Characterization of Two Novel Variants of the Steroidogenic Acute Regulatory Protein Identified in a Girl with Classic Lipoid Congenital Adrenal Hyperplasia

 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Results
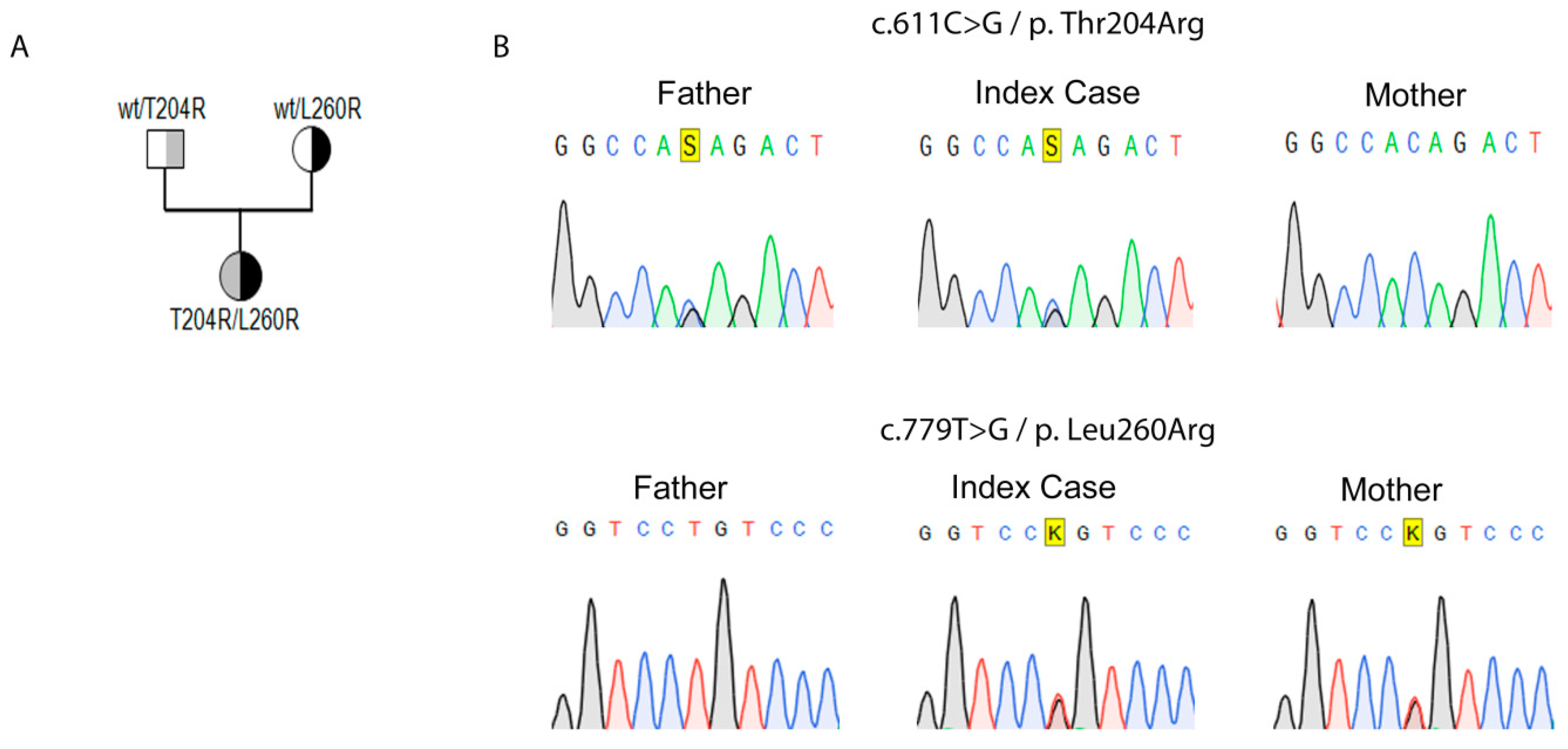
2.1. Genetic Identification of Novel Variants in StAR
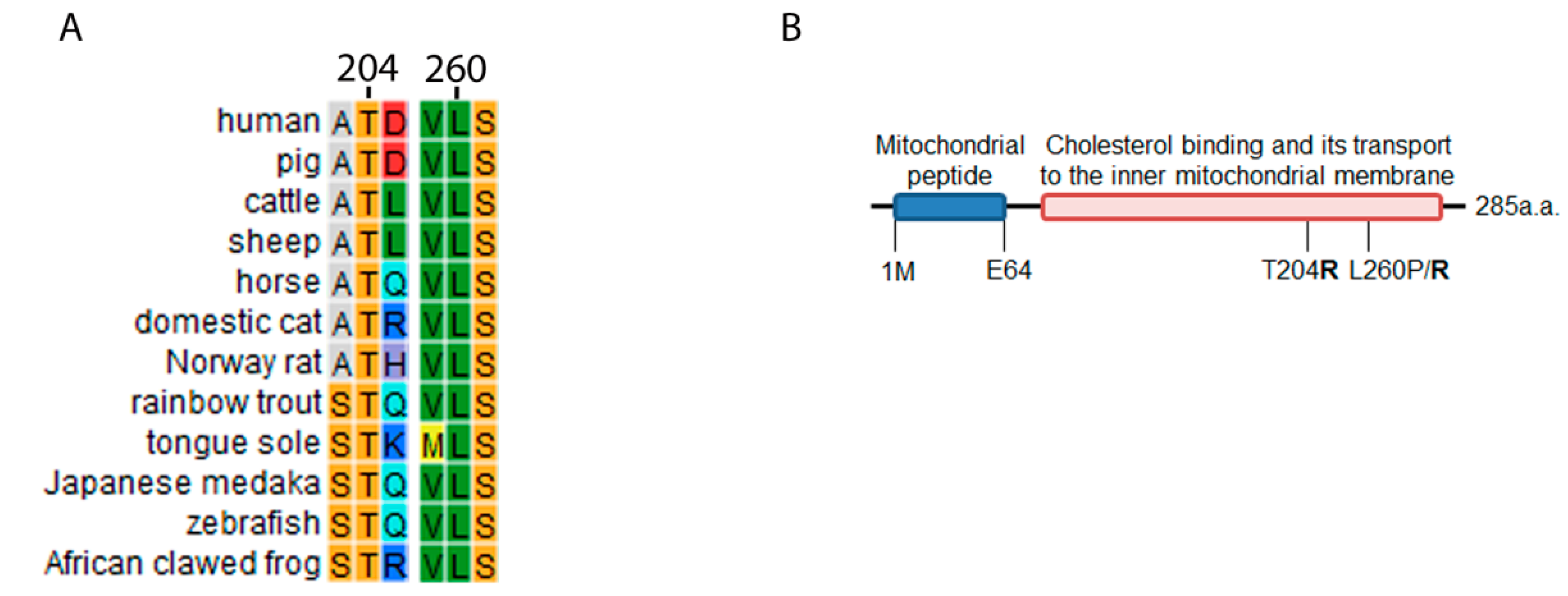
2.2. STAR Conservation Analysis and Mutagenesis Prediction for T204R and L260R
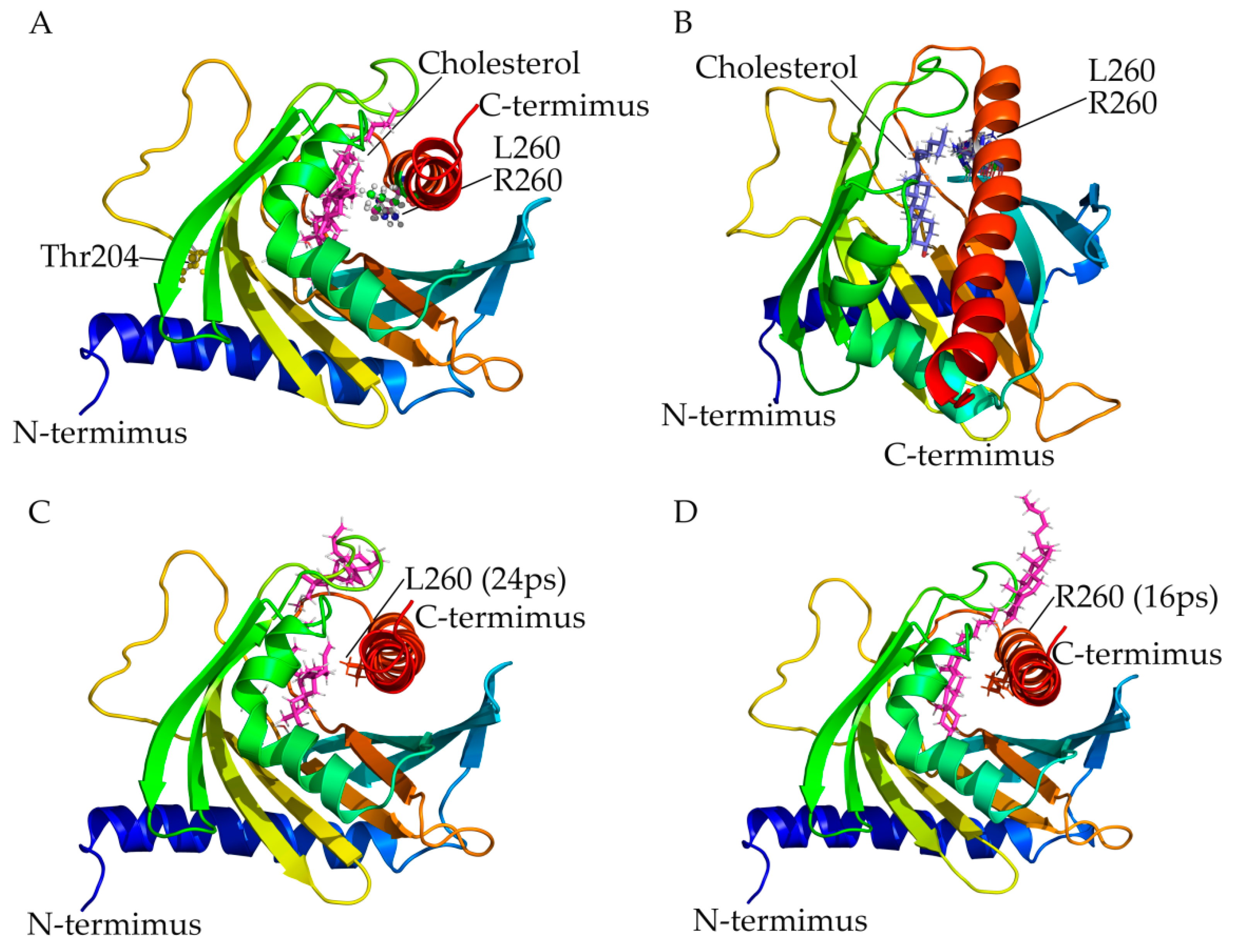
2.3. Use of StAR Structural Model, Computational Mutagenesis and Simulation of Interaction with Cholesterol for Variant Prediction
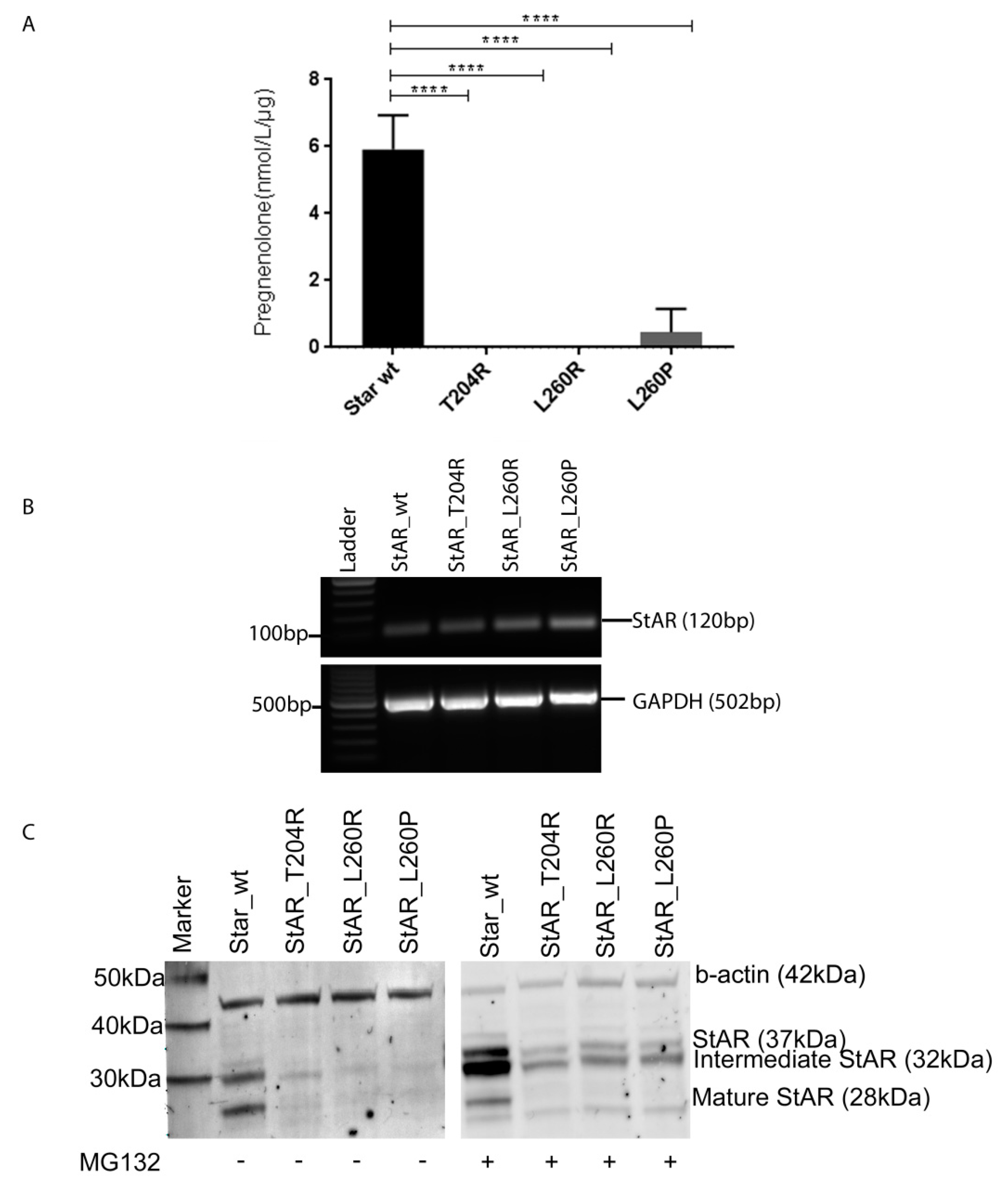
2.4. Functional Testing of Novel StAR Variants In Vitro
3. Discussion and Conclusions
4. Materials and Methods
4.1. Case Report
4.2. Genetic Analysis
4.3. Bioinformatic Studies for Structural and Functional Characterization of Novel Variants Identified in StAR
4.4. Functional Studies of Identified Novel StAR Variants In Vitro
4.5. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| StAR | Steroidogenic acute regulatory protein |
| CAH | Congenital adrenal hyperplasia |
| POR | P450 oxidoreductase |
| AI | Adrenal insufficiency |
| IMM | Inner mitochondrial membrane |
| OMM | Outer mitochondrial membrane |
| FGD | Familial glucocorticoid deficiency |
| ACTH | Adrenocorticotropic hormone |
| MC2R | Melanocortin receptor 2 |
| MC1R | Melanocortin receptor 1 |
| START | StAR-related lipid transfer |
| NGS | Next generation sequencing |
| SNARE | Soluble NSF attachment receptor protein |
| BMI | Body mass index |
| FSH | Follicle-stimulating hormone |
| LH | Luteinizing hormone |
| ACMG-AMP | American College of Medical Genetics and Genomics and the Association for Molecular Pathology |
References
- De Crecchio, L. Sopra un caso di apparenze virili in una donna. IL Morgagni 1865, 7, 151–189. [Google Scholar]
- Fluck, C.E. Mechanisms in endocrinology: Update on pathogenesis of primary adrenal insufficiency: Beyond steroid enzyme deficiency and autoimmune adrenal destruction. Eur. J. Endocrinol. 2017, 177, R99–R111. [Google Scholar] [CrossRef] [PubMed]
- Prader, A.; Gurtner, H.P. The syndrome of male pseudohermaphrodism in congenital adrenocortical hyperplasia without overproduction of androgens (adrenal male pseudohermaphrodism). Helv. Paediatr. Acta 1955, 10, 397–412. [Google Scholar] [PubMed]
- Lin, D.; Sugawara, T.; Strauss, J.F., 3rd; Clark, B.J.; Stocco, D.M.; Saenger, P.; Rogol, A.; Miller, W.L. Role of steroidogenic acute regulatory protein in adrenal and gonadal steroidogenesis. Science 1995, 267, 1828–1831. [Google Scholar] [CrossRef]
- Miller, W.L. Congenital lipoid adrenal hyperplasia: The human gene knockout for the steroidogenic acute regulatory protein. J. Mol. Endocrinol. 1997, 19, 227–240. [Google Scholar] [CrossRef] [Green Version]
- Tuckey, R.C.; Headlam, M.J.; Bose, H.S.; Miller, W.L. Transfer of cholesterol between phospholipid vesicles mediated by the steroidogenic acute regulatory protein (StAR). J. Biol. Chem. 2002, 277, 47123–47128. [Google Scholar] [CrossRef] [Green Version]
- Rone, M.B.; Midzak, A.S.; Issop, L.; Rammouz, G.; Jagannathan, S.; Fan, J.; Ye, X.; Blonder, J.; Veenstra, T.; Papadopoulos, V. Identification of a Dynamic Mitochondrial Protein Complex Driving Cholesterol Import, Trafficking, and Metabolism to Steroid Hormones. Mol. Endocrinol. 2012, 26, 1868–1882. [Google Scholar] [CrossRef] [Green Version]
- Miller, W.L. Disorders in the initial steps of steroid hormone synthesis. J. Steroid Biol. Mol. Endocrinol. 2017, 165, 18–37. [Google Scholar] [CrossRef]
- Bose, H.S. Mechanistic sequence of mitochondrial cholesterol transport by star proteins. J. Proteins Proteom. 2011, 2, 1–9. [Google Scholar]
- Soccio, R.E.; Breslow, J.L. StAR-related lipid transfer (START) proteins: Mediators of intracellular lipid metabolism. J. Biol. Chem. 2003, 278, 22183–22186. [Google Scholar] [CrossRef] [Green Version]
- Sugawara, T.; Holt, J.A.; Driscoll, D.; Strauss, J.F., 3rd; Lin, D.; Miller, W.L.; Patterson, D.; Clancy, K.P.; Hart, I.M.; Clark, B.J.; et al. Human steroidogenic acute regulatory protein: Functional activity in COS-1 cells, tissue-specific expression, and mapping of the structural gene to 8p11.2 and a pseudogene to chromosome 13. Proc. Natl. Acad. Sci. USA 1995, 92, 4778–4782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alpy, F.; Stoeckel, M.E.; Dierich, A.; Escola, J.M.; Wendling, C.; Chenard, M.P.; Vanier, M.T.; Gruenberg, J.; Tomasetto, C.; Rio, M.C. The steroidogenic acute regulatory protein homolog MLN64, a late endosomal cholesterol-binding protein. J. Biol. Chem. 2001, 276, 4261–4269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bose, H.S.; Whittal, R.M.; Huang, M.C.; Baldwin, M.A.; Miller, W.L. N-218 MLN64, a protein with StAR-like steroidogenic activity, is folded and cleaved similarly to StAR. Biochemistry 2000, 39, 11722–11731. [Google Scholar] [CrossRef] [PubMed]
- Watari, H.; Arakane, F.; Moog-Lutz, C.; Kallen, C.B.; Tomasetto, C.; Gerton, G.L.; Rio, M.C.; Baker, M.E.; Strauss, J.F., 3rd. MLN64 contains a domain with homology to the steroidogenic acute regulatory protein (StAR) that stimulates steroidogenesis. Proc. Natl. Acad. Sci. USA 1997, 94, 8462–8467. [Google Scholar] [CrossRef] [Green Version]
- Rigotti, A.; Cohen, D.E.; Zanlungo, S. STARTing to understand MLN64 function in cholesterol transport. J. Lipid Res. 2010, 51, 2015–2017. [Google Scholar] [CrossRef] [Green Version]
- Bose, H.S.; Sugawara, T.; Strauss, J.F., 3rd; Miller, W.L. The pathophysiology and genetics of congenital lipoid adrenal hyperplasia. N. Engl. J. Med. 1996, 335, 1870–1878. [Google Scholar] [CrossRef]
- Sandison, A.T. A form of lipoidosis of the adrenal cortex in an infant. Arch. Dis. Child. 1955, 30, 538–541. [Google Scholar] [CrossRef] [Green Version]
- Baker, B.Y.; Lin, L.; Kim, C.J.; Raza, J.; Smith, C.P.; Miller, W.L.; Achermann, J.C. Nonclassic congenital lipoid adrenal hyperplasia: A new disorder of the steroidogenic acute regulatory protein with very late presentation and normal male genitalia. J. Clin. Endocrinol. Metab. 2006, 91, 4781–4785. [Google Scholar] [CrossRef]
- Metherell, L.A.; Naville, D.; Halaby, G.; Begeot, M.; Huebner, A.; Nurnberg, G.; Nurnberg, P.; Green, J.; Tomlinson, J.W.; Krone, N.P.; et al. Nonclassic lipoid congenital adrenal hyperplasia masquerading as familial glucocorticoid deficiency. J. Clin. Endocrinol. Metab. 2009, 94, 3865–3871. [Google Scholar] [CrossRef] [Green Version]
- Hauffa, B.P.; Miller, W.L.; Grumbach, M.M.; Conte, F.A.; Kaplan, S.L. Congenital adrenal hyperplasia due to deficient cholesterol side-chain cleavage activity (20, 22-desmolase) in a patient treated for 18 years. Clin. Endocrinol. 1985, 23, 481–493. [Google Scholar] [CrossRef]
- Flück, C.E.; Pandey, A.V. Testicular steroidogenesis. In Endocrinology of the Testis and Male Reproduction; Simoni, M., Huhtaniemi, I.T., Eds.; Springer International Publishing: New York, NY, USA, 2017; pp. 343–371. [Google Scholar]
- Khoury, K.; Barbar, E.; Ainmelk, Y.; Ouellet, A.; Lavigne, P.; LeHoux, J.G. Thirty-Eight-Year Follow-Up of Two Sibling Lipoid Congenital Adrenal Hyperplasia Patients Due to Homozygous Steroidogenic Acute Regulatory (STARD1) Protein Mutation. Molecular Structure and Modeling of the STARD1 L275P Mutation. Front. Neurosci. 2016, 10, 527. [Google Scholar] [CrossRef] [Green Version]
- Miller, W.L.; Bose, H.S. Early steps in steroidogenesis: Intracellular cholesterol trafficking. J. Lipid Res. 2011, 52, 2111–2135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flück, C.E. Update on genetics of primary adrenal insufficiency. Curr. Opin. Endocr. Metab. Res. 2019, 8, 96–103. [Google Scholar] [CrossRef]
- Fluck, C.E.; Maret, A.; Mallet, D.; Portrat-Doyen, S.; Achermann, J.C.; Leheup, B.; Theintz, G.E.; Mullis, P.E.; Morel, Y. A novel mutation L260P of the steroidogenic acute regulatory protein gene in three unrelated patients of Swiss ancestry with congenital lipoid adrenal hyperplasia. J. Clin. Endocrinol. Metab. 2005, 90, 5304–5308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Nakae, J.; Tajima, T.; Sugawara, T.; Arakane, F.; Hanaki, K.; Hotsubo, T.; Igarashi, N.; Igarashi, Y.; Ishii, T.; Koda, N.; et al. Analysis of the steroidogenic acute regulatory protein (StAR) gene in Japanese patients with congenital lipoid adrenal hyperplasia. Hum. Mol. Genet. 1997, 6, 571–576. [Google Scholar] [CrossRef] [Green Version]
- Kaur, J.; Casas, L.; Bose, H.S. Lipoid congenital adrenal hyperplasia due to STAR mutations in a Caucasian patient. Endocrinol. Diabetes Metab. Case Rep. 2016, 2016, 150119. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Robbins, J. Proteasomal and lysosomal protein degradation and heart disease. J. Mol. Cell Cardiol. 2014, 71, 16–24. [Google Scholar] [CrossRef] [Green Version]
- Fluck, C.E.; Pandey, A.V.; Dick, B.; Camats, N.; Fernandez-Cancio, M.; Clemente, M.; Gussinye, M.; Carrascosa, A.; Mullis, P.E.; Audi, L. Characterization of novel StAR (steroidogenic acute regulatory protein) mutations causing non-classic lipoid adrenal hyperplasia. PLoS ONE 2011, 6, e20178. [Google Scholar] [CrossRef] [Green Version]
- Tanae, A.; Katsumata, N.; Sato, N.; Horikawa, R.; Tanaka, T. Genetic and endocrinological evaluations of three 46,XX patients with congenital lipoid adrenal hyperplasia previously reported as having presented spontaneous puberty. Endocr. J. 2000, 47, 629–634. [Google Scholar] [CrossRef] [Green Version]
- Sertedaki, A.; Pantos, K.; Vrettou, C.; Kokkali, G.; Christofidou, C.; Kanavakis, E.; Dacou-Voutetakis, C. Conception and pregnancy outcome in a patient with 11-bp deletion of the steroidogenic acute regulatory protein gene. Fertil. Steril. 2009, 91, 934.e915–934.e918. [Google Scholar] [CrossRef] [PubMed]
- Chatziaggelou, A.; Sakkas, E.G.; Votino, R.; Papagianni, M.; Mastorakos, G. Assisted Reproduction in Congenital Adrenal Hyperplasia. Front. Endocriol. 2019, 10, 723. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, P.H.; Gouveia, G.R.; Costa, E.M.; Domenice, S.; Martin, R.M.; de Carvalho, L.C.; Pelaes, T.; Inacio, M.; Codarin, R.R.; Sator de Faria, M.B.; et al. Successful Live Birth in a Woman With 17alpha-Hydroxylase Deficiency Through IVF Frozen-Thawed Embryo Transfer. J. Clin. Endocrinol. Metab. 2016, 101, 345–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iaea, D.B.; Spahr, Z.R.; Singh, R.K.; Chan, R.B.; Zhou, B.; Bareja, R.; Elemento, O.; Di Paolo, G.; Zhang, X.; Maxfield, F.R. Stable reduction of STARD4 alters cholesterol regulation and lipid homeostasis. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158609. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Agudo, D.; Malacrida, L.; Kakiyama, G.; Sparrer, T.; Fortes, C.; Maceyka, M.; Subler, M.A.; Windle, J.J.; Gratton, E.; Pandak, W.M.; et al. StarD5: An ER stress protein regulates plasma membrane and intracellular cholesterol homeostasis. J. Lipid Res. 2019, 60, 1087–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soccio, R.E.; Adams, R.M.; Romanowski, M.J.; Sehayek, E.; Burley, S.K.; Breslow, J.L. The cholesterol-regulated StarD4 gene encodes a StAR-related lipid transfer protein with two closely related homologues, StarD5 and StarD6. Proc. Natl. Acad. Sci. USA 2002, 99, 6943–6948. [Google Scholar] [CrossRef] [Green Version]
- Bertino, E.; Spada, E.; Occhi, L.; Coscia, A.; Giuliani, F.; Gagliardi, L.; Gilli, G.; Bona, G.; Fabris, C.; De Curtis, M.; et al. Neonatal anthropometric charts: The Italian neonatal study compared with other European studies. J. Pediatr. Gastroenterol. Nutr. 2010, 51, 353–361. [Google Scholar] [CrossRef] [Green Version]
- Tanner, J.M.; Whitehouse, R.H. Clinical longitudinal standards for height, weight, height velocity, weight velocity, and stages of puberty. Arch. Dis. Child. 1976, 51, 170–179. [Google Scholar] [CrossRef] [Green Version]
- WHO Multicentre Growth Reference Study Group. WHO Child Growth Standards: Length/Height-for-Age, Weight-for-Age, Weight-for-Length, Weight-for-Height and Body Mass Index-for-Age: Methods and Development; World Health Organization: Geneva, Switzerland, 2006. [Google Scholar]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Thorsell, A.G.; Lee, W.H.; Persson, C.; Siponen, M.I.; Nilsson, M.; Busam, R.D.; Kotenyova, T.; Schüler, H.; Lehtiö, L. Comparative structural analysis of lipid binding START domains. PLoS ONE 2011, 6, e19521. [Google Scholar] [CrossRef] [Green Version]
- Krieger, E.; Darden, T.; Nabuurs, S.B.; Finkelstein, A.; Vriend, G. Making optimal use of empirical energy functions: Force-field parameterization in crystal space. Proteins 2004, 57, 678–683. [Google Scholar] [CrossRef] [PubMed]
- The PyMOL Molecular Graphics System, Version 1.2r3pre. Schrödinger, LLC. Available online: www.pymol.org (accessed on 24 July 2020).
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Case, D.A.; Belfon, K.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E.; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Giambasu, G.; et al. Amber20; University of California: San Francisco, CA, USA, 2020. [Google Scholar]
- Ponder, J.W.; Case, D.A. Force fields for protein simulations. Adv. Protein Chem. 2003, 66, 27–85. [Google Scholar]
- Pandurangan, A.P.; Ochoa-Montano, B.; Ascher, D.B.; Blundell, T.L. SDM: A server for predicting effects of mutations on protein stability. Nucleic Acids Res. 2017, 45, W229–W235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salgado, D.; Desvignes, J.P.; Rai, G.; Blanchard, A.; Miltgen, M.; Pinard, A.; Lévy, N.; Collod-Béroud, G.; Béroud, C. UMD-Predictor: A High-Throughput Sequencing Compliant System for Pathogenicity Prediction of any Human cDNA Substitution. Hum. Mutat. 2016, 37, 439–446. [Google Scholar] [CrossRef] [Green Version]
- Sim, N.-L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef]
- Choi, Y.; Chan, A.P. Provean web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef] [Green Version]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harikrishna, J.A.; Black, S.M.; Szklarz, G.D.; Miller, W.L. Construction and function of fusion enzymes of the human cytochrome P450scc system. DNA Cell Biol. 1993, 12, 371–379. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pregnenolone Production | wt StAR | StAR-T204R | StAR-L260R | StAR-L260P |
|---|---|---|---|---|
| Pregnenolone (nmol/L/µg) | 5.92 | 0 | 0 | 0.46 |
| % of wt StAR | 100 | 0 | 0 | 8 |
| Biochemical Test | Age | Reference Values | |
|---|---|---|---|
| 1.5 m * | 6 m | ||
| Na (mEq/mL) | 133 | 137 | 129–143 |
| K (mEq/mL) | 7.4 | 5.2 | 3.7–5.8 |
| ACTHA (pg/mL) | 15,001 | 119.1 | 6–48 |
| Renin (µU/mL) | 21.15 | 160.1 | 5.3–99.1 |
| Aldosterone (pg/mL) | 34 | - | 5–90 |
| DHEAS (µg/mL) | nd | - | <1.12 */<0.49 |
| 17OH-progesterone (ng/mL) | 0.03 | <0.03 | 0.13–1.06 |
| Androstenedione (ng/mL) | 0.02 | - | 0.1–0.37 |
| Testosterone (ng/mL) | 0.04 | <0.03 | 0.14–3.63 */0.03–0.12 |
| Cortisol (ng/mL) | - | 1.1 | 1–12 |
| Cortisone (ng/mL) | - | 1 | 6.3–56.7 |
| Progesterone (ng/mL) | - | <0.03 | 0.1–0.26 |
| 11-deoxycortisone (ng/mL) | - | <0.03 | 0.07–0.4 |
| Corticosterone (ng/mL) | - | <0.03 | 0.8–15 |
| 11-deoxycortisol (ng/mL) | - | <0.03 | <10 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Katharopoulos, E.; Di Iorgi, N.; Fernandez-Alvarez, P.; Pandey, A.V.; Groessl, M.; Dubey, S.; Camats, N.; Napoli, F.; Patti, G.; Lezzi, M.; et al. Characterization of Two Novel Variants of the Steroidogenic Acute Regulatory Protein Identified in a Girl with Classic Lipoid Congenital Adrenal Hyperplasia. Int. J. Mol. Sci. 2020, 21, 6185. https://doi.org/10.3390/ijms21176185
Katharopoulos E, Di Iorgi N, Fernandez-Alvarez P, Pandey AV, Groessl M, Dubey S, Camats N, Napoli F, Patti G, Lezzi M, et al. Characterization of Two Novel Variants of the Steroidogenic Acute Regulatory Protein Identified in a Girl with Classic Lipoid Congenital Adrenal Hyperplasia. International Journal of Molecular Sciences. 2020; 21(17):6185. https://doi.org/10.3390/ijms21176185
Chicago/Turabian StyleKatharopoulos, Efstathios, Natascia Di Iorgi, Paula Fernandez-Alvarez, Amit V. Pandey, Michael Groessl, Shraddha Dubey, Núria Camats, Flavia Napoli, Giuseppa Patti, Marilea Lezzi, and et al. 2020. "Characterization of Two Novel Variants of the Steroidogenic Acute Regulatory Protein Identified in a Girl with Classic Lipoid Congenital Adrenal Hyperplasia" International Journal of Molecular Sciences 21, no. 17: 6185. https://doi.org/10.3390/ijms21176185
APA StyleKatharopoulos, E., Di Iorgi, N., Fernandez-Alvarez, P., Pandey, A. V., Groessl, M., Dubey, S., Camats, N., Napoli, F., Patti, G., Lezzi, M., Maghnie, M., & Flück, C. E. (2020). Characterization of Two Novel Variants of the Steroidogenic Acute Regulatory Protein Identified in a Girl with Classic Lipoid Congenital Adrenal Hyperplasia. International Journal of Molecular Sciences, 21(17), 6185. https://doi.org/10.3390/ijms21176185