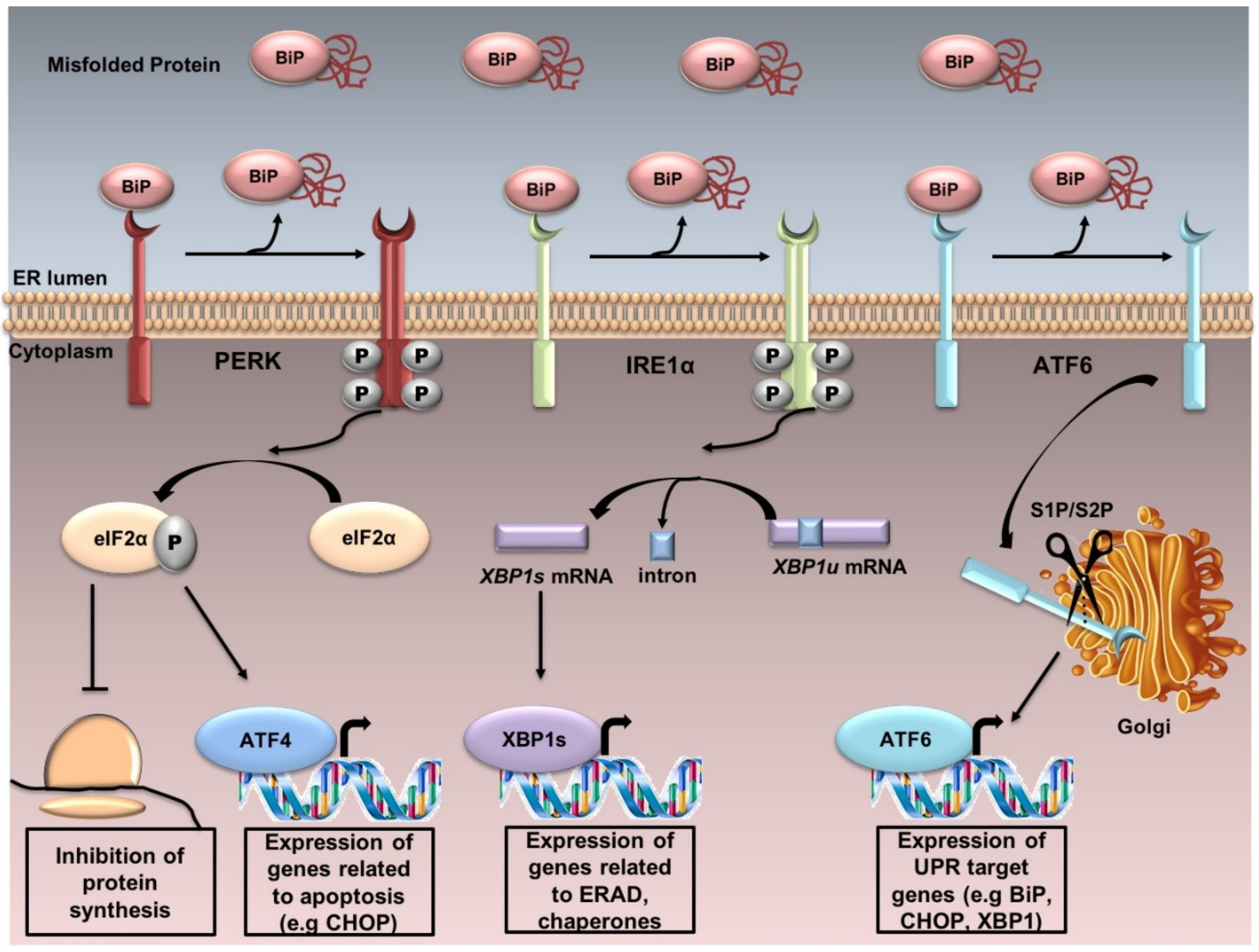
2. ER Stress and the Unfolded Protein Response (UPR)
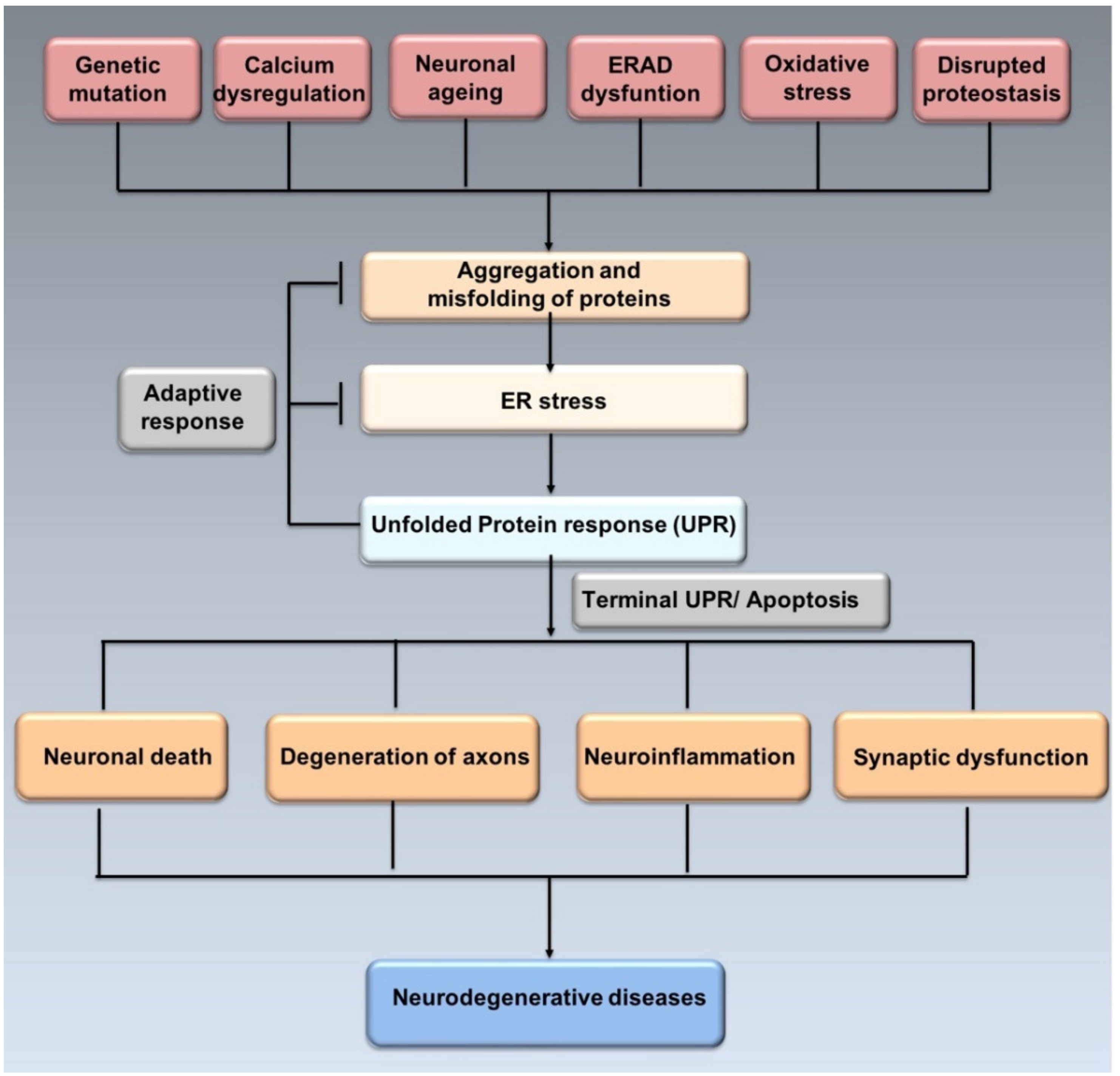
2.1. Interaction between UPR, Protein Aggregation and Neurodegeneration
2.2. Interaction between ER Stress, Autophagy and Neurodegeneration
3. ER Stress in Neurodegenerative Diseases
3.1. Alzheimer’s Disease (AD)
3.1.1. ER Stress in AD
3.1.2. ER Stress, Neuroinflammation and AD
3.2. Parkinson’s Disease (PD)
3.3. Amyotrophic Lateral Sclerosis (ALS)
3.4. Prion Diseases
4. Therapeutic Approaches: Chemical Compounds Targeting the UPR Pathways
4.1. Chemical Chaperones
4.2. GlaxoSmithKline(GSK) 2606414
4.3. ISRIB
4.4. Guanabenz
4.5. Sephin1
4.6. Trazodone Hydrochloride and Dibenzoylmethane
4.7. Salubrinal
4.8. Kinase Inhibiting RNase Attenuators (KIRA)
4.9. N-[2-hydroxy-5-methylphenyl)-3-phenylpropanamide
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 4-PBA | 4-Phenylbutyrate |
| AD | Alzheimer’s disease |
| ADAM10 | A disintegrin and metalloprotease domain 10 |
| ALS | Amyloid lateral sclerosis |
| AMPAR | AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptor |
| APP | Amyloid β-precursor protein |
| ASK1 | Apoptotic-signaling kinase-1 |
| ATF4 | Activating transcription factor 4 |
| ATF6 | Activating transcription factor 6 |
| ATG | AuTophaGy(ATG)-related proteins |
| Aβ | Amyloid-β |
| BACE1 | β-APP cleaving enzyme 1 |
| BECN1 | Beclin-1 |
| BiX | BIP/GRP78 inducer X |
| BSE | Bovine spongiform encephalopathy |
| C9orf72 | Chromosome 9 open reading frame 72 |
| CaBP-9k | Calbindin-D9K |
| Ca2+ | Calcium |
| CHOP | C/EBPα-homologous protein |
| CJD | Creutzfeldt–Jakob disease |
| CNS | Central nervous system |
| COPII | Coat protein complex II |
| CRAC | Ca2+-release-activated Ca2+ |
| CREBH | Cyclic adenosine monophosphate (cAMP)-responsive element-binding protein H |
| CWD | Chronic wasting disease |
| DAPK1 | Death-associated kinase 1 |
| eIF2α | Eukaryotic translation initiation factor α |
| ER | Endoplasmic reticulum |
| ERAD | ER-associated degradation |
| ERK | Extracellular signal-regulated kinase |
| ERMES | ER-mitochondria encounter structures |
| ERp | ER protein |
| FoxO1 | Forkhead box protein O1 |
| FTD | Frontotemporal dementia |
| FUS | Fused in sarcoma |
| GADD34 | Growth arrest and DNA-damage-inducible 34 |
| GCN2 | General control nonderepressible 2 |
| GRP17 | Glycine rich protein 17 |
| GRP78 | 78 KDa glucose-regulated protein |
| GSK-3β | Glycogen Synthase Kinase 3β |
| HD | Huntington disease |
| Herp | Homocysteine-inducible ER stress protein |
| HIPK2 | Homeodomain interacting protein kinase 2 |
| HRD1 | HMG-CoA reductase degradation protein 1 |
| HRI | Heme-regulated inhibitor |
| Hsp | Heat shock proteins |
| IBTs | 2-phenylimidazo[2,1-b]benzothiazole |
| IFN-γ | Interferon-gamma |
| IKK | IκB kinase |
| IL | Interleukin |
| IP3R | IP3 receptor |
| iPSCs | Induced pluripotent stem cells |
| IRE | Inositol-requiring transmembrane kinase/endoribonuclease |
| ISRIB | Integrated stress response inhibitor |
| JAK/STAT JNK | Janus kinase/signal transducers and activators of transcription Jun-N-terminal kinase |
| KIRA | Kinase inhibiting RNase attenuators |
| LBs | Lewy bodies |
| LNs | Lewy neurites |
| LRRK2 | Leucine-rich repeat kinase 2 |
| MAM | Mitochondria-associated ER membrane |
| MAP1LC3B | Microtubule associated protein 1 light chain 3 beta |
| Mfn2 | Mitochondrial fusion protein mitofusin 2 |
| NE | Nuclear envelope |
| NF-κB | Nuclear factor-κB |
| NINDS | National Institute of Neurological Disorders and Stroke |
| NMDA-Rs | N-methyl-d-aspartate receptors |
| Osh | Oxysterol-binding homology |
| p38 MAPK | p38 mitogen-activated protein kinase |
| PARK | Parkinson’s disease protein |
| PC12 | Pheochromocytoma of the rat adrenal medulla |
| PD | Parkinson’s disease |
| PDI | Protein disulfide isomerase |
| PE | Phosphatidylethanolamine |
| PERK | PKR-like ER kinase |
| PI3K/AKT PI4P | Phosphoinositide 3-kinase/ Protein kinase B Phosphatidylinositol 4-phosphate |
| PINK1 | PTEN-induced putative kinase 1 |
| PM | Plasma membrane |
| PMDs | Protein misfolding disorders |
| Pp1 | Protein phosphatase 1 |
| PRKN | Parkin |
| PrP | Prion protein |
| PS | Phosphatidylserine |
| PS1/2 | Presenilin-1/2 (PS1/2) |
| PTSD | Post-traumatic stress disorder |
| RAB1 | Ras-associated binding 1 |
| RIDD | Regulated IRE1-dependent decay |
| ROS | Reactive oxygen species |
| RTK | Receptor tyrosine kinase |
| S1P | Site 1 protease |
| S2P | Site 2 protease |
| SERCA | Sarco/endoplasmic reticulum Ca2+-ATPase |
| Sigr1 | Sigma-1 receptor |
| SNCA | Synuclein Alpha gene |
| SNpc | Substantia nigra pars compacta |
| SNPs | Single nucleotide polymorphisms |
| SOCE | Store operated Ca2+ entry |
| SOD1 | Superoxide dismutase1 |
| STIM | Stromal-interacting molecule |
| TARDBP or TDP-43 | TAR DNA-binding protein |
| TNF | Tumor necrosis factor |
| TRAF2 | TNF receptor-associated factor 2 |
| TSE | Transmissible spongiform encephalopathies |
| TUDCA | Tauroursodeoxycholic acid |
| uORF | Upstream open reading frame |
| UPR | Unfolded protein response |
| VAP VAPB | Vesicle-associated membrane protein-associated protein Vesicle-associated membrane protein-associated protein B |
| VDAC1 | Voltage-dependent anion selective channel protein 1 |
| VMP1 | Vacuole membrane protein 1 |
| WT | Wild type |
| XBP1 | X-box binding protein 1 |
| α-SYN | α-synuclein |
References
- English, A.R.; Voeltz, G.K. Endoplasmic reticulum structure and interconnections with other organelles. Cold Spring Harb. Perspect. Biol. 2013, 5, a013227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell. Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgoyne, T.; Patel, S.; Eden, E.R. Calcium signaling at ER membrane contact sites. Biochim. Biophys. Acta-Mol. Cell Res. 2014, 1853, 2012–2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefan, C.J.; Manford, A.G.; Baird, D.; Yamada-Hanff, J.; Mao, Y.; Emr, S.D. Osh proteins regulate phosphoinositide metabolism at ER-plasma membrane contact sites. Cell 2011, 144, 389–401. [Google Scholar] [CrossRef] [Green Version]
- Martinvalet, D. The role of the mitochondria and the endoplasmic reticulum contact sites in the development of the immune responses. Cell Death Dis. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Xia, M.F.; Zhang, Y.Z.; Jin, K.; Lu, Z.T.; Zeng, Z.; Xiong, W. Communication between mitochondria and other organelles: A brand-new perspective on mitochondria in cancer. Cell Biosci. 2019, 9, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER tubules mark sites of mitochondrial division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef] [Green Version]
- Area-Gomez, E.; De Groof, A.; Bonilla, E.; Montesinos, J.; Tanji, K.; Boldogh, I.; Pon, L.; Schon, E.A. A key role for MAM in mediating mitochondrial dysfunction in Alzheimer disease. Cell Death Dis. 2018, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Suaga, P.; Bravo-San Pedro, J.M.; González-Polo, R.A.; Fuentes, J.M.; Niso-Santano, M. ER-mitochondria signaling in Parkinson’s disease review-article. Cell Death Dis. 2018, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Uddin, M.S.; Tewari, D.; Sharma, G.; Kabir, M.T.; Barreto, G.E.; Bin-Jumah, M.N.; Perveen, A.; Abdel-Daim, M.M.; Ashraf, G.M. Molecular Mechanisms of ER Stress and UPR in the Pathogenesis of Alzheimer’s Disease. Mol. Neurobiol. 2020, 57, 1–18. [Google Scholar] [CrossRef]
- Carrara, M.; Prischi, F.; Nowak, P.R.; Kopp, M.C.; Ali, M.M.U. Noncanonical binding of BiP ATPase domain to Ire1 and Perk is dissociated by unfolded protein CH1 to initiate ER stress signaling. Elife 2015, 4, e03522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, H.; Tian, M.; Ding, C.; Yu, S. The C/EBP homologous protein (CHOP) transcription factor functions in endoplasmic reticulum stress-induced apoptosis and microbial infection. Front. Immunol. 2019, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Xiang, C.; Wang, Y.; Zhang, H.; Han, F. The role of endoplasmic reticulum stress in neurodegenerative disease. Apoptosis 2017, 22, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Hubbard, S.R. How IRE1 Reacts to ER Stress. Cell 2008, 132, 24–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollien, J.; Weissman, J.S. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 2006, 313, 104–107. [Google Scholar] [CrossRef] [Green Version]
- Hollien, J.; Lin, J.H.; Li, H.; Stevens, N.; Walter, P.; Weissman, J.S. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol. 2009, 186, 323–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hillary, R.F.; Fitzgerald, U. A lifetime of stress: ATF6 in development and homeostasis. J. Biomed. Sci. 2018, 25, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Rutkowski, D.T.; Dubois, M.; Swathirajan, J.; Saunders, T.; Wang, J.; Song, B.; Yau, G.D.Y.; Kaufman, R.J. ATF6α Optimizes Long-Term Endoplasmic Reticulum Function to Protect Cells from Chronic Stress. Dev. Cell 2007, 13, 351–364. [Google Scholar] [CrossRef] [Green Version]
- Soto, C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 2003, 4, 49–60. [Google Scholar] [CrossRef]
- Ashraf, G.; Greig, N.; Khan, T.; Hassan, I.; Tabrez, S.; Shakil, S.; Sheikh, I.; Zaidi, S.; Akram, M.; Jabir, N.; et al. Protein Misfolding and Aggregation in Alzheimer’s Disease and Type 2 Diabetes Mellitus. CNS Neurol. Disord. Drug Targets 2014, 13, 1280–1293. [Google Scholar] [CrossRef]
- Hetz, C.; Saxena, S. ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 2017, 13, 477–491. [Google Scholar] [CrossRef]
- Remondelli, P.; Renna, M. The endoplasmic reticulum unfolded protein response in neurodegenerative disorders and its potential therapeutic significance. Front. Mol. Neurosci. 2017, 10, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweeney, P.; Park, H.; Baumann, M.; Dunlop, J.; Frydman, J.; Kopito, R.; McCampbell, A.; Leblanc, G.; Venkateswaran, A.; Nurmi, A.; et al. Protein misfolding in neurodegenerative diseases: Implications and strategies. Transl. Neurodegener. 2017, 6, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Hughes, D.; Mallucci, G.R. The unfolded protein response in neurodegenerative disorders—Therapeutic modulation of the PERK pathway. FEBS J. 2019, 286, 342–355. [Google Scholar] [CrossRef]
- Cabral-Miranda, F.; Hetz, C. ER stress in neurodegenerative disease: From disease mechanisms to therapeutic interventions. Endoplasmic Reticulum Stress Dis. 2017, 4, 11–26. [Google Scholar] [CrossRef]
- García-González, P.; Cabral-Miranda, F.; Hetz, C.; Osorio, F. Function in the development of neurodegenerative diseases. Front. Immunol. 2018, 9, 1–10. [Google Scholar] [CrossRef]
- Gerakis, Y.; Hetz, C. Emerging roles of ER stress in the etiology and pathogenesis of Alzheimer’s disease. FEBS J. 2018, 285, 995–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colla, E. Linking the endoplasmic reticulum to Parkinson’s disease and alpha-synucleinopathy. Front. Neurosci. 2019, 13, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Prell, T.; Stubendorff, B.; Le, T.T.; Gaur, N.; Tadić, V.; Rödiger, A.; Witte, O.W.; Grosskreutz, J. Reaction to endoplasmic reticulum stress via ATF6 in amyotrophic lateral sclerosis deteriorates with aging. Front. Aging Neurosci. 2019, 11, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindholm, D.; Korhonen, L.; Eriksson, O.; Kõks, S. Recent insights into the role of unfolded protein response in ER stress in health and disease. Front. Cell Dev. Biol. 2017, 5, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Marotta, D.; Tinelli, E.; Mole, S.E. NCLs and ER: A stressful relationship. Biochim. Biophys. Acta-Mol. Basis Dis. 2017, 1863, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Stutzbach, L.D.; Xie, S.X.; Naj, A.C.; Albin, R.; Gilman, S.; Lee, V.M.Y.; Trojanowski, J.Q.; Devlin, B.; Schellenberg, G.D. The unfolded protein response is activated in disease-affected brain regions in progressive supranuclear palsy and Alzheimer’s disease. Acta Neuropathol. Commun. 2013, 1, 31. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Ackerman, S.L. Endoplasmic reticulum stress in health and disease. Curr. Opin. Cell Biol. 2006, 18, 444–452. [Google Scholar] [CrossRef]
- Sadleir, K.R.; Popovic, J.; Vassar, R. ER stress is not elevated in the 5XFAD mouse model of Alzheimer’s disease. J. Biol. Chem. 2018, 293, 18434–18443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, E.; Cuervo, A.M. Autophagy gone awry in neurodegenerative diseases. Nat. Neurosci. 2010, 13, 805–811. [Google Scholar] [CrossRef] [Green Version]
- Tettamanti, G.; Carata, E.; Montali, A.; Dini, L.; Fimia, G.M. Autophagy in development and regeneration: Role in tissue remodelling and cell survival. Eur. Zool. J. 2019, 86, 113–131. [Google Scholar] [CrossRef] [Green Version]
- Son, J.H.; Shim, J.H.; Kim, K.H.; Ha, J.Y.; Han, J.Y. Neuronal autophagy and neurodegenerative diseases. Exp. Mol. Med. 2012, 44, 89–98. [Google Scholar] [CrossRef]
- Fujikake, N.; Shin, M.; Shimizu, S. Association between autophagy and neurodegenerative diseases. Front. Neurosci. 2018, 12, 255. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Arikkath, J.; Yang, L.; Guo, M.L.; Periyasamy, P.; Buch, S. Interplay of endoplasmic reticulum stress and autophagy in neurodegenerative disorders. Autophagy 2016, 12, 225–244. [Google Scholar] [CrossRef]
- Birdsall, V.; Waites, C.L. Autophagy at the synapse. Neurosci. Lett. 2019, 697, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Negrete-Hurtado, A.; Overhoff, M.; Bera, S.; De Bruyckere, E.; Schätzmüller, K.; Kye, M.J.; Qin, C.; Lammers, M.; Kondylis, V.; Neundorf, I.; et al. Autophagy lipidation machinery regulates axonal microtubule dynamics but is dispensable for survival of mammalian neurons. Nat. Commun. 2020, 11, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Yan, D.Y.; Wang, C.; Ma, Z.; Deng, Y.; Liu, W.; Xu, B. Manganese activates autophagy to alleviate endoplasmic reticulum stress–induced apoptosis via PERK pathway. J. Cell. Mol. Med. 2020, 24, 328–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, W.; Xie, W.; Yin, D.; Luo, R.; Liu, M.; Guo, F. ATG5 and ATG7 induced autophagy interplays with UPR via PERK signaling. Cell Commun. Signal. 2019, 17, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- B’Chir, W.; Maurin, A.C.; Carraro, V.; Averous, J.; Jousse, C.; Muranishi, Y.; Parry, L.; Stepien, G.; Fafournoux, P.; Bruhat, A. The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013, 41, 7683–7699. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.L.; Yang, S.F.; Hung, T.W.; Lin, C.L.; Hsieh, Y.H.; Chiou, H.L. Inhibition of eIF2α dephosphorylation accelerates pterostilbene-induced cell death in human hepatocellular carcinoma cells in an ER stress and autophagy-dependent manner. Cell Death Dis. 2019, 10, 418. [Google Scholar] [CrossRef]
- Hosoi, T.; Nomura, J.; Tanaka, K.; Ozawa, K.; Nishi, A.; Nomura, Y. Link between endoplasmic reticulum stress and autophagy in neurodegenerative diseases. Endoplasmic Reticulum Stress Dis. 2017, 4, 37–45. [Google Scholar] [CrossRef]
- Kishino, A.; Hayashi, K.; Hidai, C.; Masuda, T.; Nomura, Y.; Oshima, T. XBP1-FoxO1 interaction regulates ER stress-induced autophagy in auditory cells. Sci. Rep. 2017, 7, 4442. [Google Scholar] [CrossRef] [Green Version]
- Yan, C.; Liu, J.; Gao, J.; Sun, Y.; Zhang, L.; Song, H.; Xue, L.; Zhan, L.; Gao, G.; Ke, Z.; et al. IRE1 promotes neurodegeneration through autophagy-dependent neuron death in the Drosophila model of Parkinson’s disease. Cell Death Dis. 2019, 10, 800–815. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Yan, D.Y.; Wang, C.; Ma, Z.; Deng, Y.; Liu, W.; Xu, B. IRE1 signaling pathway mediates protective autophagic response against manganese-induced neuronal apoptosis in vivo and in vitro. Sci. Total Environ. 2020, 712, 136480. [Google Scholar] [CrossRef]
- Gade, P.; Ramachandran, G.; Maachani, U.B.; Rizzo, M.A.; Okada, T.; Prywes, R.; Cross, A.S.; Mori, K.; Kalvakolanu, D.V. An IFN-γ-stimulated ATF6-C/EBP-β-signaling pathway critical for the expression of death associated protein kinase 1 and induction of autophagy. Proc. Natl. Acad. Sci. USA 2012, 109, 10316–10321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, D.M. Endoplasmic Reticulum Stress and Related Pathological Processes. J. Diagn. Tech. Biomed. Anal. 2013, 1, 1000107. [Google Scholar] [CrossRef]
- Wang, P.; Kou, D.; Le, W. Roles of VMP1 in Autophagy and ER–Membrane Contact: Potential Implications in Neurodegenerative Disorders. Front. Mol. Neurosci. 2020, 13, 42. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.L.; Freeman, O.J.; Butcher, A.J.; Holmqvist, S.; Humoud, I.; Schätzl, T.; Hughes, D.T.; Verity, N.C.; Swinden, D.P.; Hayes, J.; et al. Astrocyte Unfolded Protein Response Induces a Specific Reactivity State that Causes Non-Cell-Autonomous Neuronal Degeneration. Neuron 2020, 105, 855–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, L.E.; Ferreira, S.T. Crosstalk between endoplasmic reticulum stress and brain inflammation in Alzheimer’s disease. Neuropharmacology 2018, 136, 350–360. [Google Scholar] [CrossRef]
- Lindholm, D.; Wootz, H.; Korhonen, L. ER stress and neurodegenerative diseases. Cell Death Differ. 2006, 13, 385–392. [Google Scholar] [CrossRef]
- Mattsson, N.; Zetterberg, H.; Janelidze, S.; Insel, P.S.; Andreasson, U.; Stomrud, E.; Palmqvist, S.; Baker, D.; Tan Hehir, C.A.; Jeromin, A.; et al. Plasma tau in Alzheimer disease. Neurology 2016, 87, 1827–1835. [Google Scholar] [CrossRef] [Green Version]
- Walsh, D.M.; Selkoe, D.J. Aβ oligomers—A decade of discovery. J. Neurochem. 2007, 101, 1172–1184. [Google Scholar] [CrossRef]
- Gendron, T.F. The role of tau in neurodegeneration. Mol. Neurodegener. 2009, 4, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Ma, Q.; Zhang, Y.; Xu, H. Proteolytic processing of APP. J. Neurochem. 2012, 120 (Suppl. 1), 9–21. [Google Scholar] [CrossRef] [Green Version]
- Haass, C. Take five-BACE and the γ-secretase quartet conduct Alzheimer’s amyloid β-peptide generation. EMBO J. 2004, 23, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Wallon, D.; Rovelet-lecrux, A.; Richard, A.; Pasquier, F.; Lacour, M.; Rollin-sillaire, A.; Martinaud, O.; Quillard-muraine, M.; De, V.; Boutoleau-bretonniere, C.; et al. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLoS Med. 2017, 1, 1–16. [Google Scholar]
- Abisambra, J.F.; Jinwal, U.K.; Blair, L.J.; O’Leary, J.C.; Li, Q.; Brady, S.; Wang, L.; Guidi, C.E.; Zhang, B.; Nordhues, B.A.; et al. Tau accumulation activates the unfolded protein response by impairing endoplasmic reticulum-associated degradation. J. Neurosci. 2013, 33, 9498–9507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Chang, L.; Song, Y.; Li, H.; Wu, Y. The role of NMDA receptors in Alzheimer’s disease. Front. Neurosci. 2019, 13, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.; Park, K.A.; Lee, W.T.; Lee, J.E. Apoptosis signal regulating kinase 1 (ASK1): Potential as a therapeutic target for Alzheimer’s disease. Int. J. Mol. Sci. 2014, 15, 2119–2129. [Google Scholar] [CrossRef] [Green Version]
- Lin, Q.; Cao, Y.; Gao, J. Serum calreticulin is a negative biomarker in patients with Alzheimer’s disease. Int. J. Mol. Sci. 2014, 15, 21740–21753. [Google Scholar] [CrossRef] [Green Version]
- Sharma, V.; Ounallah-Saad, H.; Chakraborty, D.; Hleihil, M.; Sood, R.; Barrera, I.; Edry, E.; Chandran, S.K.; de Leon, S.B.T.; Kaphzan, H.; et al. Local inhibition of PERK enhances memory and reverses age-related deterioration of cognitive and neuronal properties. J. Neurosci. 2018, 38, 648–658. [Google Scholar] [CrossRef] [Green Version]
- Hugon, J.; Mouton-Liger, F.; Dumurgier, J.; Paquet, C. PKR involvement in Alzheimer’s disease. Alzheimer’s Res. Ther. 2017, 9, 83. [Google Scholar] [CrossRef]
- Devi, L.; Ohno, M. PERK mediates eIF2α phosphorylation responsible for BACE1 elevation, CREB dysfunction and neurodegeneration in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2014, 35, 2272–2281. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, T.; Sadleir, K.R.; Maus, E.; Velliquette, R.A.; Zhao, J.; Cole, S.L.; Eimer, W.A.; Hitt, B.; Bembinster, L.A.; Lammich, S.; et al. Phosphorylation of the Translation Initiation Factor eIF2α Increases BACE1 Levels and Promotes Amyloidogenesis. Neuron 2008, 60, 988–1009. [Google Scholar] [CrossRef] [Green Version]
- Pasini, S.; Corona, C.; Liu, J.; Greene, L.A.; Shelanski, M.L. Specific downregulation of hippocampal ATF4 reveals a necessary role in synaptic plasticity and memory. Cell Rep. 2015, 11, 183–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duran-Aniotz, C.; Cornejo, V.H.; Espinoza, S.; Ardiles, Á.O.; Medinas, D.B.; Salazar, C.; Foley, A.; Gajardo, I.; Thielen, P.; Iwawaki, T.; et al. IRE1 signaling exacerbates Alzheimer’s disease pathogenesis. Acta Neuropathol. 2017, 134, 489–506. [Google Scholar] [CrossRef] [PubMed]
- Martínez, G.; Vidal, R.L.; Mardones, P.; Serrano, F.G.; Ardiles, A.O.; Wirth, C.; Valdés, P.; Thielen, P.; Schneider, B.L.; Kerr, B.; et al. Regulation of Memory Formation by the Transcription Factor XBP1. Cell Rep. 2016, 14, 1382–1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peron, R.; Vatanabe, I.P.; Manzine, P.R.; Camins, A.; Cominetti, M.R. Alpha-secretase ADAM10 regulation: Insights into Alzheimer’s disease treatment. Pharmaceuticals 2018, 11, 12. [Google Scholar] [CrossRef] [Green Version]
- Cissé, M.; Duplan, E.; Checler, F. The transcription factor XBP1 in memory and cognition: Implications in Alzheimer’s disease. Mol. Med. 2017, 22, 905–917. [Google Scholar] [CrossRef]
- Lee, A.-H.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 Regulates a Subset of Endoplasmic Reticulum Resident Chaperone Genes in the Unfolded Protein Response. Mol. Cell. Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.; Liu, X.; Zhu, X.; Liu, Y.; Wang, X.; Wu, X. Activating transcription factor 6 reduces Aβ1–42 and restores memory in Alzheimer’s disease model mice. Int. J. Neurosci. 2020, 1–9. [Google Scholar] [CrossRef]
- Li, Y.; Guo, Y.; Tang, J.; Jiang, J.; Chen, Z. New insights into the roles of CHOP-induced apoptosis in ER stress Structure and Properties of C/EBP Homologous Protein Roles of CHOP in ER Stress-Mediated Apoptosis. Acta Biochim. Biophys. Sin. 2014, 46, 629–640. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Li, X.; Zhou, W.; Lou, D.; Huang, D.; Li, Y.; Kang, Y.; Xiang, Y.; Li, T.; Zhou, W.; et al. Regulation of SET Gene Expression by NFkB. Mol. Neurobiol. 2017, 54, 4477–4485. [Google Scholar] [CrossRef]
- Mohammed-Ali, Z.; Cruz, G.L.; Dickhout, J.G. Crosstalk between the unfolded protein response and NF-κ B-mediated inflammation in the progression of chronic kidney disease. J. Immunol. Res. 2015, 2015. [Google Scholar] [CrossRef] [Green Version]
- Vukic, V.; Callaghan, D.; Walker, D.; Lue, L.F.; Liu, Q.Y.; Couraud, P.O.; Romero, I.A.; Weksler, B.; Stanimirovic, D.B.; Zhang, W. Expression of inflammatory genes induced by beta-amyloid peptides in human brain endothelial cells and in Alzheimer’s brain is mediated by the JNK-AP1 signaling pathway. Neurobiol. Dis. 2009, 34, 95–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meares, G.P.; Liu, Y.; Rajbhandari, R.; Qin, H.; Nozell, S.E.; Mobley, J.A.; Corbett, J.A.; Benveniste, E.N. PERK-Dependent Activation of JAK1 and STAT3 Contributes to Endoplasmic Reticulum Stress-Induced Inflammation. Mol. Cell. Biol. 2014, 34, 3911–3925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llorens-Martín, M.; Jurado, J.; Hernández, F.; Ávila, J. GSK-3β, a pivotal kinase in Alzheimer disease. Front. Mol. Neurosci. 2014, 7, 46. [Google Scholar] [PubMed] [Green Version]
- Hirtz, D.; Thurman, D.J.; Gwinn-Hardy, K.; Mohamed, M.; Chaudhuri, A.R.; Zalutsky, R. How common are the “common” neurologic disorders? Neurology 2007, 68, 326–337. [Google Scholar] [CrossRef]
- Zhang, Z.X.; Roman, G.C.; Hong, Z.; Wu, C.B.; Qu, Q.M.; Huang, J.B.; Zhou, B.; Geng, Z.P.; Wu, J.X.; Wen, H.B.; et al. Parkinson’s disease in China: Prevalence in Beijing, Xian, and Shanghai. Lancet 2005, 365, 595–597. [Google Scholar] [CrossRef]
- Tan, L.C.S.; Venketasubramanian, N.; Jamora, R.D.G.; Heng, D. Incidence of Parkinson’s disease in Singapore. Park. Relat. Disord. 2007, 13, 40–43. [Google Scholar] [CrossRef]
- Tian, Y.Y.; Tang, C.J.; Wu, J.; Zhou, J.S. Parkinson’s disease in China. Neurol. Sci. 2011, 32, 23–30. [Google Scholar] [CrossRef]
- Elbaz, A.; Carcaillon, L.; Kab, S.; Moisan, F. Epidemiology of Parkinson’s disease. Rev. Neurol. 2016, 172, 14–26. [Google Scholar] [CrossRef]
- Corti, O.; Lesage, S.; Brice, A. What genetics tells us about the causes and mechanisms of Parkinson’s disease. Physiol. Rev. 2011, 91, 1161–1218. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G.; Del Tredici, K.; Braak, H. 100 years of Lewy pathology. Nat. Rev. Neurol. 2013, 9, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Witt, S.N. Molecular chaperones, α-synuclein, and neurodegeneration. Mol. Neurobiol. 2013, 47, 552–560. [Google Scholar] [CrossRef] [Green Version]
- Malek, N.; Swallow, D.; Grosset, K.A.; Anichtchik, O.; Spillantini, M.; Grosset, D.G. Alpha-synuclein in peripheral tissues and body fluids as a biomarker for Parkinson’s disease—A systematic review. Acta Neurol. Scand. 2014, 130, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Golbe, L.I.; Di Iorio, G.; Bonavita, V.; Miller, D.C.; Duvoisin, R.C. A large kindred with autosomal dominant Parkinson’s disease. Ann. Neurol. 1990, 27, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Zarranz, J.J.; Alegre, J.; Gómez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atarés, B.; et al. The New Mutation, E46K, of α-Synuclein Causes Parkinson and Lewy Body Dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Krüger, R.; Kuhn, W.; Müller, T.; Woitalla, D.; Graeber, M.; Kösel, S.; Przuntek, H.; Epplen, J.T.; Schöls, L.; Riess, O. Ala30Pro mutation in the gene encoding α-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef]
- Soto, C. Transmissible proteins: Expanding the prion heresy. Cell 2012, 149, 968–977. [Google Scholar] [CrossRef] [Green Version]
- Bellucci, A.; Navarria, L.; Zaltieri, M.; Falarti, E.; Bodei, S.; Sigala, S.; Battistin, L.; Spillantini, M.; Missale, C.; Spano, P. Induction of the unfolded protein response by α-synuclein in experimental models of Parkinson’s disease. J. Neurochem. 2011, 116, 588–605. [Google Scholar] [CrossRef]
- Bellani, S.; Mescola, A.; Ronzitti, G.; Tsushima, H.; Tilve, S.; Canale, C.; Valtorta, F.; Chieregatti, E. GRP78 clustering at the cell surface of neurons transduces the action of exogenous alpha-synuclein. Cell Death Differ. 2014, 21, 1971–1983. [Google Scholar] [CrossRef]
- Hoozemans, J.J.M.; van Haastert, E.S.; Eikelenboom, P.; de Vos, R.A.I.; Rozemuller, J.M.; Scheper, W. Activation of the unfolded protein response in Parkinson’s disease. Biochem. Biophys. Res. Commun. 2007, 354, 707–711. [Google Scholar] [CrossRef]
- Colla, E.; Coune, P.; Liu, Y.; Pletnikova, O.; Troncoso, J.C.; Iwatsubo, T.; Schneider, B.L.; Lee, M.K. Endoplasmic reticulum stress is important for the manifestations of α-synucleinopathy in vivo. J. Neurosci. 2012, 32, 3306–3320. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.W.; Jiang, H.; Pei, Z.; Tanaka, Y.; Morita, H.; Sawa, A.; Dawson, V.L.; Dawson, T.M.; Ross, C.A. Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha-synuclein-induced toxicity. Hum. Mol. Genet. 2005, 14, 3801–3811. [Google Scholar] [CrossRef] [PubMed]
- Boyce, M.; Bryant, K.F.; Jousse, C.; Long, K.; Harding, H.P.; Scheuner, D.; Kaufman, R.J.; Ma, D.; Coen, D.M.; Ron, D.; et al. A selective inhibitor of elF2α dephosphorylation protects cells from ER stress. Science 2005, 307, 935–939. [Google Scholar] [CrossRef] [PubMed]
- Valdés, P.; Mercado, G.; Vidal, R.L.; Molina, C.; Parsons, G.; Court, F.A.; Martinez, A.; Galleguillos, D.; Armentano, D.; Schneider, B.L.; et al. Control of dopaminergic neuron survival by the unfolded protein response transcription factor XBP1. Proc. Natl. Acad. Sci. USA 2014, 111, 6804–6809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, A.A.; Gitler, A.D.; Cashikar, A.; Haynes, C.M.; Hill, K.J.; Bhullar, B.; Liu, K.; Xu, K.; Strathearn, K.E.; Liu, F.; et al. α-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 2006, 313, 324–328. [Google Scholar] [CrossRef] [Green Version]
- Gitler, A.D.; Bevis, B.J.; Shorter, J.; Strathearn, K.E.; Hamamichi, S.; Su, L.J.; Caldwell, K.A.; Caldwell, G.A.; Rochet, J.C.; McCaffery, J.M.; et al. The Parkinson’s disease protein α-synuclein disrupts cellular Rab homeostasis. Proc. Natl. Acad. Sci. USA 2008, 105, 145–150. [Google Scholar] [CrossRef] [Green Version]
- Mazzulli, J.R.; Zunke, F.; Isacson, O.; Studer, L.; Krainc, D. α-Synuclein-induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proc. Natl. Acad. Sci. USA 2016, 113, 1931–1936. [Google Scholar] [CrossRef] [Green Version]
- Credle, J.J.; Forcelli, P.A.; Delannoy, M.; Oaks, A.W.; Permaul, E.; Berry, D.L.; Duka, V.; Wills, J.; Sidhu, A. α-Synuclein-mediated inhibition of ATF6 processing into COPII vesicles disrupts UPR signaling in Parkinson’s disease. Neurobiol. Dis. 2015, 76, 112–125. [Google Scholar] [CrossRef]
- Betzer, C.; Lassen, L.B.; Olsen, A.; Kofoed, R.H.; Reimer, L.; Gregersen, E.; Zheng, J.; Calì, T.; Gai, W.; Chen, T.; et al. Alpha-synuclein aggregates activate calcium pump SERCA leading to calcium dysregulation. EMBO Rep. 2018, 19, e44617. [Google Scholar] [CrossRef]
- Jung, E.M.; Yoo, Y.M.; Park, S.Y.; Ahn, C.; Jeon, B.H.; Hong, E.J.; Kim, W.Y.; Jeung, E.B. Calbindin-D9k is a novel risk gene for neurodegenerative disease. Cell. Physiol. Biochem. 2020, 54, 438–456. [Google Scholar]
- Matus, S.; Valenzuela, V.; Medinas, D.B.; Hetz, C. ER dysfunction and protein folding stress in ALS. Int. J. Cell Biol. 2013, 2013, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atkin, J.D.; Farg, M.A.; Walker, A.K.; McLean, C.; Tomas, D.; Horne, M.K. Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol. Dis. 2008, 30, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Thielen, P.; Matus, S.; Nassif, M.; Court, F.; Kiffin, R.; Martinez, G.; Cuervo, A.M.; Brown, R.H.; Glimcher, L.H. XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 2009, 23, 2294–2306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, Y.; Yamada, M.; Tanaka, H.; Aida, K.; Tsuruma, K.; Shimazawa, M.; Hozumi, I.; Inuzuka, T.; Takahashi, H.; Hara, H. Involvement of CHOP, an ER-stress apoptotic mediator, in both human sporadic ALS and ALS model mice. Neurobiol. Dis. 2009, 36, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S. Endoplasmic Reticulum Stress in Motor Neurons of the Spinal Cord in Sporadic Amyotrophic Lateral Sclerosis. J. Neuropathol. Exp. Neurol. 2010, 69, 346–355. [Google Scholar] [CrossRef] [Green Version]
- Dion, P.A.; Daoud, H.; Rouleau, G.A. Genetics of motor neuron disorders: New insights into pathogenic mechanisms. Nat. Rev. Genet. 2009, 10, 769–782. [Google Scholar] [CrossRef]
- Tang, L.; Ma, Y.; Liu, X.L.; Chen, L.; Fan, D.S. Better survival in female SOD1-mutant patients with ALS: A study of SOD1-related natural history. Transl. Neurodegener. 2019, 8, 1–10. [Google Scholar]
- Chen, D.; Wang, Y.; Chin, E.R. Activation of the endoplasmic reticulum stress response in skeletal muscle of G93a*SOD1 amyotrophic lateral sclerosis mice. Front. Cell. Neurosci. 2015, 9, 170. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, H.; Almer, G.; Yamashita, S.; Guégan, C.; Nagai, M.; Xu, Z.; Sosunov, A.A.; McKhann, G.M.; Przedborski, S. Spinal cord endoplasmic reticulum stress associated with a microsomal accumulation of mutant superoxide dismutase-1 in an ALS model. Proc. Natl. Acad. Sci. USA 2006, 103, 6025–6030. [Google Scholar] [CrossRef] [Green Version]
- Lautenschlaeger, J.; Prell, T.; Grosskreutz, J. Endoplasmic reticulum stress and the ER mitochondrial calcium cycle in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2012, 13, 166–177. [Google Scholar] [CrossRef]
- Nishitoh, H.; Kadowaki, H.; Nagai, A.; Maruyama, T.; Yokota, T.; Fukutomi, H.; Noguchi, T.; Matsuzawa, A.; Takeda, K.; Ichijo, H. ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1. Genes Dev. 2008, 22, 1451–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Popko, B.; Roos, R.P. The unfolded protein response in familial amyotrophic lateral sclerosis. Hum. Mol. Genet. 2011, 20, 1008–1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Popko, B.; Roos, R.P. An enhanced integrated stress response ameliorates mutant SOD1-induced ALS. Hum. Mol. Genet. 2014, 23, 2629–2638. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.Q.; Ren, M.; Jiang, H.Z.; Wang, J.; Zhang, J.; Yin, X.; Wang, S.Y.; Qi, Y.; Wang, X.D.; Feng, H.L. Guanabenz delays the onset of disease symptoms, extends lifespan, improves motor performance and attenuates motor neuron loss in the SOD1 G93A mouse model of amyotrophic lateral sclerosis. Neuroscience 2014, 277, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Dzhashiashvili, Y.; Monckton, C.P.; Shah, H.S.; Kunjamma, R.B.; Popko, B. The UPR-PERK pathway is not a promising therapeutic target for mutant SOD1-induced ALS. Neurobiol. Dis. 2019, 127, 527–544. [Google Scholar] [CrossRef] [PubMed]
- Matus, S.; Lopez, E.; Valenzuela, V.; Nassif, M.; Hetz, C. Functional Contribution of the Transcription Factor ATF4 to the Pathogenesis of Amyotrophic Lateral Sclerosis. PLoS ONE 2013, 8, e66672. [Google Scholar] [CrossRef]
- Lee, S.; Shang, Y.; Redmond, S.A.; Urisman, A.; Tang, A.A.; Li, K.H.; Burlingame, A.L.; Pak, R.A.; Jovičić, A.; Gitler, A.D.; et al. Activation of HIPK2 Promotes ER Stress-Mediated Neurodegeneration in Amyotrophic Lateral Sclerosis. Neuron 2016, 91, 41–55. [Google Scholar] [CrossRef] [Green Version]
- Clark, E.M.; Nonarath, H.J.T.; Bostrom, J.R.; Link, B.A. Establishment and validation of an endoplasmic reticulum stress reporter to monitor zebrafish ATF6 activity in development and disease. DMM Dis. Model. Mech. 2020, 13, dmm041426. [Google Scholar] [CrossRef] [Green Version]
- Walker, A.K.; Atkin, J.D. Mechanisms of neuroprotection by protein disulphide isomerase in amyotrophic lateral sclerosis. Neurol. Res. Int. 2011, 2011. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Perez, P.; Woehlbier, U.; Chian, R.J.; Sapp, P.; Rouleau, G.A.; Leblond, C.S.; Daoud, H.; Dion, P.A.; Landers, J.E.; Hetz, C.; et al. Identification of rare protein disulfide isomerase gene variants in amyotrophic lateral sclerosis patients. Gene 2015, 566, 158–165. [Google Scholar] [CrossRef]
- Woehlbier, U.; Colombo, A.; Saaranen, M.J.; Pérez, V.; Ojeda, J.; Bustos, F.J.; Andreu, C.I.; Torres, M.; Valenzuela, V.; Medinas, D.B.; et al. ALS -linked protein disulfide isomerase variants cause motor dysfunction. EMBO J. 2016, 35, 845–865. [Google Scholar] [CrossRef]
- Walker, A.K.; Soo, K.Y.; Sundaramoorthy, V.; Parakh, S.; Ma, Y.; Farg, M.A.; Wallace, R.H.; Crouch, P.J.; Turner, B.J.; Horne, M.K.; et al. ALS-associated TDP-43 induces endoplasmic reticulum stress, which drives cytoplasmic TDP-43 accumulation and stress granule formation. PLoS ONE 2013, 8, e81170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.J.; Raphael, A.R.; Ladow, E.S.; Mcgurk, L.; Weber, R.A.; Trojanowski, J.Q.; Lee, V.M.Y.; Finkbeiner, S.; Gitler, A.D.; Bonini, N.M. Therapeutic modulation of eIF2α phosphorylation rescues TDP-43 toxicity in amyotrophic lateral sclerosis disease models. Nat. Genet. 2014, 46, 152–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farg, M.A.; Soo, K.Y.; Walker, A.K.; Pham, H.; Orian, J.; Horne, M.K.; Warraich, S.T.; Williams, K.L.; Blair, I.P.; Atkin, J.D. Mutant FUS induces endoplasmic reticulum stress in amyotrophic lateral sclerosis and interacts with protein disulfide-isomerase. Neurobiol. Aging 2012, 33, 2855–2868. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, B.T.; Livesey, M.R.; Zhao, C.; Gregory, J.M.; James, O.T.; Cleary, E.M.; Chouhan, A.K.; Gane, A.B.; Perkins, E.M.; Dando, O.; et al. C9ORF72 repeat expansion causes vulnerability of motor neurons to Ca2+-permeable AMPA receptor-mediated excitotoxicity. Nat. Commun. 2018, 9, 347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregory, J.M.; Livesey, M.R.; McDade, K.; Selvaraj, B.T.; Barton, S.K.; Chandran, S.; Smith, C. Dysregulation of AMPA receptor subunit expression in sporadic ALS post-mortem brain. J. Pathol. 2020, 250, 67–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreser, A.; Vollrath, J.T.; Sechi, A.; Johann, S.; Roos, A.; Yamoah, A.; Katona, I.; Bohlega, S.; Wiemuth, D.; Tian, Y.; et al. The ALS-linked E102Q mutation in Sigma receptor-1 leads to ER stress-mediated defects in protein homeostasis and dysregulation of RNA-binding proteins. Cell Death Differ. 2017, 24, 1655–1671. [Google Scholar] [CrossRef]
- Gkogkas, C.; Middleton, S.; Kremer, A.M.; Wardrope, C.; Hannah, M.; Gillingwater, T.H.; Skehel, P. VAPB interacts with and modulates the activity of ATF6. Hum. Mol. Genet. 2008, 17, 1517–1526. [Google Scholar] [CrossRef]
- Hall, C.E.; Yao, Z.; Choi, M.; Tyzack, G.E.; Serio, A.; Luisier, R.; Harley, J.; Preza, E.; Arber, C.; Crisp, S.J.; et al. Progressive Motor Neuron Pathology and the Role of Astrocytes in a Human Stem Cell Model of VCP-Related ALS. Cell Rep. 2017, 19, 1739–1749. [Google Scholar] [CrossRef] [Green Version]
- Belzil, V.V.; Katzman, R.B.; Petrucelli, L. ALS and FTD: An epigenetic perspective. Acta Neuropathol. 2016, 132, 487–502. [Google Scholar] [CrossRef]
- Liscic, R.M.; Alberici, A.; Cairns, N.J.; Romano, M.; Buratti, E. From basic research to the clinic: Innovative therapies for ALS and FTD in the pipeline. Mol. Neurodegener. 2020, 15, 31. [Google Scholar] [CrossRef] [PubMed]
- Satterfield, T.; Pritchett, J.; Cruz, S.; Kemp, K. Prion disease and endoplasmic reticulum stress pathway correlations and treatment pursuits. Endoplasmic Reticulum Stress Dis. 2017, 4, 27–36. [Google Scholar] [CrossRef] [Green Version]
- Mays, C.E.; Soto, C. The stress of prion disease. Brain Res. 2016, 1648, 553–560. [Google Scholar] [CrossRef]
- Hughes, D.; Halliday, M. What is our current understanding of prpsc-associated neurotoxicity and its molecular underpinnings? Pathogens 2017, 6, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakrabarti, O.; Ashok, A.; Hegde, R.S. Prion protein biosynthesis and its emerging role in neurodegeneration. Trends Biochem. Sci. 2009, 34, 287–295. [Google Scholar] [CrossRef] [Green Version]
- Thapa, S.; Abdelaziz, D.H.; Abdulrahman, B.A.; Schatzl, H.M. Sephin1 Reduces Prion Infection in Prion-Infected Cells and Animal Model. Mol. Neurobiol. 2020, 57, 1–14. [Google Scholar] [CrossRef]
- Orsi, A.; Fioriti, L.; Chiesa, R.; Sitia, R. Conditions of endoplasmic reticulum stress favor the accumulation of cytosolic prion protein. J. Biol. Chem. 2006, 281, 30431–30438. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Castilla, J.; Soto, C. Perturbation of endoplasmic reticulum homeostasis facilitates prion replication. J. Biol. Chem. 2007, 282, 12725–12733. [Google Scholar] [CrossRef] [Green Version]
- Rane, N.S.; Kang, S.W.; Chakrabarti, O.; Feigenbaum, L.; Hegde, R.S. Reduced Translocation of Nascent Prion Protein During ER Stress Contributes to Neurodegeneration. Dev. Cell 2008, 15, 359–370. [Google Scholar] [CrossRef] [Green Version]
- Nunziante, M.; Ackermann, K.; Dietrich, K.; Wolf, H.; Gädtke, L.; Gilch, S.; Vorberg, I.; Groschup, M.; Schätzl, H.M. Proteasomal dysfunction and endoplasmic reticulum stress enhance trafficking of prion protein aggregates through the secretory pathway and increase accumulation of pathologic prion protein. J. Biol. Chem. 2011, 286, 33942–33953. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Russelakis-Carneiro, M.; Wälchli, S.; Carboni, S.; Vial-Knecht, E.; Maundrell, K.; Castilla, J.; Soto, C. The disulfide isomerase Grp58 is a protective factor against prion neurotoxicity. J. Neurosci. 2005, 25, 2793–2802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hetz, C.; Russelakis-Carneiro, M.; Maundrell, K.; Castilla, J.; Soto, C. Caspase-12 and endoplasmic reticulum stress mediate neurotoxicity of pathological prion protein. EMBO J. 2003, 22, 5435–5445. [Google Scholar] [CrossRef] [Green Version]
- Mays, C.E.; Armijo, E.; Morales, R.; Kramm, C.; Flores, A.; Tiwari, A.; Bian, J.; Telling, G.C.; Pandita, T.K.; Hunt, C.R.; et al. Prion disease is accelerated in mice lacking stress-induced heat shock protein 70 (HSP70). J. Biol. Chem. 2019, 294, 13619–13628. [Google Scholar] [CrossRef] [PubMed]
- Park, K.W.; Eun Kim, G.; Morales, R.; Moda, F.; Moreno-Gonzalez, I.; Concha-Marambio, L.; Lee, A.S.; Hetz, C.; Soto, C. The Endoplasmic Reticulum Chaperone GRP78/BiP Modulates Prion Propagation in vitro and in vivo. Sci. Rep. 2017, 7, 44723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallardo, G. Unfolding the mystery of UPR in astrocytes. Sci. Transl. Med. 2020, 12, 14. [Google Scholar] [CrossRef]
- Moreno, J.A.; Radford, H.; Peretti, D.; Steinert, J.R.; Verity, N.; Martin, M.G.; Halliday, M.; Morgan, J.; Dinsdale, D.; Ortori, C.A.; et al. Sustained translational repression by eIF2α-P mediates prion neurodegeneration. Nature 2012, 485, 507–511. [Google Scholar] [CrossRef] [Green Version]
- Moreno, J.A.; Halliday, M.; Molloy, C.; Radford, H.; Verity, N.; Axten, J.M.; Ortori, C.A.; Willis, A.E.; Fischer, P.M.; Barrett, D.A.; et al. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci. Transl. Med. 2013, 5, 206ra138. [Google Scholar] [CrossRef]
- Halliday, M.; Radford, H.; Sekine, Y.; Moreno, J.; Verity, N.; Le Quesne, J.; Ortori, C.A.; Barrett, D.A.; Fromont, C.; Fischer, P.M.; et al. Partial restoration of protein synthesis rates by the small molecule ISRIB prevents neurodegeneration without pancreatic toxicity. Cell Death Dis. 2015, 6, e1672. [Google Scholar] [CrossRef] [Green Version]
- Urano, F.; Wang, X.Z.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Lee, A.H.; Gonzalez-Romero, D.; Thielen, P.; Castilla, J.; Soto, C.; Glimcher, L.H. Unfolded protein response transcription factor XBP-1 does not influence prion replication or pathogenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 757–762. [Google Scholar] [CrossRef] [Green Version]
- Casas-Tinto, S.; Zhang, Y.; Sanchez-Garcia, J.; Gomez-Velazquez, M.; Rincon-Limas, D.E.; Fernandez-Funez, P. The ER stress factor XBP1s prevents amyloid-β neurotoxicity. Hum. Mol. Genet. 2011, 20, 2144–2160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, S.; Yalcin, A.; Lee, G.Y.; Li, P.; Fan, J.; Arruda, A.P.; Pers, B.M.; Yilmaz, M.; Eguchi, K.; Hotamisligil, G.S. Phenotypic assays identify azoramide as a small-molecule modulator of the unfolded protein response with antidiabetic activity. Sci. Transl. Med. 2015, 7, 292ra98. [Google Scholar] [CrossRef] [Green Version]
- Kudo, T.; Kanemoto, S.; Hara, H.; Morimoto, N.; Morihara, T.; Kimura, R.; Tabira, T.; Imaizumi, K.; Takeda, M. A molecular chaperone inducer protects neurons from ER stress. Cell Death Differ. 2008, 15, 364–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitakaze, K.; Taniuchi, S.; Kawano, E.; Hamada, Y.; Miyake, M.; Oyadomari, M.; Kojima, H.; Kosako, H.; Kuribara, T.; Yoshida, S.; et al. Cell-based hts identifies a chemical chaperone for preventing er protein aggregation and proteotoxicity. Elife 2019, 8, e43302. [Google Scholar] [CrossRef] [PubMed]
- Nunes, A.F.; Amaral, J.D.; Lo, A.C.; Fonseca, M.B.; Viana, R.J.S.; Callaerts-Vegh, Z.; D’Hooge, R.; Rodrigues, C.M.P. TUDCA, a bile acid, attenuates amyloid precursor protein processing and amyloid-β deposition in APP/PS1 mice. Mol. Neurobiol. 2012, 45, 440–454. [Google Scholar] [CrossRef]
- Rozpędek-Kamińska, W.; Siwecka, N.; Wawrzynkiewicz, A.; Wojtczak, R.; Pytel, D.; Diehl, J.A.; Majsterek, I. The PERK-dependent molecular mechanisms as a novel therapeutic target for neurodegenerative diseases. Int. J. Mol. Sci. 2020, 21, 2108. [Google Scholar] [CrossRef] [Green Version]
- Grande, V.; Ornaghi, F.; Comerio, L.; Restelli, E.; Masone, A.; Corbelli, A.; Tolomeo, D.; Capone, V.; Axten, J.M.; Laping, N.J.; et al. PERK inhibition delays neurodegeneration and improves motor function in a mouse model of Marinesco-Sjögren syndrome. Hum. Mol. Genet. 2018, 27, 2477–2489. [Google Scholar] [CrossRef]
- Mercado, G.; Castillo, V.; Soto, P.; López, N.; Axten, J.M.; Sardi, S.P.; Hoozemans, J.J.M.; Hetz, C. Targeting PERK signaling with the small molecule GSK2606414 prevents neurodegeneration in a model of Parkinson’s disease. Neurobiol. Dis. 2018, 112, 136–148. [Google Scholar] [CrossRef]
- Radford, H.; Moreno, J.A.; Verity, N.; Halliday, M.; Mallucci, G.R. PERK inhibition prevents tau-mediated neurodegeneration in a mouse model of frontotemporal dementia. Acta Neuropathol. 2015, 130, 633–642. [Google Scholar] [CrossRef] [Green Version]
- Mahameed, M.; Wilhelm, T.; Darawshi, O.; Obiedat, A.; Tommy, W.S.; Chintha, C.; Schubert, T.; Samali, A.; Chevet, E.; Eriksson, L.A.; et al. The unfolded protein response modulators GSK2606414 and KIRA6 are potent KIT inhibitors. Cell Death Dis. 2019, 10, 300. [Google Scholar] [CrossRef]
- Li, A.; Song, N.J.; Riesenberg, B.P.; Li, Z. The Emerging Roles of Endoplasmic Reticulum Stress in Balancing Immunity and Tolerance in Health and Diseases: Mechanisms and Opportunities. Front. Immunol. 2020, 10, 3154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidrauski, C.; McGeachy, A.M.; Ingolia, N.T.; Walter, P. The small molecule ISRIB reverses the effects of eIF2α phosphorylation on translation and stress granule assembly. Elife 2015, 4, e05033. [Google Scholar] [CrossRef] [PubMed]
- Rabouw, H.H.; Langereis, M.A.; Anand, A.A.; Visser, L.J.; De Groot, R.J.; Walter, P.; Van Kuppeveld, F.J.M. Small molecule ISRIB suppresses the integrated stress response within a defined window of activation. Proc. Natl. Acad. Sci. USA 2019, 116, 2097–2102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bugallo, R.; Marlin, E.; Baltanás, A.; Toledo, E.; Ferrero, R.; Vinueza-Gavilanes, R.; Larrea, L.; Arrasate, M.; Aragón, T. Fine tuning of the unfolded protein response by ISRIB improves neuronal survival in a model of amyotrophic lateral sclerosis. Cell Death Dis. 2020, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Markov, N.S.; Lu, Z.; Aillon, R.P.; Soberanes, S.; Runyan, C.E.; Ren, Z.; Grant, R.A.; Maciel, M.; Abdala-Valencia, H.; et al. Resetting proteostasis with ISRIB prevents pulmonary fibrosis. bioRxiv 2020. [Google Scholar] [CrossRef]
- Sidrauski, C.; Acosta-Alvear, D.; Khoutorsky, A.; Vedantham, P.; Hearn, B.R.; Li, H.; Gamache, K.; Gallagher, C.M.; Ang, K.K.H.; Wilson, C.; et al. Pharmacological brake-release of mRNA translation enhances cognitive memory. Elife 2013, 2, e00498. [Google Scholar] [CrossRef]
- Halliday, M.; Radford, H.; Zents, K.A.M.; Molloy, C.; Moreno, J.A.; Verity, N.C.; Smith, E.; Ortori, C.A.; Barrett, D.A.; Bushell, M.; et al. Repurposed drugs targeting eIF2α-P-mediated translational repression prevent neurodegeneration in mice. Brain 2017, 140, 1768–1783. [Google Scholar] [CrossRef] [Green Version]
- Tsaytler, P.; Harding, H.P.; Ron, D.; Bertolotti, A. Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science 2011, 332, 91–94. [Google Scholar] [CrossRef]
- Wang, L.; Popko, B.; Tixier, E.; Roos, R.P. Guanabenz, which enhances the unfolded protein response, ameliorates mutant SOD1-induced amyotrophic lateral sclerosis. Neurobiol. Dis. 2014, 71, 317–324. [Google Scholar] [CrossRef] [Green Version]
- Das, I.; Krzyzosiak, A.; Schneider, K.; Wrabetz, L.; D’Antonio, M.; Barry, N.; Sigurdardottir, A.; Bertolotti, A. Preventing proteostasis diseases by selective inhibition of a phosphatase regulatory subunit. Science 2015, 348, 239–242. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Podojil, J.R.; Kunjamma, R.B.; Jones, J.; Weiner, M.; Lin, W.; Miller, S.D.; Popko, B. Sephin1, which prolongs the integrated stress response, is a promising therapeutic for multiple sclerosis. Brain 2019, 142, 344–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crespillo-Casado, A.; Chambers, J.E.; Fischer, P.M.; Marciniak, S.J.; Ron, D. PPP1R15A-mediated dephosphorylation of eIF2a is unaffected by sephin1 or guanabenz. Elife 2017, 6, e26109. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Lee, K.S.; Lee, H.J.; Kim, D.H.; Noh, Y.H.; Yu, K.; Jung, H.Y.; Lee, S.H.; Lee, J.Y.; Youn, Y.C.; et al. Activation of PERK signaling attenuates Aβ-mediated ER stress. PLoS ONE 2010, 5, e10489. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Luo, N.; Zhao, H.R.; Gao, Q.; Lu, J.; Pan, Y.; Shi, J.P.; Tian, Y.Y.; Zhang, Y.D. Salubrinal protects against rotenone-induced SH-SY5Y cell death via ATF4-parkin pathway. Brain Res. 2014, 1549, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Niso-Santano, M.; Pedro, J.M.B.S.; Gómez-Sánchez, R.; Climent, V.; Soler, G.; Fuentes, J.M.; González-Polo, R.A. ASK1 overexpression accelerates paraquat-induced autophagy via endoplasmic reticulum stress. Toxicol. Sci. 2011, 119, 158–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, C.J.; Kopp, M.C.; Larburu, N.; Nowak, P.R.; Ali, M.M.U. Structure and molecular mechanism of ER stress signaling by the unfolded protein response signal activator IRE1. Front. Mol. Biosci. 2019, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Carlesso, A.; Chintha, C.; Gorman, A.M.; Samali, A.; Eriksson, L.A. Effect of kinase inhibiting rnase attenuator (Kira) compounds on the formation of face-to-face dimers of inositol-requiring enzyme 1: Insights from computational modeling. Int. J. Mol. Sci. 2019, 20, 5538. [Google Scholar] [CrossRef] [Green Version]
- Plate, L.; Cooley, C.B.; Chen, J.J.; Paxman, R.J.; Gallagher, C.M.; Madoux, F.; Genereux, J.C.; Dobbs, W.; Garza, D.; Spicer, T.P.; et al. Small molecule proteostasis regulators that reprogram the ER to reduce extracellular protein aggregation. Elife 2016, 5, 75. [Google Scholar] [CrossRef]
- Paxman, R.; Plate, L.; Blackwood, E.A.; Glembotski, C.; Powers, E.T.; Wiseman, R.L.; Kelly, J.W. Pharmacologic ATF6 activating compounds are metabolically activated to selectively modify endoplasmic reticulum proteins. Elife 2018, 7, e37168. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghemrawi, R.; Khair, M. Endoplasmic Reticulum Stress and Unfolded Protein Response in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 6127. https://doi.org/10.3390/ijms21176127
Ghemrawi R, Khair M. Endoplasmic Reticulum Stress and Unfolded Protein Response in Neurodegenerative Diseases. International Journal of Molecular Sciences. 2020; 21(17):6127. https://doi.org/10.3390/ijms21176127
Chicago/Turabian StyleGhemrawi, Rose, and Mostafa Khair. 2020. "Endoplasmic Reticulum Stress and Unfolded Protein Response in Neurodegenerative Diseases" International Journal of Molecular Sciences 21, no. 17: 6127. https://doi.org/10.3390/ijms21176127
APA StyleGhemrawi, R., & Khair, M. (2020). Endoplasmic Reticulum Stress and Unfolded Protein Response in Neurodegenerative Diseases. International Journal of Molecular Sciences, 21(17), 6127. https://doi.org/10.3390/ijms21176127