Elucidation of Melanogenesis Cascade for Identifying Pathophysiology and Therapeutic Approach of Pigmentary Disorders and Melanoma

 , , , and
, , , and
Abstract
:1. Introduction
2. Melanocyte Development and Differentiation
2.1. Melanocyte Development from Neural Crest
2.2. A Master Role of MITF in Melanocyte Development and Differentiation
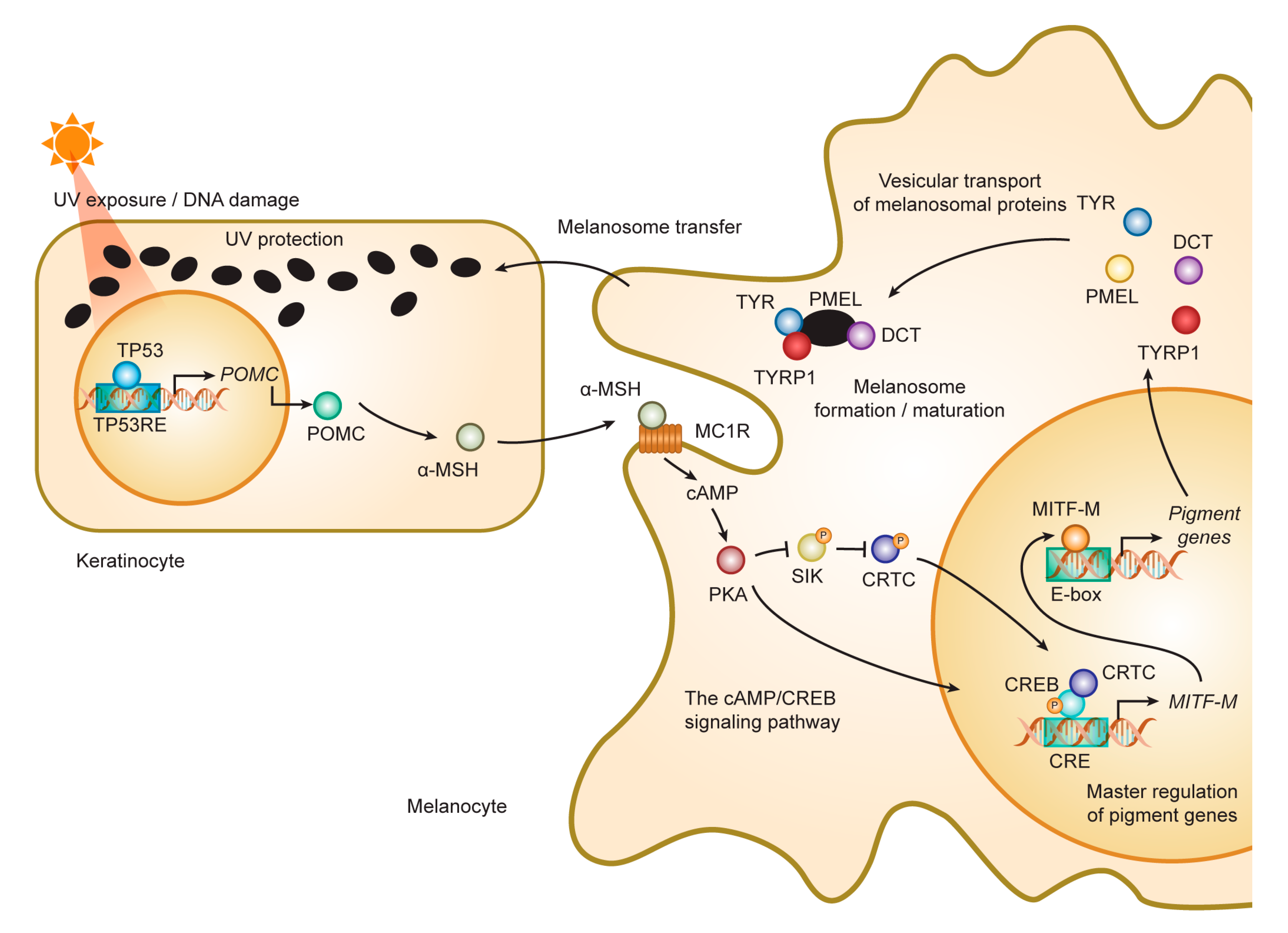
3. Melanogenesis and Intracellular Trafficking for Melanosome Biosynthesis
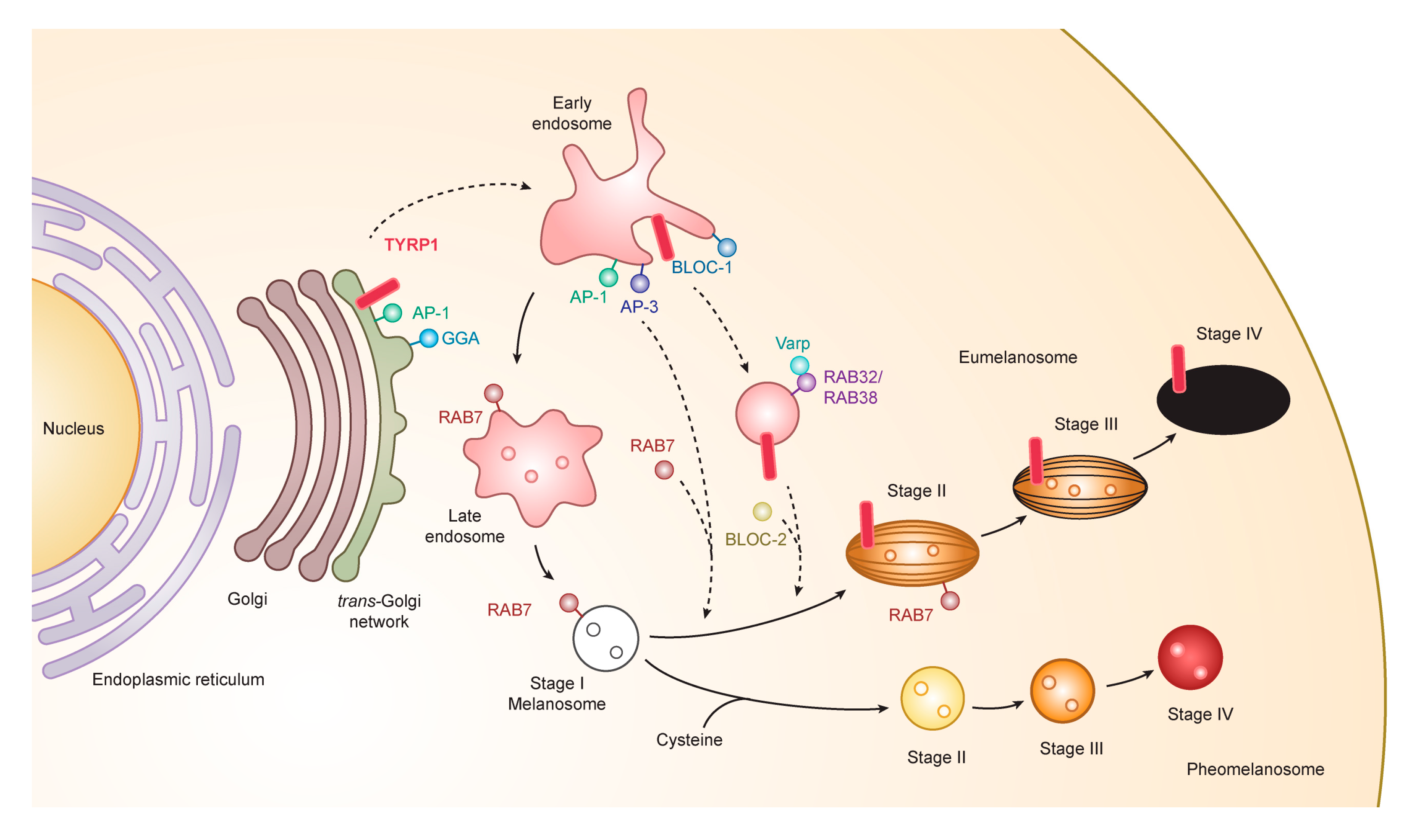
3.1. Formation and Maturation of Melanosomes
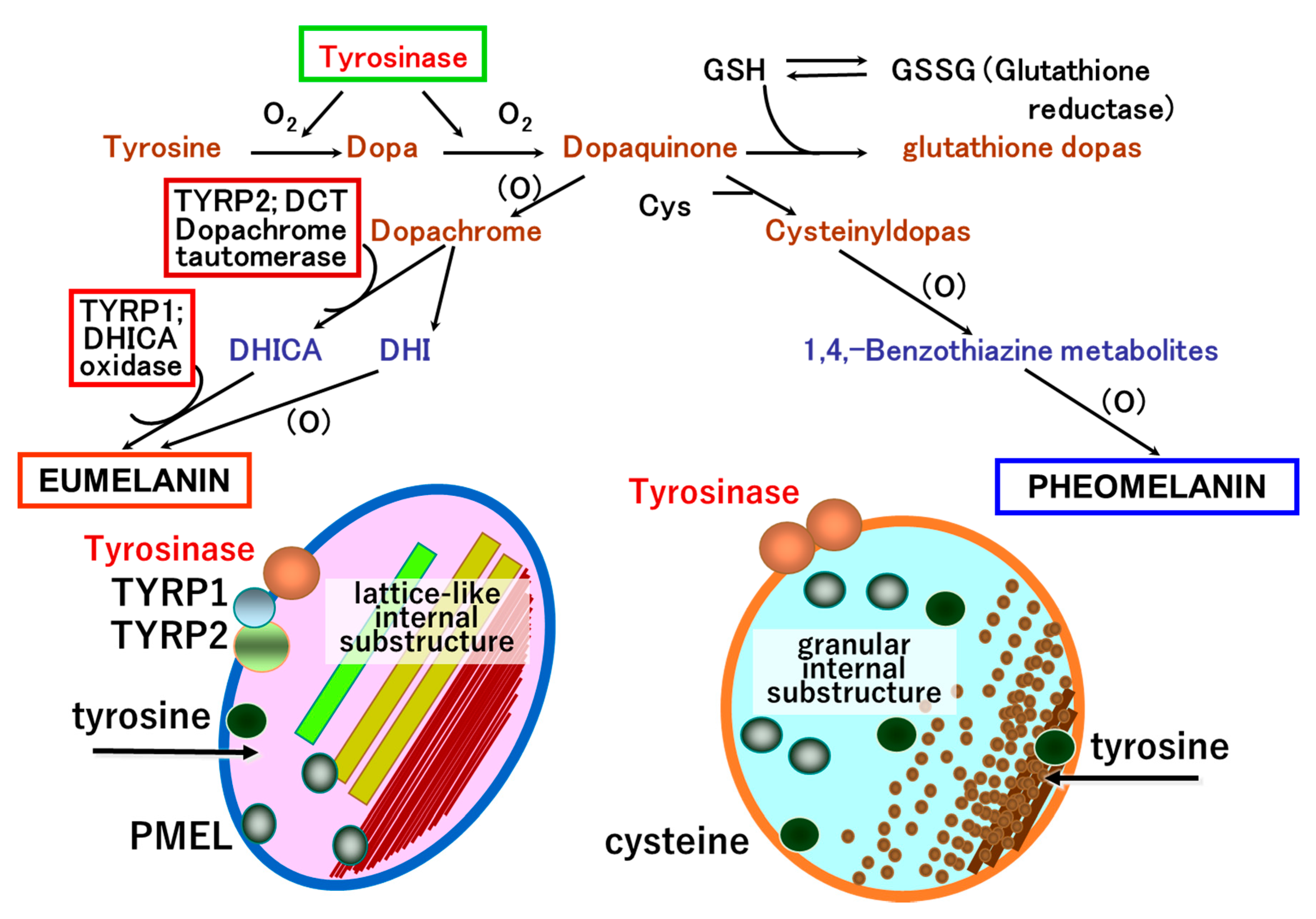
3.2. Roles and Signaling of Tyrosinase and Related Proteins in Melanogenesis
3.3. Transport to Melanosomes and Sorting of Tyrosinase and Tyrosinase-Related Proteins
3.4. Novel Functional Motif of TYRP1 in the Early Stage of Melanogenesis
4. Melanin Pigmentation and Pigment-Type Switching
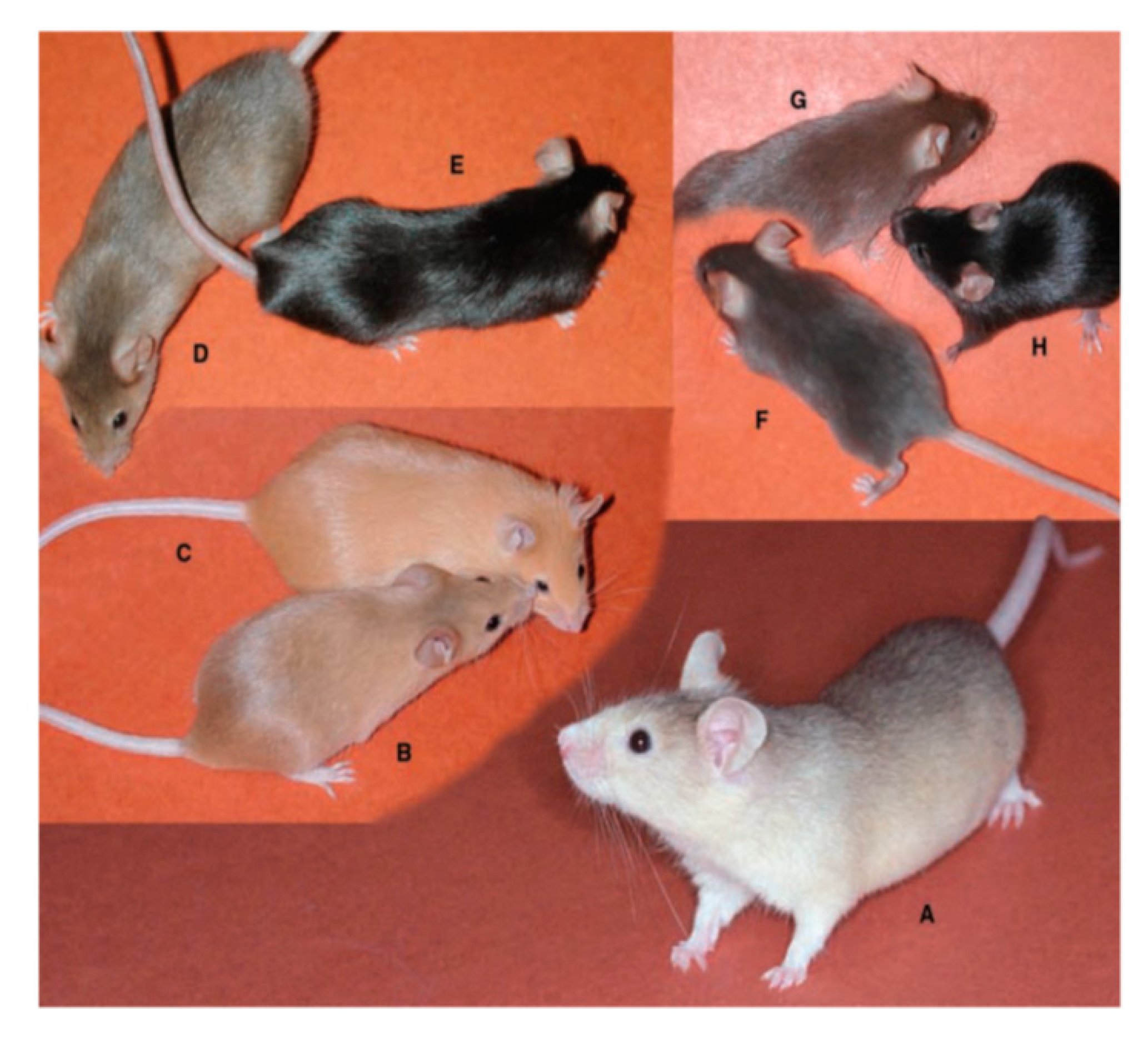
4.1. Coat Color and Melanin Pigmentation
4.2. Mechanism of Pigment-Type Switching
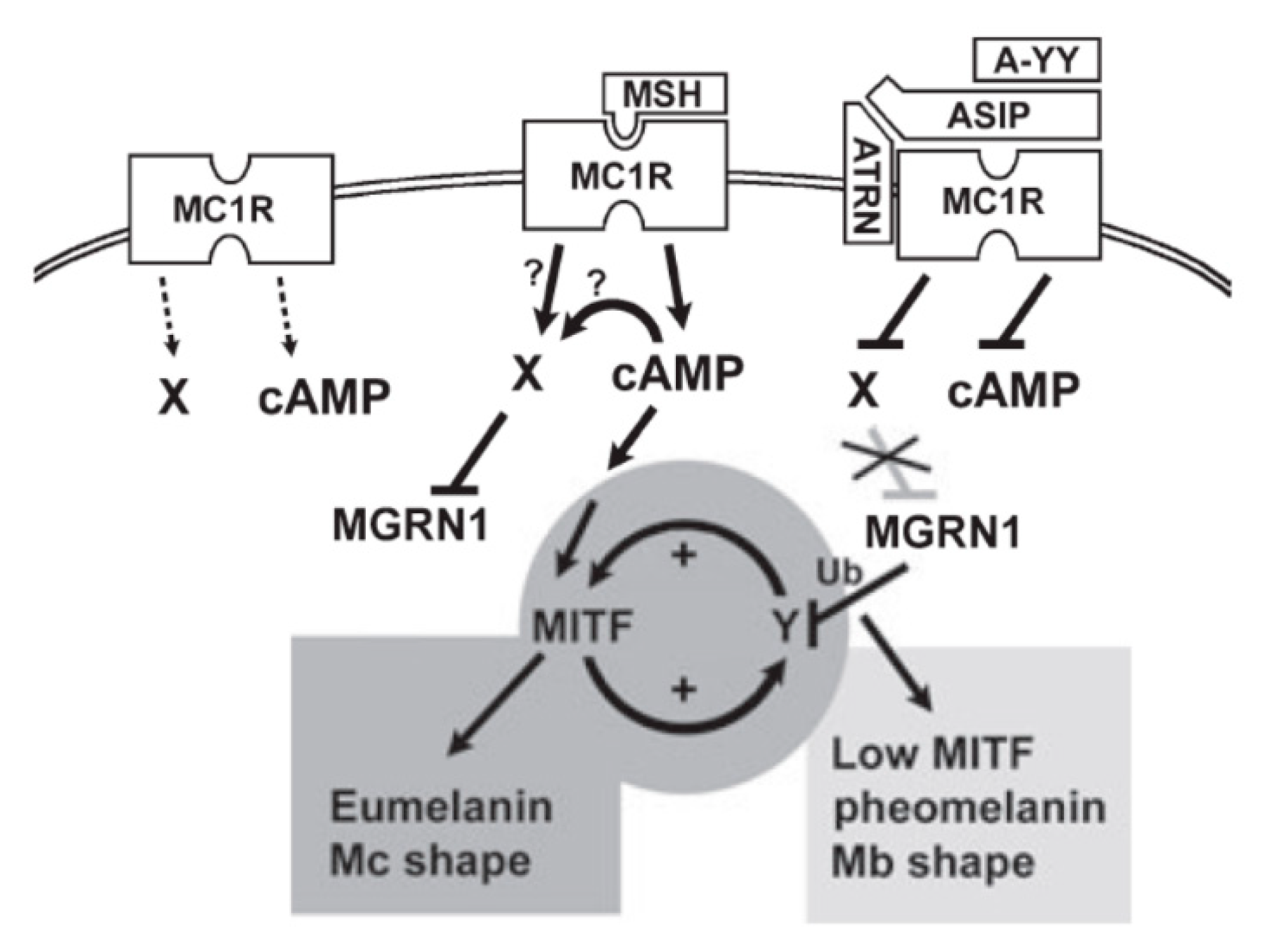
4.3. Accessory Factors of Signaling Through MC1R
5. Development of a Novel Therapeutic Approach for Pigmentary Disorders and Malignant Melanoma by Elucidation of the Melanogenesis Cascade
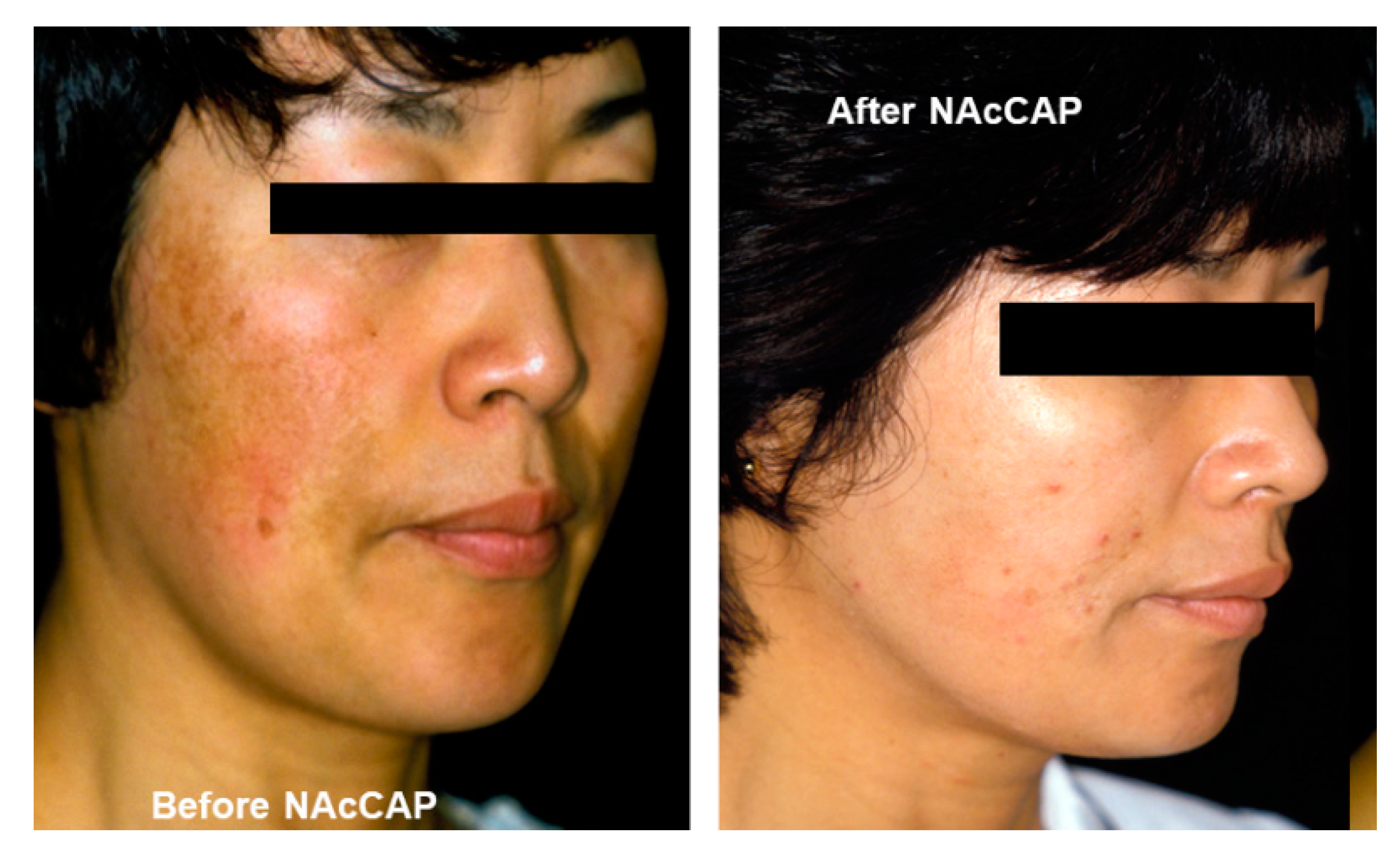
5.1. Melanogenesis Elucidation and Therapeutic Approach for Pigmentary Disorders and Malignant Melanoma
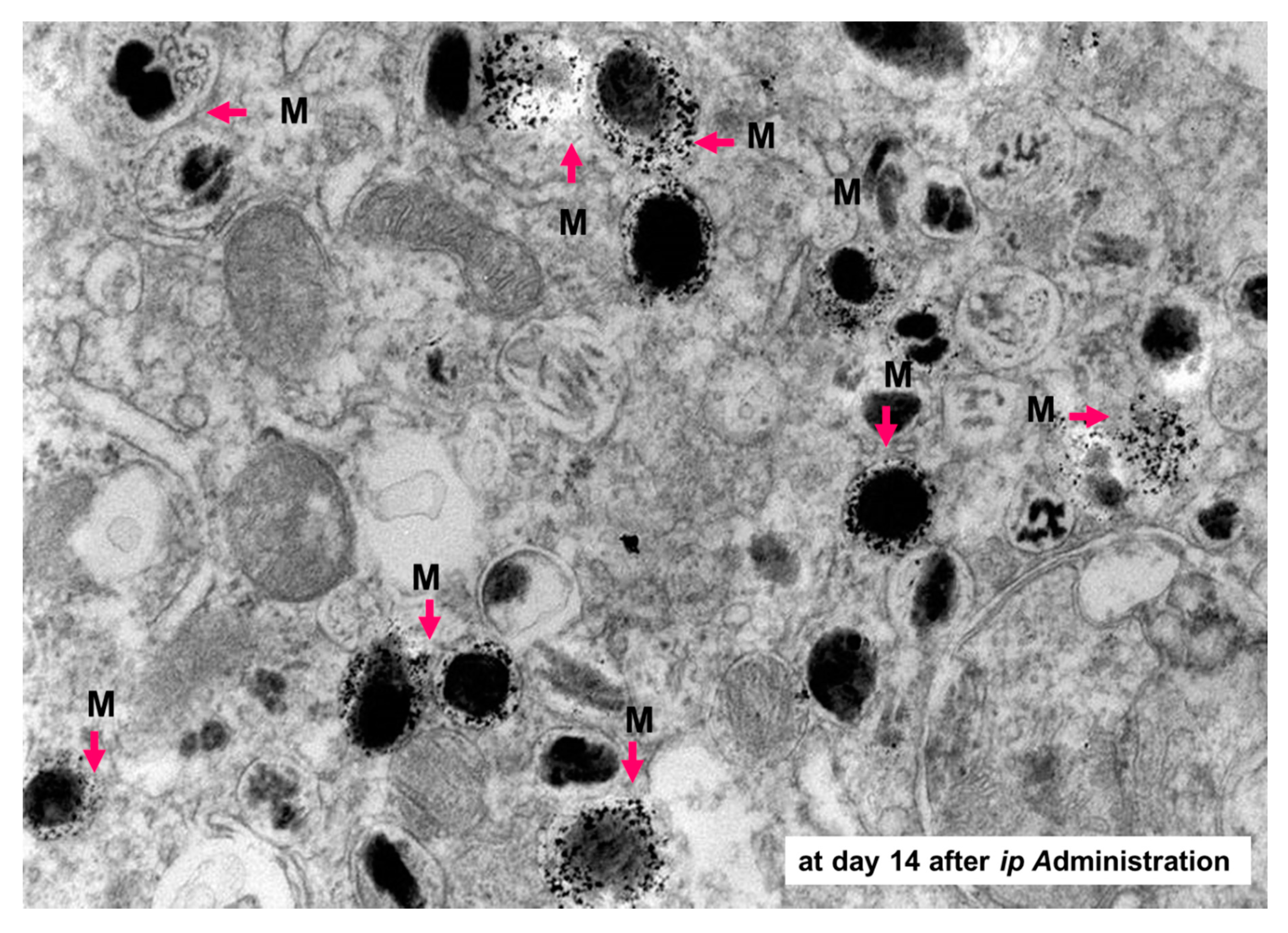
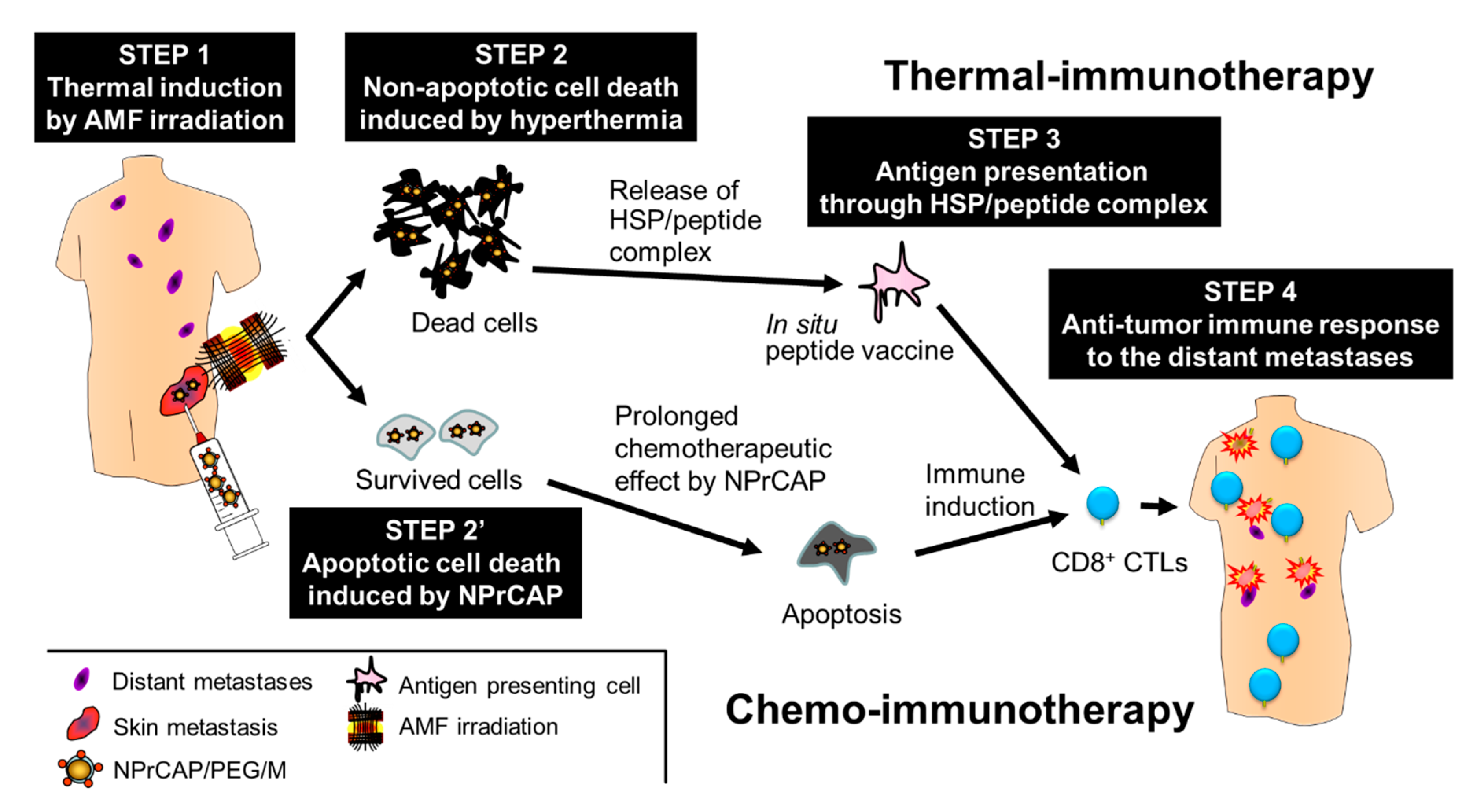
5.2. Melanogenesis-Targeted Melanoma Treatment Based on Chemotherapy and Thermo-Immunotherapy
5.3. Development of Therapeutic Protocol for Melanoma Chemo-Thermo-Immunotherapy
5.4. Acquisition of Anti-Melanoma Immunity by CTI Therapy
5.5. Preliminary study of CTI therapy in Advanced Melanoma Patients
6. Ethics Approval
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACTH | adrenocorticotrophic hormone |
| AMF | alternative magnetic field |
| AP | adaptor-related protein |
| ARF | ADP-ribosylation factor |
| ASIP | agouti signaling protein |
| ATRN | attractin |
| cAMP | cyclic adenosine monophosphate |
| CML | cationic magnetic liposome particles |
| CI-M6PR | cation-independent mannose 6-phosphate receptor |
| CRE | cAMP response element |
| CREB | CRE-binding protein |
| CRTC | CREB-regulated transcriptional coactivator |
| CTI | chemo-thermo-immuno |
| DCT | dopachrome tautomerase |
| EMU | epidermal melanin unit |
| GGA | Golgi-localized γ-ear-containing ADP-ribosylation factor-binding protein |
| HSP | heat shock protein |
| ILV | intralumenal vesicles |
| i.p. | intraperitoneal |
| M | magnetite |
| MC1R | melanocortin 1 receptor |
| MITF | microphthalmia-associated transcription factor |
| MSH | melanocyte-stimulating hormone |
| MW | molecular weight |
| NAcCAP | N-acetyl-4-S-cysteaminylphenol |
| NPrCAP | N-propionyl-4-S-cysteaminylphenol |
| NML | neutral magnetic liposome particles |
| OCA | oculocutaneous albinism |
| PEG | polyethylene glycol |
| PI3K | phosphoinositide-3 kinase |
| PKA | protein kinase A |
| POMC | proopiomelanocortin |
| SIK | salt-inducible kinase |
| TGN | trans-Golgi network |
| TIL | tumor-infiltrating lymph nodes |
| TYRP | tyrosinase-related protein |
References
- Jimbow, K.; Prota, G.; Quevedo, W.C.; Fitzpatrick, T.B. Biology of Melanocytes. In Fitzpatrick’s Dermatology in General Medicine, 5th ed.; Freedberg, I.M., Eisen, A.Z., Wolff, E., Austen, K.F., Goldsmith, L.A., Katz, S.I., Fitzpatrick, T.B., Eds.; McGraw-Hill: New York, NY, USA, 1998; pp. 192–220. [Google Scholar]
- Kawakami, A.; Fisher, D.E. The master role of microphthalmia-associated transcription factor in melanocyte and melanoma biology. Lab. Investig. 2017, 97, 649–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyofuku, K.; Wada, I.; Hirosaki, K.; Park, J.S.; Hori, Y.; Jimbow, K. Promotion of tyrosinase folding in Cos 7 cells by calnexin. J. Biochem. 1999, 125, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Jimbow, K.; Park, J.S.; Kato, F.; Hirosaki, K.; Toyofuku, K.; Hua, C.; Yamashita, T. Assembly, target-signaling and intracellular transport of tyrosinase gene family protein in the initial stage of melanosome biogenesis. Pigment Cell Res. 2000, 13, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Kamada, A.; Nagaya, H.; Tamura, T.; Kinjo, M.; Jin, H.Y.; Yamashita, T.; Jimbow, K.; Kanoh, H.; Wada, I. Regulation of immature protein dynamics in the endoplasmic reticulum. J. Biol. Chem. 2004, 279, 21533–21542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez, P.F.; Luo, D.; Hirosaki, K.; Shinoda, K.; Yamashita, T.; Suzuki, J.; Otsu, K.; Ishikawa, K.; Jimbow, K. Identification of Rab7 as a melanosome-associated protein involved in the intracellular transport of tyrosinase-related protein 1. J. Investig. Dermatol. 2001, 117, 81–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riley, P.A. Melanin. Int. J. Biochem. Cell Biol. 1997, 29, 1235–1239. [Google Scholar] [CrossRef]
- Hirosaki, K.; Yamashita, T.; Wada, I.; Jin, H.Y.; Jimbow, K. Tyrosinase and tyrosinase-related protein 1 require Rab7 for their intracellular transport. J. Investig. Dermatol. 2002, 119, 475–480. [Google Scholar] [CrossRef]
- Kawakami, A.; Sakane, F.; Imai, S.; Yasuda, S.; Kai, M.; Kanoh, H.; Jin, H.Y.; Hirosaki, K.; Yamashita, T.; Fisher, D.E.; et al. Rab7 regulates maturation of melanosomal matrix protein gp100/Pmel17/Silv. J. Investig. Dermatol. 2008, 128, 143–150. [Google Scholar] [CrossRef] [Green Version]
- Prota, G. Regulatory mechanisms of melanogenesis: Beyond the tyrosinase concept. J. Investig. Dermatol. 1993, 100, 156S–161S. [Google Scholar] [CrossRef] [Green Version]
- Simon, J.D.; Peles, D.; Wakamatsu, K.; Ito, S. Current challenges in understanding melanogenesis: Bridging chemistry, biological control, morphology, and function. Pigment Cell Melanoma Res. 2009, 22, 563–579. [Google Scholar] [CrossRef]
- Jimbow, K.; Takada, T.; Osai, Y.; Thomas, P.D.; Sato, M.; Sato, A.; Kamiya, T.; Ono, I.; Tamura, Y.; Sato, N.; et al. Melanogenesis exploitation and melanoma nanomedicine; Utilization of melanogenesis substrate, NPrCAP for exploiting melanoma-targeting drug and its conjunction magnetite nanoparticles for developing melanoma chemo-thermo-immunotherapy. Open Conf. Proc. J. 2011, 2, 5–16. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, A.; Fisher, D.E. Key discoveries in melanocyte development. J. Investig. Dermatol. 2011, 131, E2–E4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adameyko, I.; Lallemend, F.; Aquino, J.B.; Pereira, J.A.; Topilko, P.; Müller, T.; Fritz, N.; Beljajeva, A.; Mochii, M.; Liste, I.; et al. Schwann cell precursors from nerve innervation are a cellular origin of melanocytes in skin. Cell 2009, 139, 366–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adameyko, I.; Lallemend, F.; Furlan, A.; Zinin, N.; Aranda, S.; Kitambi, S.S.; Blanchart, A.; Favaro, R.; Nicolis, S.; Lübke, M.; et al. Sox2 and Mitf cross-regulatory interactions consolidate progenitor and melanocyte lineages in the cranial neural crest. Development 2012, 139, 397–410. [Google Scholar] [CrossRef] [Green Version]
- Steel, K.P.; Davidson, D.R.; Jackson, I.J. TRP-2/DT, a new early melanoblast marker, shows that steel growth factor (c-kit ligand) is a survival factor. Development 1992, 115, 1111–1119. [Google Scholar]
- Wehrle-Haller, B.; Weston, J.A. Soluble and cell-bound forms of steel factor activity play distinct roles in melanocyte precursor dispersal and survival on the lateral neural crest migration pathway. Development 1995, 121, 731–742. [Google Scholar]
- Shin, M.K.; Levorse, J.M.; Ingram, R.S.; Tilghman, S.M. The temporal requirement for endothelin receptor-B signalling during neural crest development. Nature 1999, 402, 496–501. [Google Scholar] [CrossRef]
- Nishimura, E.K.; Jordan, S.A.; Oshima, H.; Yoshida, H.; Osawa, M.; Moriyama, M.; Jackson, I.J.; Barrandon, Y.; Miyachi, Y.; Nishikawa, S. Dominant role of the niche in melanocyte stem-cell fate determination. Nature 2002, 416, 854–860. [Google Scholar] [CrossRef]
- Nishimura, E.K.; Granter, S.R.; Fisher, D.E. Mechanisms of hair graying: Incomplete melanocyte stem cell maintenance in the niche. Science 2005, 307, 720–724. [Google Scholar] [CrossRef] [Green Version]
- Inomata, K.; Aoto, T.; Binh, N.T.; Okamoto, N.; Tanimura, S.; Wakayama, T.; Iseki, S.; Hara, E.; Masunaga, T.; Shimizu, H.; et al. Genotoxic stress abrogates renewal of melanocyte stem cells by triggering their differentiation. Cell 2009, 137, 1088–1099. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Ma, S.; Rachmin, I.; He, M.; Baral, P.; Choi, S.; Gonçalves, W.A.; Shwartz, Y.; Fast, E.M.; Su, Y.; et al. Hyperactivation of sympathetic nerves drives depletion of melanocyte stem cells. Nature 2020, 577, 676–681. [Google Scholar] [CrossRef]
- Hertwig, P. Neue Mutationen und Kopplungsgruppen bei der Hausmaus. Z. Induct. Abstammungs-Vererbungsl. 1942, 80, 220–246. [Google Scholar]
- Hodgkinson, C.A.; Moore, K.J.; Nakayama, A.; Steingrímsson, E.; Copeland, N.G.; Jenkins, N.A.; Arnheiter, H. Mutations at the mouse microphthalmia locus are associated with defects in a gene encoding a novel basic-helix-loop-helix-zipper protein. Cell 1993, 74, 395–404. [Google Scholar] [CrossRef]
- Steingrímsson, E.; Moore, K.J.; Lamoreux, M.L.; Ferré-D’Amaré, A.R.; Burley, S.K.; Zimring, D.C.; Skow, L.C.; Hodgkinson, C.A.; Arnheiter, H.; Copeland, N.G.; et al. Molecular basis of mouse microphthalmia (mi) mutations helps explain their developmental and phenotypic consequences. Nat. Genet. 1994, 8, 256–263. [Google Scholar] [CrossRef]
- Tassabehji, M.; Newton, V.E.; Read, A.P. Waardenburg syndrome type 2 caused by mutations in the human microphthalmia (MITF) gene. Nat. Genet. 1994, 8, 251–255. [Google Scholar] [CrossRef]
- Hemesath, T.J.; Steingrímsson, E.; McGill, G.; Hansen, M.J.; Vaught, J.; Hodgkinson, C.A.; Arnheiter, H.; Copeland, N.G.; Jenkins, N.A.; Fisher, D.E. Microphthalmia, a critical factor in melanocyte development, defines a discrete transcription factor family. Genes Dev. 1994, 28, 179–183. [Google Scholar] [CrossRef] [Green Version]
- Hemesath, T.J.; Price, E.R.; Takemoto, C.; Badalian, T.; Fisher, D.E. MAP kinase links the transcription factor Microphthalmia to c-Kit signalling in melanocytes. Nature 1998, 391, 298–301. [Google Scholar] [CrossRef]
- Yasumoto, K.; Yokoyama, K.; Shibata, K.; Tomita, Y.; Shibahara, S. Microphthalmia-associated transcription factor as a regulator for melanocyte-specific transcription of the human tyrosinase gene. Mol. Cell Biol. 1994, 14, 8058–8070. [Google Scholar] [CrossRef]
- Bertolotto, C.; Buscà, R.; Abbe, P.; Bille, K.; Aberdam, E.; Ortonne, J.P.; Ballotti, R. Different cis-acting elements are involved in the regulation of TRP1 and TRP2 promoter activities by cyclic AMP: Pivotal role of M boxes (GTCATGTGCT) and of microphthalmia. Mol. Cell. Biol. 1998, 18, 694–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, J.; Miller, A.J.; Widlund, H.R.; Horstmann, M.A.; Ramaswamy, S.; Fisher, D.E. MLANA/MART1 and SILV/PMEL17/GP100 are transcriptionally regulated by MITF in melanocytes and melanoma. Am. J. Pathol. 2003, 163, 333–343. [Google Scholar] [CrossRef]
- Bertolotto, C.; Abbe, P.; Hemesath, T.J.; Bille, K.; Fisher, D.E.; Ortonne, J.P.; Ballotti, R. Microphthalmia gene product as a signal transducer in cAMP-induced differentiation of melanocytes. J. Cell Biol. 1998, 142, 827–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, E.R.; Horstmann, M.A.; Wells, A.G.; Weilbaecher, K.N.; Takemoto, C.M.; Landis, M.W.; Fisher, D.E. alpha-Melanocyte-stimulating hormone signaling regulates expression of microphthalmia, a gene deficient in Waardenburg syndrome. J. Biol. Chem. 1998, 273, 33042–33047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, K.; Yasumoto, K.; Takada, R.; Takada, S.; Watanabe, K.; Udono, T.; Saito, H.; Takahashi, K.; Shibahara, S. Induction of melanocyte-specific microphthalmia-associated transcription factor by Wnt-3a. J. Biol. Chem. 2000, 275, 14013–14016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, A.; Takeda, K.; Ploplis, B.; Tachibana, M. Epistatic relationship between Waardenburg syndrome genes MITF and PAX3. Nat. Genet. 1998, 18, 283–286. [Google Scholar] [CrossRef] [PubMed]
- Cui, R.; Widlund, H.R.; Feige, E.; Lin, J.Y.; Wilensky, D.L.; Igras, V.E.; D’Orazio, J.; Fung, C.Y.; Schanbacher, C.F.; Granter, S.R.; et al. Central role of p53 in the suntan response and pathologic hyperpigmentation. Cell 2007, 128, 853–864. [Google Scholar] [CrossRef] [Green Version]
- D’Orazio, J.A.; Nobuhisa, T.; Cui, R.; Arya, M.; Spry, M.; Wakamatsu, K.; Igras, V.; Kunisada, T.; Granter, S.R.; Nishimura, E.K.; et al. Topical drug rescue strategy and skin protection based on the role of Mc1r in UV-induced tanning. Nature 2006, 443, 340–344. [Google Scholar] [CrossRef]
- Mujahid, N.; Liang, Y.; Murakami, R.; Choi, H.G.; Dobry, A.S.; Wang, J.; Suita, Y.; Weng, Q.Y.; Allouche, J.; Kemeny, L.V.; et al. A UV-Independent Topical Small-Molecule Approach for Melanin Production in Human Skin. Cell Rep. 2017, 19, 2177–2184. [Google Scholar] [CrossRef] [Green Version]
- Yun, W.J.; Kim, E.Y.; Park, J.E.; Jo, S.Y.; Bang, S.H.; Chang, E.J.; Chang, S.E. Microtubule-associated protein light chain 3 is involved in melanogenesis via regulation of MITF expression in melanocytes. Sci. Rep. 2016, 6, 19914. [Google Scholar] [CrossRef]
- Djehal, A.; Krayem, M.; Najem, A.; Hammoud, H.; Cresteil, T.; Nebigil, C.G.; Wang, D.; Yu, P.; Bentouhami, E.; Ghanem, G.E.; et al. Targeting prohibitin with small molecules to promote melanogenesis and apoptosis in melanoma cells. Eur. J. Med. Chem. 2018, 155, 880–888. [Google Scholar] [CrossRef]
- Theos, A.C.; Truschel, S.T.; Raposo, G.; Marks, M.S. The Silver locus product Pmel17/gp100/Silv/ME20: Controversial in name and in function. Pigment Cell Res. 2005, 18, 322–336. [Google Scholar] [CrossRef] [Green Version]
- Kushimoto, T.; Basrur, V.; Valencia, J.; Matsunaga, J.; Vieira, W.D.; Ferrans, V.J.; Muller, J.; Appella, E.; Hearing, V.J. A model for melanosome biogenesis based on the purification and analysis of early melanosomes. Proc. Natl. Acad. Sci. USA 2001, 98, 10698–10703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raposo, G.; Tenza, D.; Murphy, D.M.; Berson, J.F.; Marks, M.S. Distinct protein sorting and localization to premelanosomes, melanosomes, and lysosomes in pigmented melanocytic cells. J. Cell Biol. 2001, 152, 809–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theos, A.C.; Berson, J.F.; Theos, S.C.; Herman, K.E.; Harper, D.C.; Tenza, D.; Sviderskaya, E.V.; Lamoreux, M.L.; Bennett, D.C.; Raposo, G.; et al. Dual loss of ER export and endocytic signals with altered melanosome morphology in the silver mutation of Pmel17. Mol. Biol. Cell 2006, 17, 3598–3612. [Google Scholar] [CrossRef] [Green Version]
- Leonhardt, R.M.; Vigneron, N.; Rahner, C.; Cresswell, P. Proprotein convertases process PMEL17 during secretion. J. Biol. Chem. 2011, 286, 9321–9337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delevoye, C.; Giordano, F.; van Niel, G.; Raposo, G. Biogenesis of melanosome—The chessboard of pigmentation. Med. Sci. 2011, 27, 153–162. [Google Scholar] [CrossRef] [Green Version]
- Basrur, V.; Yang, F.; Kushimoto, T.; Higashimoto, Y.; Yasumoto, K.; Valencia, J.; Muller, J.; Vieira, W.D.; Watabe, H.; Shabanowitz, J.; et al. Proteomic analysis of early melanosomes: Identification of novel melanosomal proteins. J. Proteome Res. 2003, 2, 69–79. [Google Scholar] [CrossRef]
- Incerti, B.; Cortese, K.; Pizzigoni, A.; Surace, E.M.; Varani, S.; Coppola, M.; Jeffery, G.; Seeliger, M.; Jaissle, G.; Bennett, D.C.; et al. Oa1 knock-out: New insights on the pathogenesis of ocular albinism type 1. Hum. Mol. Genet. 2000, 9, 2781–2788. [Google Scholar] [CrossRef] [Green Version]
- Lopez, V.M.; Decatur, C.L.; Stamer, W.D.; Lynch, R.M.; McKay, B.S. L-DOPA is an endogenous ligand for OA1. PLoS Biol. 2008, 6, e236. [Google Scholar] [CrossRef] [Green Version]
- Giordano, F.; Bonetti, C.; Surace, E.M.; Marigo, V.; Raposo, G. The ocular albinism type 1 (OA1) G-protein-coupled receptor functions with MART-1 at early stages of melanogenesis to control melanosome identity and composition. Hum. Mol. Genet. 2009, 18, 4530–4545. [Google Scholar] [CrossRef] [Green Version]
- Raposo, G.; Marks, M.S. Melanosomes–dark organelles enlighten endosomal membrane transport. Nat. Rev. Mol. Cell Biol. 2007, 8, 786–797. [Google Scholar] [CrossRef] [Green Version]
- Sitaram, A.; Marks, M.S. Mechanisms of protein delivery to melanosomes in pigment cells. Physiology 2012, 27, 85–99. [Google Scholar] [CrossRef] [Green Version]
- Pennamen, P.; Le, L.; Tingaud-Sequeira, A.; Fiore, M.; Bauters, A.; Van Duong Béatrice, N.; Coste, V.; Bordet, J.C.; Plaisant, C.; Diallo, M.; et al. BLOC1S5 pathogenic variants cause a new type of Hermansky-Pudlak syndrome. Genet. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Di Pietro, S.M.; Falcón-Pérez, J.M.; Tenza, D.; Setty, S.R.; Marks, M.S.; Raposo, G.; Dell’Angelica, E.C. BLOC-1 interacts with BLOC-2 and the AP-3 complex to facilitate protein trafficking on endosomes. Mol. Biol. Cell 2006, 17, 4027–4038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Setty, S.R.; Tenza, D.; Truschel, S.T.; Chou, E.; Sviderskaya, E.V.; Theos, A.C.; Lamoreux, M.L.; Di Pietro, S.M.; Starcevic, M.; Bennett, D.C.; et al. BLOC-1 is required for cargo-specific sorting from vacuolar early endosomes toward lysosome-related organelles. Mol. Biol. Cell 2007, 18, 768–780. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Salopek, T.G.; Jimbow, K. The role of phosphoinositide 3-kinase in the sorting and transport of newly synthesized tyrosinase-related protein-1 (TRP-1). J. Investig. Dermatol. Symp. Proc. 2001, 6, 105–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theos, A.C.; Tenza, D.; Martina, J.A.; Hurbain, I.; Peden, A.A.; Sviderskaya, E.V.; Stewart, A.; Robinson, M.S.; Bennett, D.C.; Cutler, D.F.; et al. Functions of adaptor protein (AP)-3 and AP-1 in tyrosinase sorting from endosomes to melanosomes. Mol. Biol. Cell 2005, 16, 5356–5372. [Google Scholar] [CrossRef]
- Nakayama, K.; Wakatsuki, S. The structure and function of GGAs, the traffic controllers at the TGN sorting crossroads. Cell Struct. Funct. 2003, 28, 431–442. [Google Scholar] [CrossRef] [Green Version]
- Boman, A.L. GGA proteins: New players in the sorting game. J. Cell Sci. 2001, 114, 3413–3418. [Google Scholar]
- Bonifacino, J.S. The GGA proteins: Adaptors on the move. Nat. Rev. Mol. Cell Biol. 2004, 5, 23–32. [Google Scholar] [CrossRef]
- Calvo, P.A.; Frank, D.W.; Bieler, B.M.; Berson, J.F.; Marks, M.S. A cytoplasmic sequence in human tyrosinase defines a second class of di-leucine-based sorting signals for late endosomal and lysosomal delivery. J. Biol. Chem. 1999, 274, 12780–12789. [Google Scholar] [CrossRef] [Green Version]
- Huizing, M.; Sarangarajan, R.; Strovel, E.; Zhao, Y.; Gahl, W.A.; Boissy, R.E. AP-3 mediates tyrosinase but not TRP-1 trafficking in human melanocytes. Mol. Biol. Cell 2001, 12, 2075–2085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinayagamoorthy, T.; Dakour, J.; Dixon, W.; Jimbow, K. cDNA-based functional domains of a calnexin-like melanosomal protein, p90. Melanoma Res. 1993, 3, 263–269. [Google Scholar]
- Hida, T.; Sohma, H.; Kokai, Y.; Kawakami, A.; Hirosaki, K.; Okura, M.; Tosa, N.; Yamashita, T.; Jimbow, K. Rab7 is a critical mediator in vesicular transport of tyrosinase-related protein 1 in melanocytes. J. Dermatol. 2011, 38, 432–441. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Hearing, V.J. Melanocytes and their diseases. Cold Spring Harb. Perspect. Med. 2014, 4, a017046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimbow, K.; Quevedo, W.C., Jr.; Fitzpatrick, T.B.; Szabo, G. Some aspects of melanin biology; 1950-1975. J. Investig. Dermatol. 1976, 67, 721–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzpatrick, T.B.; Jimbow, K.; Donaldson, D.D. Dominant oculocutaneous albinism. Br. J. Dermatol. 1975, 91, 23. [Google Scholar] [CrossRef]
- Ito, S.; Wakamatsu, K. Chemistry of mixed melanogenesis--pivotal roles of dopaquinone. Photochem. Photobiol. 2008, 84, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Wakamatsu, K.; Kavanagh, R.; Kadekaro, A.L.; Terzieva, S.; Sturm, R.A.; Leachman, S.; Abdel-Malek, Z.; Ito, S. Diversity of pigmentation in cultured human melanocytes is due to differences in the type as well as quantity of melanin. Pigment Cell Res. 2006, 19, 154–162. [Google Scholar] [CrossRef]
- Ito, S.; Wakamatsu, K. Quantitative analysis of eumelanin and pheomelanin in humans, mice, and other animals: A comparative review. Pigment Cell Res. 2003, 16, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Silvers, W.K. The Coat Colors of Mice: A Model for Mammalian Gene Action and Interaction; Springer: New York, NY, USA, 1979; Available online: http://www.informatics.jax.org/wksilvers/index.shtml (accessed on 4 July 2020).
- Bennett, D.C.; Lamoreux, M.L. The color loci of mice--a genetic century. Pigment Cell Res. 2003, 16, 333–344. [Google Scholar] [CrossRef] [Green Version]
- Mountjoy, K.G.; Robbins, L.S.; Mortrud, M.T.; Cone, R.D. The cloning of a family of genes that encode the melanocortin receptors. Science 1992, 257, 1248–1251. [Google Scholar] [CrossRef] [PubMed]
- Chhajlani, V.; Wikberg, J.E. Molecular cloning and expression of the human melanocyte stimulating hormone receptor cDNA. FEBS Lett. 1992, 309, 417–420. [Google Scholar] [CrossRef] [Green Version]
- García-Borrón, J.C.; Abdel-Malek, Z.; Jiménez-Cervantes, C. MC1R, the cAMP pathway, and the response to solar UV: Extending the horizon beyond pigmentation. Pigment Cell Melanoma Res. 2014, 27, 699–720. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.K.; Funasaka, Y.; Slominski, A.; Ermak, G.; Hwang, J.; Pawelek, J.M.; Ichihashi, M. Production and release of proopiomelanocortin (POMC) derived peptides by human melanocytes and keratinocytes in culture: Regulation by ultraviolet B. Biochim. Biophys. Acta 1996, 1313, 130–138. [Google Scholar] [CrossRef] [Green Version]
- Slominski, A.; Plonka, P.M.; Pisarchik, A.; Smart, J.L.; Tolle, V.; Wortsman, J.; Low, M.J. Preservation of eumelanin hair pigmentation in proopiomelanocortin-deficient mice on a nonagouti (a/a) genetic background. Endocrinology 2005, 146, 1245–1253. [Google Scholar] [CrossRef] [Green Version]
- Hida, T.; Wakamatsu, K.; Sviderskaya, E.V.; Donkin, A.J.; Montoliu, L.; Lynn Lamoreux, M.; Yu, B.; Millhauser, G.L.; Ito, S.; Barsh, G.S.; et al. Agouti protein, mahogunin, and attractin in pheomelanogenesis and melanoblast-like alteration of melanocytes: A cAMP-independent pathway. Pigment Cell Melanoma Res. 2009, 22, 623–634. [Google Scholar] [CrossRef] [Green Version]
- Herraiz, C.; Garcia-Borron, J.C.; Jiménez-Cervantes, C.; Olivares, C. MC1R signaling. Intracellular partners and pathophysiological implications. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2448–2461. [Google Scholar] [CrossRef]
- Chiaverini, C.; Beuret, L.; Flori, E.; Busca, R.; Abbe, P.; Bille, K.; Bahadoran, P.; Ortonne, J.P.; Bertolotto, C.; Ballotti, R. Microphthalmia-associated transcription factor regulates RAB27A gene expression and controls melanosome transport. J. Biol. Chem. 2008, 283, 12635–12642. [Google Scholar] [CrossRef] [Green Version]
- Ollmann, M.M.; Lamoreux, M.L.; Wilson, B.D.; Barsh, G.S. Interaction of Agouti protein with the melanocortin 1 receptor in vitro and in vivo. Genes Dev. 1998, 12, 316–330. [Google Scholar] [CrossRef] [Green Version]
- Sulem, P.; Gudbjartsson, D.F.; Stacey, S.N.; Helgason, A.; Rafnar, T.; Jakobsdottir, M.; Steinberg, S.; Gudjonsson, S.A.; Palsson, A.; Thorleifsson, G.; et al. Two newly identified genetic determinants of pigmentation in Europeans. Nat. Genet. 2008, 40, 835–837. [Google Scholar] [CrossRef]
- Liu, F.; Visser, M.; Duffy, D.L.; Hysi, P.G.; Jacobs, L.C.; Lao, O.; Zhong, K.; Walsh, S.; Chaitanya, L.; Wollstein, A.; et al. Genetics of skin color variation in Europeans: Genome-wide association studies with functional follow-up. Hum. Genet. 2015, 134, 823–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunn, T.M.; Miller, K.A.; He, L.; Hyman, R.W.; Davis, R.W.; Azarani, A.; Schlossman, S.F.; Duke-Cohan, J.S.; Barsh, G.S. The mouse mahogany locus encodes a transmembrane form of human attractin. Nature 1999, 398, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Gunn, T.M.; Inui, T.; Kitada, K.; Ito, S.; Wakamatsu, K.; He, L.; Bouley, D.M.; Serikawa, T.; Barsh, G.S. Molecular and phenotypic analysis of Attractin mutant mice. Genetics 2001, 158, 1683–1695. [Google Scholar] [PubMed]
- Gunn, T.M.; Silvius, D.; Bagher, P.; Sun, K.; Walker, K.K. MGRN1-dependent pigment-type switching requires its ubiquitination activity but not its interaction with TSG101 or NEDD4. Pigment Cell Melanoma Res. 2013, 26, 263–268. [Google Scholar] [CrossRef]
- Srivastava, D.; Chakrabarti, O. Mahogunin-mediated α-tubulin ubiquitination via noncanonical K6 linkage regulates microtubule stability and mitotic spindle orientation. Cell Death Dis. 2014, 5, e1064. [Google Scholar] [CrossRef] [Green Version]
- Abrisqueta, M.; Olivares, C.; Herraiz, C.; Castejón-Griñán, M.; Sirés-Campos, J.; García-Borrón, J.C.; Jiménez-Cervantes, C. Human melanocortin 1 receptor-mediated ubiquitination of nonvisual arrestins. Role of Mahogunin Ring Finger 1 E3 ligase. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 76–94. [Google Scholar] [CrossRef]
- Pérez-Oliva, A.B.; Olivares, C.; Jiménez-Cervantes, C.; García-Borrón, J.C. Mahogunin ring finger-1 (MGRN1) E3 ubiquitin ligase inhibits signaling from melanocortin receptor by competition with Galphas. J. Biol. Chem. 2009, 284, 31714–31725. [Google Scholar] [CrossRef] [Green Version]
- Kerns, J.A.; Cargill, E.J.; Clark, L.A.; Candille, S.I.; Berryere, T.G.; Olivier, M.; Lust, G.; Todhunter, R.J.; Schmutz, S.M.; Murphy, K.E.; et al. Linkage and segregation analysis of black and brindle coat color in domestic dogs. Genetics 2007, 176, 1679–1689. [Google Scholar] [CrossRef] [Green Version]
- Candille, S.I.; Kaelin, C.B.; Cattanach, B.M.; Yu, B.; Thompson, D.A.; Nix, M.A.; Kerns, J.A.; Schmutz, S.M.; Millhauser, G.L.; Barsh, G.S. A defensin mutation causes black coat color in domestic dogs. Science 2007, 318, 1418–1423. [Google Scholar] [CrossRef] [Green Version]
- Swope, V.B.; Jameson, J.A.; McFarland, K.L.; Supp, D.M.; Miller, W.E.; McGraw, D.W.; Patel, M.A.; Nix, M.A.; Millhauser, G.L.; Babcock, G.F.; et al. Defining MC1R regulation in human melanocytes by its agonist α-melanocortin and antagonists agouti signaling protein and β-defensin 3. J. Investig. Dermatol. 2012, 132, 2255–2262. [Google Scholar] [CrossRef] [Green Version]
- Nix, M.A.; Kaelin, C.B.; Ta, T.; Weis, A.; Morton, G.J.; Barsh, G.S.; Millhauser, G.L. Molecular and functional analysis of human β-defensin 3 action at melanocortin receptors. Chem. Biol. 2013, 20, 784–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domingues, B.; Lopes, J.M.; Soares, P.; Populo, H. Melanoma treatment in review. Immunotargets Ther. 2018, 7, 35–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, J.R.; Kim, N.H.; Cho, J.; Choi, K.C. Current treatments for advanced melanoma and introduction of a promising novel gene therapy for melanoma. Oncol. Rep. 2016, 36, 1779–1786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimbow, K. N-acetyl-4-S-cysteaminylphenol as a new type of depigmenting agent for the melanoderma of patients with melasma. Arch. Dermatol. 1991, 127, 1528–1534. [Google Scholar] [CrossRef] [PubMed]
- Alena, F.; Iwashina, T.; Gili, A.; Jimbow, K. Selective in vivo accumulation of N-acetyl-4-S-cysteaminylphenol in B16F10 murine melanoma and enhancement of its in vitro and in vivo antimelanoma effect by combination of buthionine sulfoximine. Cancer Res. 1994, 54, 2661–2666. [Google Scholar] [PubMed]
- Minamitsuji, Y.; Toyofuku, K.; Sugiyama, S.; Yamada, K.; Jimbow, K. Sulfur containing tyrosine analogs can cause selective melanocytotoxicity involving tyrosinase-mediated apoptosis. J. Investig. Dermatol. Symp. Proc. 1999, 4, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Kakimi, K.; Nakayama, E.; Jimbow, K. Antitumor immunity by magnetic nanoparticle-mediated hyperthermia. Nanomedicine 2014, 9, 1715–1726. [Google Scholar] [CrossRef] [Green Version]
- Sato, M.; Yamashita, T.; Ohkura, M.; Osai, Y.; Sato, A.; Takada, T.; Matsusaka, H.; Ono, I.; Tamura, Y.; Sato, N.; et al. N-propionyl-cysteaminylphenol-magnetite conjugate (NPrCAP/M) is a nanoparticle for the targeted growth suppression of melanoma cells. J. Investig. Dermatol. 2009, 129, 2233–2241. [Google Scholar] [CrossRef] [Green Version]
- Algan, O.; Fosmire, H.; Hynynen, K.; Dalkin, B.; Cui, H.; Drach, G.; Stea, B.; Cassady, J.R. External beam radiotherapy and hyperthermia in the treatment of patients with locally advanced prostate carcinoma. Cancer 2000, 89, 399–403. [Google Scholar] [CrossRef]
- Takada, T.; Yamashita, T.; Sato, M.; Sato, A.; Ono, I.; Tamura, Y.; Sato, N.; Miyamoto, A.; Ito, A.; Honda, H.; et al. Growth inhibition of re-challenge B16 melanoma transplant by conjugates of melanogenesis substrate and magnetite nanoparticles as the basis for developing melanoma-targeted chemo-thermo-immunotherapy. J. Biomed. Biotechnol. 2009, 2009, 457936. [Google Scholar] [CrossRef]
- Ishii-Osai, Y.; Yamashita, T.; Tamura, Y.; Sato, N.; Ito, A.; Honda, H.; Wakamatsu, K.; Ito, S.; Nakayama, E.; Okura, M.; et al. N-propionyl-4-S-cysteaminylphenol induces apoptosis in B16F1 cells and mediates tumor-specific T-cell immune responses in a mouse melanoma model. J. Dermatol. Sci. 2012, 67, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Tamura, Y.; Sato, N.; Yamashita, T.; Takada, T.; Sato, M.; Osai, Y.; Okura, M.; Ono, I.; Ito, A.; et al. Melanoma-targeted chemo-thermo-immuno (CTI)-therapy using N-propionyl-4-S-cysteaminylphenol-magnetite nanoparticles elicits CTL response via heat shock protein-peptide complex release. Cancer Sci. 2010, 101, 1939–1946. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Yamaguchi, M.; Okamoto, N.; Sanematsu, Y.; Kawabe, Y.; Wakamatsu, K.; Ito, S.; Honda, H.; Kobayashi, T.; Nakayama, E.; et al. T-cell receptor repertoires of tumor-infiltrating lymphocytes after hyperthermia using functionalized magnetite nanoparticles. Nanomedicine 2013, 8, 891–902. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Human Disease | Major Clinical Features | Mutated Gene | Gene Action in Humans |
|---|---|---|---|
| GENERALIZED | |||
| Oculocutaneous albinism type 1 | Hypopigmentation, nystagmus | TYR | Key enzyme for melanin biosynthesis |
| Oculocutaneous albinism type 2 | OCA2 | Melanosome biogenesis and size | |
| Oculocutaneous albinism type 3 | TYRP1 | Melanosomal enzyme; stabilizing factor | |
| Oculocutaneous albinism type 4 | SLC45A2 | Solute transporter; previously named MATP | |
| Oculocutaneous albinism type 5 | (4q24) | Responsible gene is not known | |
| Oculocutaneous albinism type 6 | SLC24A5 | Predominant sodium-calcium exchanger in melanocytes | |
| Oculocutaneous albinism type 7 | LRMDA | Required for melanocyte differentiation; previously named C10orf11 | |
| Ocular albinism type 1 | Iris hypopigmentation, nystagmus | GPR143 | G protein-coupled receptor localized at melanosomal membrane |
| Hermansky–Pudlak syndrome type 1 | Hypopigmentation, bleeding, immunodeficiency | HPS1 | Component of BLOC-3, which acts as a guanine exchange factor; organelle biogenesis and size |
| Hermansky–Pudlak syndrome type 2 | AP3B1 | β1 subunit of AP-3 complex; organelle protein routing | |
| Hermansky–Pudlak syndrome type 3 | HPS3 | BLOC-2 subunit 1; organelle biogenesis | |
| Hermansky–Pudlak syndrome type 4 | HPS4 | Component of BLOC-3; organelle biogenesis and size | |
| Hermansky–Pudlak syndrome type 5 | HPS5 | BLOC-2 subunit 2; organelle biogenesis | |
| Hermansky–Pudlak syndrome type 6 | HPS6 | BLOC-2 subunit 3; organelle biogenesis | |
| Hermansky–Pudlak syndrome type 7 | DTNBP1 | Dysbindin, component of BLOC-1 | |
| Hermansky–Pudlak syndrome type 8 | BLOC1S3 | BLOC-1 subunit 3 | |
| Hermansky–Pudlak syndrome type 9 | BLOC1S6 | BLOC-1 subunit 6 | |
| Hermansky–Pudlak syndrome type 10 | AP3D1 | δ1 subunit of AP-3 complex; organelle protein routing | |
| Hermansky–Pudlak syndrome type 11 | BLOC1S5 | BLOC-1 subunit 5 | |
| Chediak–Higashi syndrome | Hypopigmentation, immunodeficiency | LYST | Protein required for sorting endosomal resident proteins into late multivesicular endosomes |
| Griscelli syndrome type 1 | Hypopigmentation, pancytopenia, immunologic disorder, central nervous system abnormalities | MYO5A | Melanosome transport; myosin type Va/dilute mice |
| Griscelli syndrome type 2 | RAB27A | Melanosome transport; RAS-associated protein/ashen mice | |
| Griscelli syndrome type 3 | MLPH | Melanosome transport; melanophilin/leaden mice | |
| Phenylketonuria | Phenylalanine hydroxylase deficiency | PAH | Phenylalanine hydroxylase |
| Charcot–Marie–Tooth disease type 4J | Pale skin, alopecia, clumped melanosomes, immune effects | FIG4 | Phosphatidyl-inositol 3,5-bisphosphate 5-phosphatase; aberrant early melanosome architecture |
| Menkes disease | Copper transport disorders, kinky hair | ATP7A | ATPase, copper-transporting α polypeptide |
| Wilson disease | Copper transport disorders, kinky hair | ATP7B | ATPase, copper-transporting β polypeptide |
| Cystinosis | Blond hair, multiple organ dysfunctions | CTNS | Cystinosin, cysteine/H+ symporter, which exports cysteine out of lysosomes |
| Tietz albinism-deafness syndrome | Congenital profound deafness, generalized hypopigmentation | MITF | Transcription factor; master regulator of melanocyte lineage |
| CIRCUMSCRIBED | |||
| Waardenburg syndrome type 1 and 3 | White forelock, premature graying, hearing loss, heterochromia, other neural crest defects | PAX3 | Transcription factor; neural tube development |
| Waardenburg syndrome type 2 | MITF, SNAI2, SOX10 | Transcription factors; master regulator of melanocyte lineage transcription factor | |
| Waardenburg syndrome type 4 | EDNRB, EDN3, SOX10 | Endothelin receptor B; melanoblast/neuroblast growth and differentiation factor; transcription factor | |
| Piebaldism | White spotting, megacolon, and other neural crest defects | KIT, SNAI2 | Receptor for SCF; required for melanoblast survival and homing; melanocyte lineage transcription factor |
| Tuberous sclerosis | White macules, angiofibromas and Koenen tumors | TSC1, TSC2 | Negative regulators of PI3K-AKT-MTOR pathway |
| Hypomelanosis of Ito | Hypopigmentation along Blaschko lines/neural disorders | Chromosomal aberration | Somatic mosaicism probably affecting keratinocytes |
| Incontinentia pigmenti | White striae along Blaschko’s lines (stage 4) | IKBKG | Nuclear factor-κB essential modulator/inhibitor of κ light polypeptide gene enhancer in B cells, kinase γ |
| Human Gene | Mouse Gene (Locus) | Function | Relevant Clinical Condition |
|---|---|---|---|
| ASIP | Agouti (a) | Reverse agonist of MC1R | Hair/skin color polymorphism |
| MC1R | Mc1r (E) | G-protein coupled receptor Hormonal regulation | Hair/skin color polymorphism Susceptibility to UV-induced damage |
| ATRN | Atrn (mg) | Modifier of MC1R-agouti binding | Darker hair (mouse) Spongiform encephalopathy (mouse) |
| MGRN1 | Mgrn1 (md) | E3 ubiquitin ligase Modifier of MC1R signaling | Darker hair (mouse) Spongiform encephalopathy (mouse) Facial dysmorphology (mouse) Curled whisker (mouse) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hida, T.; Kamiya, T.; Kawakami, A.; Ogino, J.; Sohma, H.; Uhara, H.; Jimbow, K. Elucidation of Melanogenesis Cascade for Identifying Pathophysiology and Therapeutic Approach of Pigmentary Disorders and Melanoma. Int. J. Mol. Sci. 2020, 21, 6129. https://doi.org/10.3390/ijms21176129
Hida T, Kamiya T, Kawakami A, Ogino J, Sohma H, Uhara H, Jimbow K. Elucidation of Melanogenesis Cascade for Identifying Pathophysiology and Therapeutic Approach of Pigmentary Disorders and Melanoma. International Journal of Molecular Sciences. 2020; 21(17):6129. https://doi.org/10.3390/ijms21176129
Chicago/Turabian StyleHida, Tokimasa, Takafumi Kamiya, Akinori Kawakami, Jiro Ogino, Hitoshi Sohma, Hisashi Uhara, and Kowichi Jimbow. 2020. "Elucidation of Melanogenesis Cascade for Identifying Pathophysiology and Therapeutic Approach of Pigmentary Disorders and Melanoma" International Journal of Molecular Sciences 21, no. 17: 6129. https://doi.org/10.3390/ijms21176129
APA StyleHida, T., Kamiya, T., Kawakami, A., Ogino, J., Sohma, H., Uhara, H., & Jimbow, K. (2020). Elucidation of Melanogenesis Cascade for Identifying Pathophysiology and Therapeutic Approach of Pigmentary Disorders and Melanoma. International Journal of Molecular Sciences, 21(17), 6129. https://doi.org/10.3390/ijms21176129