The Role of the JC Virus in Central Nervous System Tumorigenesis




Abstract
:1. Introduction
2. History and Epidemiology of JC Virus
3. JC Virus Characteristics
4. JCV Early Gene Products and Agno Protein and Their Oncogenic Potential
5. JCV Oncogenicity in Animal Models

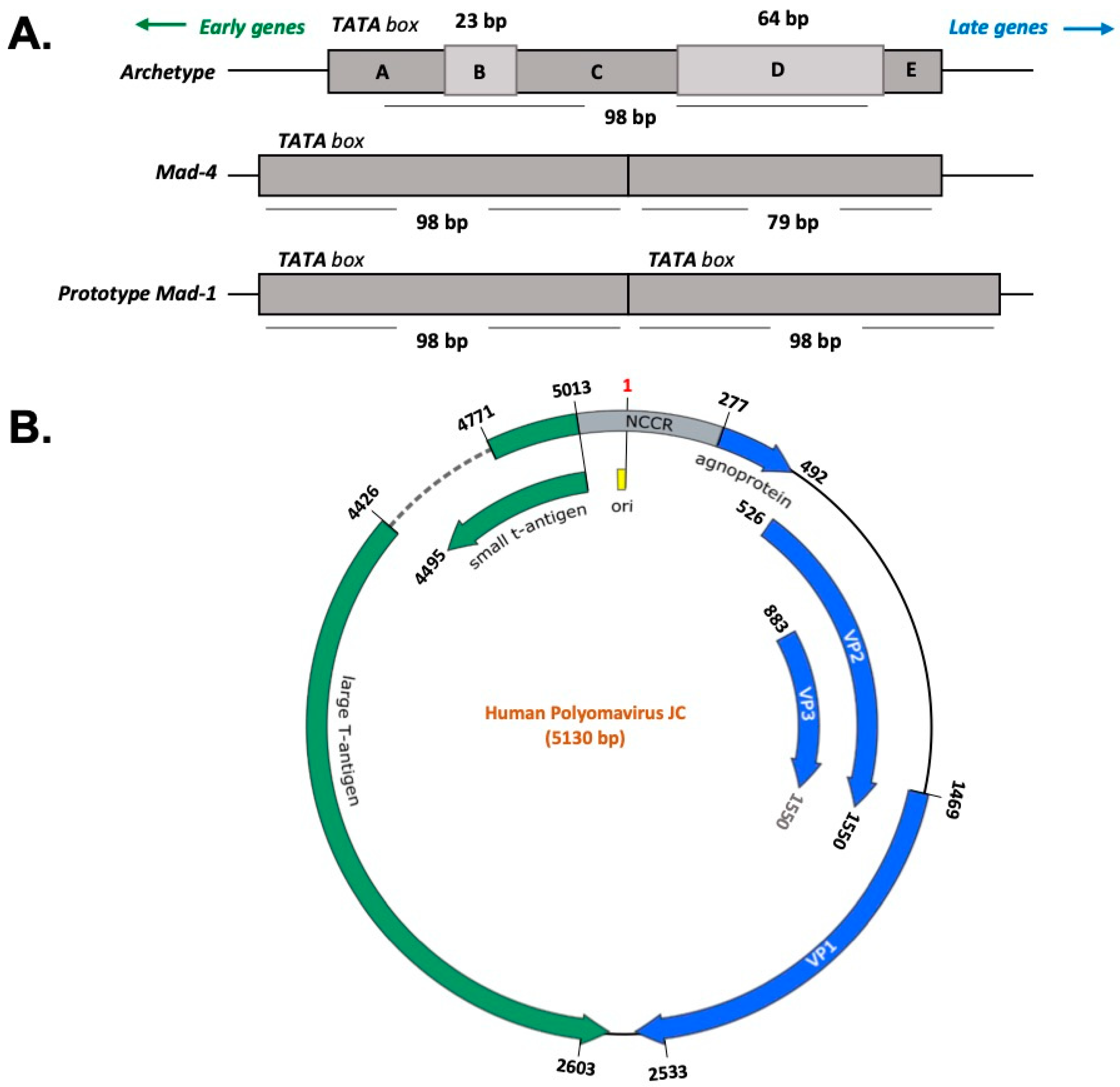{kind=link}
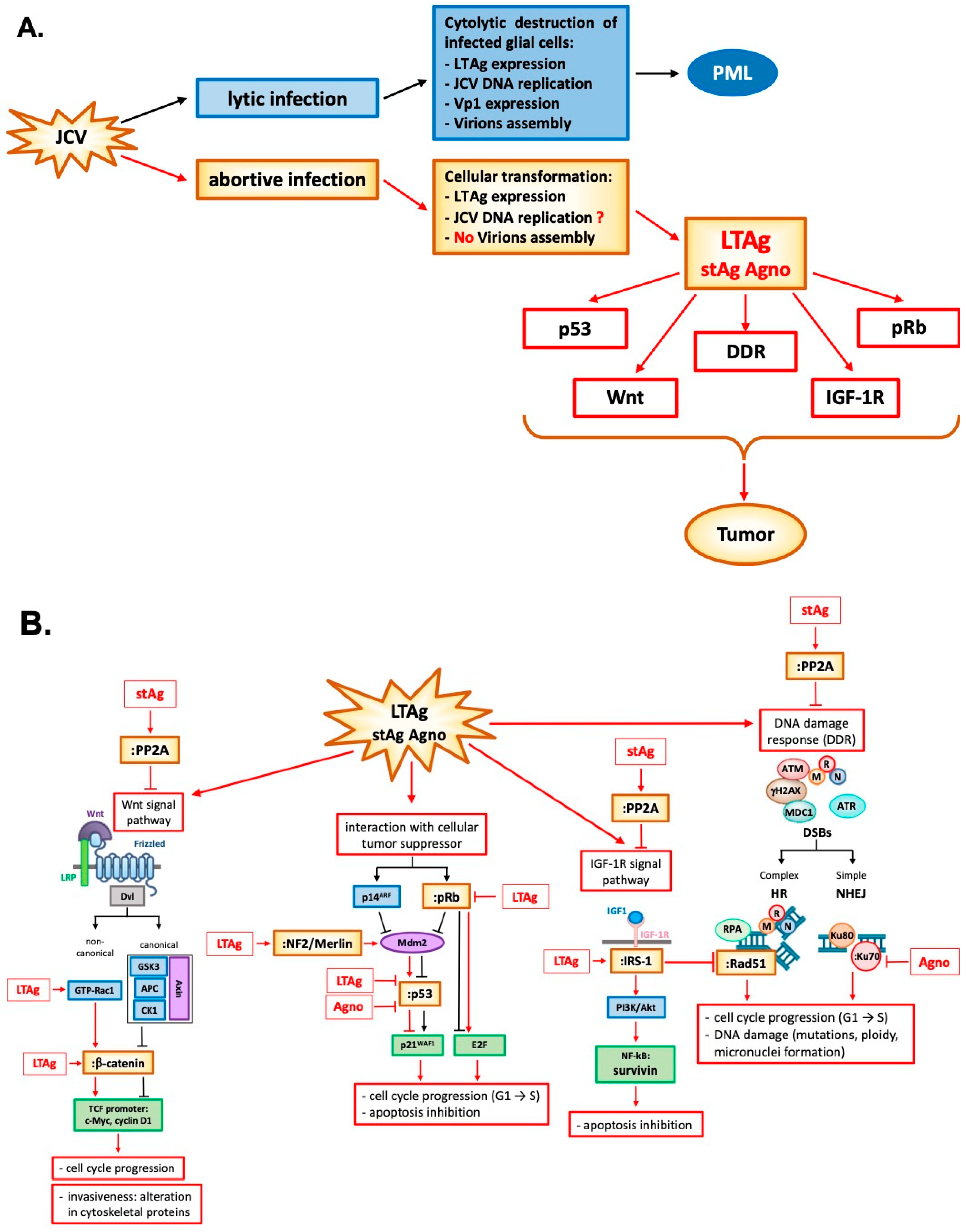{kind=link}
| Signaling Pathway | Cellular Factor | JCV Factor | Oncogenic Effect | References |
|---|---|---|---|---|
| Tumor suppressors | p53, p21WAF1 | Agno | cell cycle arrest in G2/M in vitro | Darbinyan A et al. 2002 [80] |
| p53, p21WAF1 | LTAg | pituitary neoplasia in LTAG transgenic mice | Gordon J et al. 2000 [152] | |
| pRb | LTAg | cell cycle progression in vitro | Dyson N et al. 1990 [95] | |
| pRb2/p130, E2F4/5 | LTAg | cell cycle progression in vitro | Caracciolo V et al. 2007 [97] | |
| p53, pRb | LTAg | cell cycle dysregulation in tumor formation in LTAg transgenic mice | Krynska B et al. 1997 [99] | |
| NF2 | LTAg | transgenic mouse model of malignant peripheral nerve sheath tumors | Shollar D et al. 2004 [103] | |
| pRb, PP2A | stAg | cell cycle dysregulation and viral DNA replication | Bollag B et al. 2010 [127]; Sariyer IK et al. 2008 [128]; Pallas DC et al. 1990 [129] | |
| Wnt | -catenin, c-Myc, Cyclin D1 | LTAg | oncogenesis of colon cancer | Enam S et al. 2002 [3]; Ripple MJ et al. 2014 [108] |
| -catenin | LTAg | mouse medulloblastoma cell line (BSB8), JCV-induced hamster astrocytoma cell line (HJC2) and human astrocytoma U-87MG cell line | Gan DD and Khalili K 2004 [105] | |
| -catenin, LEF-1/TCF promoter | LTAg | murine medulloblastoma cell line (BsB8) | Gan DD et al. 2001 [111] | |
| Rac1 GTPase | LTAg | -catenin stabilization and cell cycle progression in vitro | Bhattacheryya R et al. 2007 [112] | |
| PP2A | stAg | Inhibition of Wnt signaling, alteration in cytoskeleton proteins and increase of invasiveness | Nunbhakdi-Craig V et al. 2003 [131] | |
| IGF-1R | IRS-1 | LTAg | translocation to the nucleus and cell cycle progression | Lassal A et al. 2002 [106] |
| survivin | LTAg | apoptosis inhibition | Piña-Oviedo S et al. 2007 [107] | |
| survivin | LTAg | apoptosis inhibition and proliferation of neural progenitors | Gualco E et al. 2010 [114] | |
| IGF-1R and DDR | IRS-1, Rad51 | LTAg | HR dysregulation and DNA damage | Trojanek J et al. 2006 [113] |
| DDR | NHEJ Ku70 | Agno | HR dysregulation and DNA damage | Darbinyan A et al. 2004 [136] |
| HR Rad51, NHEJ Ku70, H2AX | LTAg, Agno | HR dysregulation and DNA damage (mutation, ploidy, and micronuclei formation) | Darbinyan A et al. 2007 [115] | |
| HR Rad51, ATM | LTAg | DNA damage | White MK et al. 2014 [73]; White MK et al. 2017 [118] | |
| PP2A | stAg | DNA damage | Huang JL et al. 2015 [130] |
| Animal Model | JCV Delivery | Tumors | Assay | References |
|---|---|---|---|---|
| Golden Syrian Hamsters (Mesocricetus auratus) | newborns inoculated intracerebrally and subcutaneously with JCV isolated from a patient with PML | malignant gliomas: most of the tumors were glioblastomas and medulloblastomas | transplantation of tumors subcutaneously and isolation of JCV from 5/7 tumors tested. Cells from four of these tumors were cultivated in vitro: intranuclear LTAg antigenically related to SV40 LTAg; JCV virions after fusion of this culture with permissive cells | Walker DL et al. 1973 [139] |
| three groups of newborns inoculated intracerebrally with three different JCV strains (Mad-2, Mad-3, and Mad-4) | cerebellar medulloblastomas with Mad-2 inoculation; pineal gland tumors and tumors in the cerebellum with Mad-4 inoculation. | histologic characterization of tumors. | Padgett BL et al. 1977b [137] | |
| one group of newborns inoculated intraocularly. Another group was inoculated subcutaneously and intraperitoneally. Both with JCV Mad-1 strain | neuroblastomas and primary tumors in the abdominal cavity with metastasis in liver, bone marrow, and lymph nodes. | two neuroblastomas were transplanted serially, and a tissue culture cell line was established from one of them. T-antigen was detected in 3/5 primary tumors tested and in the transplanted tumors. | Varakis J et al. 1978 [140] | |
| newborns inoculated intracerebrally and subcutaneously with JCV isolated from a patient with PML | medulloblastoma involved the internal granular layer of the cerebellum: lesion comparable to childhood human medulloblastoma | LTAg IF and histology | ZuRhein GM et al. 1979 [138] | |
| newborns inoculated intracerebrally with Tokio-1 JCV strain (isolated form a patient with PML, serologically identical to Mad-1 strain). | cerebellar medulloblastoma | LTAg IF and histology (Homer-Wright rosettes) | Nagashima K et al. 1984 [148] | |
| Owl Monkeys (Aotus trivirgatus) | two animals inoculated intracerebrally, subcutaneously, and intravenously with JCV isolated from a patient with PML | astrocytoma (resembling human glioblastoma multiforme) and a malignant tumor containing both glial and neuronal cells | TAg IF and histology | London WT et al. 1978 [142] |
| Squirrel Monkeys (Saimiri sciureus) | six animals inoculated intracerebrally, subcutaneously, and intravenously with JCV isolated from a patient with PML | astrocytomas in 4/6 animals. | histologic characterization of tumors | London WT et al. 1983 [146] |
| Sprague-DawleyRats | newborns inoculated intracranially with Tokyo-1 JCV strain. | brain tumors in the cerebrum: undifferentiated neuroectodermal nature and pseudo-rosettes. | LTAg IHC and histology. Neuronal differentiation was not proved. Glial differentiation was confirmed by subcutaneous transplantation of cultured tumor cells | Oshumi et al. 1985 [149]; Oshumi et al. 1986 [150]. |
| Transgenic Mice | transgenic mice for the early region of JCV Archetype strain | primitive tumors originating from the cerebellum: close resemblance of human medulloblastoma/primitive neuroectodermal tumors (PNETs) | RT-PCR for LTAg mRNA, IHC for LTAg and p53, IP for LTAg and p53 and Archetype NCCR sequencing | Krynska B et al. 1999b [151] |
| transgenic mice for the early region of JCV Mad-4 strain | pituitary neoplasia | IHC for LTAg and p53, IP for LTAg, p53 and p21WAF1 | Gordon J et al. 2000 [152] | |
| transgenic mice for the early region of JCV Mad-4 strain | pituitary neoplasia and signs resembling malignant peripheral nerve sheath tumors. | IHC for LTAg, NF-1, NF2,p53, and p21WAF1 and IP for LTAg, NF-1, NF2 and p53, | Shollar D et al. 2004 [103] |
6. Evidence of JCV Infectivity in Human Tumor Tissues
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Moore, P.; Chang, Y. Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nat. Rev. Cancer 2010, 10, 878–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parkin, D.M. The global health burden of infection-associated cancers in the year 2002. Int. J. Cancer 2006, 118, 3030–3044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enam, S.; Del Valle, L.; Lara, C.; Gan, D.-D.; Ortiz-Hidalgo, C.; Palazzo, J.P.; Khalili, K. Association of human polyomavirus JCV with colon cancer: Evidence for interaction of viral T-antigen and beta-catenin. Cancer Res. 2002, 62, 7093–7101. [Google Scholar]
- Kassem, A.; Technau, K.; Kurz, A.K.; Pantulu, D.; Löning, M.; Kayser, G.; Stickeler, E.; Weyers, W.; Díaz, C.; Werner, M.; et al. Merkel cell polyomavirus sequences are frequently detected in nonmelanoma skin cancer of immunosuppressed patients. Int. J. Cancer 2009, 125, 356–361. [Google Scholar] [CrossRef]
- Reiss, K.; Khalili, K. Viruses and cancer: Lessons from the human polyomavirus, JCV. Oncogene 2003, 22, 6517–6523. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.; Langhoff, E. Polyomavirus in Human Cancer Development. Adv. Exp. Med. Biol. 2007, 577, 310–318. [Google Scholar] [CrossRef]
- Pinto, M.; Dobson, S. BK and JC virus: A review. J. Infect. 2014, 68 (Suppl. 1), S2–S8. [Google Scholar] [CrossRef]
- Knowles, W.A. Discovery and epidemiology of the human polyomaviruses BK virus (BKV) and JC virus (JCV). Adv. Exp. Med. Biol. 2007, 577, 19–45. [Google Scholar] [CrossRef]
- Tan, C.S.; Koralnik, I.J. Progressive multifocal leukoencephalopathy and other disorders caused by JC virus: Clinical features and pathogenesis. Lancet Neurol. 2010, 9, 425–437. [Google Scholar] [CrossRef] [Green Version]
- Arthur, R.R.; Dagostin, S.; Shah, K.V. Detection of BK virus and JC virus in urine and brain tissue by the polymerase chain reaction. J. Clin. Microbiol. 1989, 27, 1174–1179. [Google Scholar] [CrossRef] [Green Version]
- Miranda-Filho, A.; Piñeros, M.; Soerjomataram, I.; Deltour, I.; Bray, F. Cancers of the brain and CNS: Global patterns and trends in incidence. Neuro-Oncology 2016, 19, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Boldorini, R.; Pagani, E.; Car, P.G.; Omodeo-Zorini, E.; Borghi, E.; Tarantini, L.; Bellotti, C.; Ferrante, P.; Monga, G. Molecular characterisation of JC virus strains detected in human brain tumours. Pathology 2003, 35, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Delbue, S.; Pagani, E.; Guerini, F.R.; Agliardi, C.; Mancuso, R.; Borghi, E.; Rossi, F.; Boldorini, R.; Veggiani, C.; Car, P.G.; et al. Distribution, characterization and significance of polyomavirus genomic sequences in tumors of the brain and its covering. J. Med. Virol. 2005, 77, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Del Valle, L.; Azizi, S.A.; Krynska, B.; Enam, S.; Croul, S.E.; Khalili, K. Reactivation of human neurotropic JC virus expressing oncogenic protein in a recurrent glioblastoma multiforme. Ann. Neurol. 2000, 48, 932–936. [Google Scholar] [CrossRef]
- Del Valle, L.; Gordon, J.; Assimakopoulou, M.; Enam, S.; Geddes, J.F.; Varakis, J.N.; Katsetos, C.D.; Croul, S.; Khalili, K. Detection of JC virus DNA sequences and expression of the viral regulatory protein T-antigen in tumors of the central nervous system. Cancer Res. 2001, 61, 4287–4293. [Google Scholar] [PubMed]
- Piña-Oviedo, S.; De León-Bojorge, B.; Cuesta-Mejías, T.; White, M.K.; Ortiz-Hidalgo, C.; Khalili, K.; Del Valle, L. Glioblastoma multiforme with small cell neuronal-like component: Association with human neurotropic JC virus. Acta Neuropathol. 2006, 111, 388–396. [Google Scholar] [CrossRef]
- Caldarelli-Stefano, R.; Boldorini, R.; Monga, G.; Meraviglia, E.; Zorini, E.O.; Ferrante, P. JC virus in human glial-derived tumors. Hum. Pathol. 2000, 31, 394–395. [Google Scholar] [CrossRef]
- Del Valle, L.; Gordon, J.; Ferrante, P.; Khalili, K. JC virus in experimental and clinical brain tumorigenesis. In Human Polyomaviruses; Khalili, K., Stoner, G., Eds.; Wiley & Sons, Inc.: New York, NY, USA, 2003; pp. 409–430. [Google Scholar]
- Shiramizu, B.; Hu, N.; Frisque, R.J.; Nerurkar, V.R. High prevalence of human polyomavirus JC VP1 gene sequences in pediatric malignancies. Cell. Mol. Biol. 2007, 53, 4–12. [Google Scholar]
- Krynska, B.; Del Valle, L.; Croul, S.; Gordon, J.; Katsetos, C.D.; Carbone, M.; Giordano, A.; Khalili, K. Detection of human neurotropic JC virus DNA sequence and expression of the viral oncogenic protein in pediatric medulloblastomas. Proc. Natl. Acad. Sci. USA 1999, 96, 11519–11524. [Google Scholar] [CrossRef] [Green Version]
- Del Valle, L.; Baehring, J.; Lorenzana, C.; Giordano, A.; Khalili, K.; Croul, S. Expression of a human polyomavirus oncoprotein and tumour suppressor proteins in medulloblastomas. Mol. Pathol. 2001, 54, 331–337. [Google Scholar] [CrossRef] [Green Version]
- Del Valle, L.; Gordon, J.; Enam, S.; Delbue, S.; Croul, S.; Abraham, S.; Radhakrishnan, S.; Assimakopoulou, M.; Katsetos, C.D.; Khalili, K. Expression of human neurotropic polyomavirus JCV late gene product agnoprotein in human medulloblastoma. J. Natl. Cancer Inst. 2002, 94, 267–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Valle, L.; Delbue, S.; Gordon, J.; Enam, S.; Croul, S.; Ferrante, P.; Khalili, K. Expression of JC virus T-antigen in a patient with MS and glioblastoma multiforme. Neurology 2002, 58, 895–900. [Google Scholar] [CrossRef] [PubMed]
- Rencic, A.; Gordon, J.; Otte, J.; Curtis, M.; Kovatich, A.; Zoltick, P.; Khalili, K.; Andrews, D. Detection of JC virus DNA sequence and expression of the viral oncoprotein, tumor antigen, in brain of immunocompetent patient with oligoastrocytoma. Proc. Natl. Acad. Sci. USA 1996, 93, 7352–7357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Valle, L.; Enam, S.; Lara, C.; Ortiz-Hidalgo, C.; Katsetos, C.D.; Khalili, K. Detection of JC polyomavirus DNA sequences and cellular localization of T-antigen and agnoprotein in oligodendrogliomas. Clin. Cancer Res. 2002, 8, 3332–3340. [Google Scholar] [PubMed]
- Padgett, B.; Walker, D.; ZuRhein, G.; Eckroade, R.; Dessel, B. Cultivation of papova-like virus from human brain with progressive multifocal leucoencephalopathy. Lancet 1971, 297, 1257–1260. [Google Scholar] [CrossRef]
- Tavazzi, E.; White, M.K.; Khalili, K. Progressive multifocal leukoencephalopathy: Clinical and molecular aspects. Rev. Med. Virol. 2011, 22, 18–32. [Google Scholar] [CrossRef] [Green Version]
- Berger, J.R.; Scott, G.; Albrecht, J.; Belman, A.L.; Tornatore, C.; Major, E.O. Progressive multifocal leukoencephalopathy in HIV-1-infected children. AIDS 1992, 6, 837–842. [Google Scholar] [CrossRef]
- Wollebo, H.S.; White, M.K.; Gordon, J.; Berger, J.R.; Khalili, K. Persistence and pathogenesis of the neurotropic polyomavirus JC. Ann. Neurol. 2015, 77, 560–570. [Google Scholar] [CrossRef]
- Morriss, M.C.; Rutstein, R.M.; Rudy, B.; DesRochers, C.; Hunter, J.V.; Zimmerman, R.A. Progressive multifocal leukoencephalopathy in an HIV-infected child. Neuroradiology 1997, 39, 142–144. [Google Scholar] [CrossRef]
- Shitrit, D.; Lev, N.; Shitrit, A.B.-G.; Kramer, M. Progressive multifocal leukoencephalopathy in transplant recipients. Transpl. Int. 2004, 17, 658–665. [Google Scholar] [CrossRef]
- Hecht, J.H.; Glenn, O.A.; Wara, D.W.; Wu, Y.W. JC Virus granule cell neuronopathy in a child with cd40 ligand deficiency. Pediatr. Neurol. 2007, 36, 186–189. [Google Scholar] [CrossRef]
- Redfearn, A.; Pennie, R.A.; Mahony, J.B.; Dent, P.B. Progressive multifocial leukoencephalopathy in a child with immunodeficiency and hyperimmunoglobulinemia M. Pediatr. Infect. Dis. J. 1993, 12, 399–401. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.T.; Frisque, R.J. Identification of JC virus variants in multiple tissues of pediatric and adult PML patients. J. Med. Virol. 1999, 58, 79–86. [Google Scholar] [CrossRef]
- Baldwin, K.J.; Hogg, J.P. Progressive multifocal leukoencephalopathy in patients with multiple sclerosis. Curr. Opin. Neurol. 2013, 26, 318–323. [Google Scholar] [CrossRef]
- Van Assche, G.; Van Ranst, M.; Sciot, R.; Dubois, B.; Vermeire, S.; Noman, M.; Verbeeck, J.; Geboes, K.; Robberecht, W.; Rutgeerts, P. Progressive multifocal leukoencephalopathy after natalizumab therapy for Crohn’s disease. N. Engl. J. Med. 2005, 353, 362–368. [Google Scholar] [CrossRef] [Green Version]
- Elia, F.; Villani, S.; Ambrogi, F.; Signorini, L.; Dallari, S.; Binda, S.; Primache, V.; Pellegrinelli, L.; Ferrante, P.; Delbue, S. JC virus infection is acquired very early in life: Evidence from a longitudinal serological study. J. NeuroVirol. 2016, 23, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, F.; Kajioka, J.; Miyamura, T. Prevalence rate and age of acquisition of antibodies against JC virus and BK virus in human sera. Microbiol. Immunol. 1982, 26, 1057–1064. [Google Scholar] [CrossRef]
- Padgett, B.L.; Rogers, C.M.; Walker, D.L. JC virus, a human polyomavirus associated with progressive multifocal leukoencephalopathy: Additional biological characteristics and antigenic relationships. Infect. Immun. 1977, 15, 656–662. [Google Scholar] [CrossRef] [Green Version]
- Egli, A.; Infanti, L.; Dumoulin, A.; Buser, A.; Samaridis, J.; Stebler, C.; Gosert, R.; Hirsch, H.H. Prevalence of polyomavirus BK and JC infection and replication in 400 healthy blood donors. J. Infect. Dis. 2009, 199, 837–846. [Google Scholar] [CrossRef] [Green Version]
- Knowles, W.A.; Pipkin, P.; Andrews, N.; Vyse, A.; Minor, P.; Brown, D.W.; Miller, E. Population-based study of antibody to the human polyomaviruses BKV and JCV and the simian polyomavirus SV40. J. Med. Virol. 2003, 71, 115–123. [Google Scholar] [CrossRef]
- Stolt, A.; Sasnauskas, K.; Koskela, P.; Lehtinen, M.; Dillner, J. Seroepidemiology of the human polyomaviruses. J. Gen. Virol. 2003, 84, 1499–1504. [Google Scholar] [CrossRef] [PubMed]
- Kean, J.M.; Rao, S.; Wang, M.; Garcea, R.L. Seroepidemiology of human polyomaviruses. PLoS Pathog. 2009, 5, e1000363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Major, E.O.; Amemiya, K.; Tornatore, C.S.; Houff, S.A.; Berger, J.R. Pathogenesis and molecular biology of progressive multifocal leukoencephalopathy, the JC virus-induced demyelinating disease of the human brain. Clin. Microbiol. Rev. 1992, 5, 49–73. [Google Scholar] [CrossRef] [PubMed]
- Bofill-Mas, S.; Clemente-Casares, P.; Major, E.O.; Curfman, B.; Gironés, R. Analysis of the excreted JC virus strains and their potential oral transmission. J. NeuroVirol. 2003, 9, 498–507. [Google Scholar] [CrossRef]
- Ault, G.S.; Stoner, G.L. Two major types of JC virus defined in progressive multifocal leukoencephalopathy brain by early and late coding region DNA sequences. J. Gen. Virol. 1992, 73, 2669–2678. [Google Scholar] [CrossRef]
- Kitamura, T.; Kunitake, T.; Guo, J.; Tominaga, T.; Kawabe, K.; Yogo, Y. Transmission of the human polyomavirus JC virus occurs both within the family and outside the family. J. Clin. Microbiol. 1994, 32, 2359–2363. [Google Scholar] [CrossRef] [Green Version]
- Berger, J.R.; Miller, C.S.; Mootoor, Y.; Avdiushko, S.A.; Kryscio, R.J.; Zhu, H. JC virus detection in bodily fluids: Clues to transmission. Clin. Infect. Dis. 2006, 43, e9–e12. [Google Scholar] [CrossRef]
- Monaco, M.C.; Atwood, W.J.; Gravell, M.; Tornatore, C.S.; Major, E.O. JC virus infection of hematopoietic progenitor cells, primary B lymphocytes, and tonsillar stromal cells: Implications for viral latency. J. Virol. 1996, 70, 7004–7012. [Google Scholar] [CrossRef] [Green Version]
- Monaco, M.C.G.; Jensen, P.N.; Hou, J.; Durham, L.C.; Major, E.O. Detection of JC virus DNA in human tonsil tissue: Evidence for site of initial viral infection. J. Virol. 1998, 72, 9918–9923. [Google Scholar] [CrossRef] [Green Version]
- Ricciardiello, L.; Laghi, L.; Ramamirtham, P.; Chang, C.L.; Chang, D.K.; Randolph, A.E.; Boland, C.R. JC virus DNA sequences are frequently present in the human upper and lower gastrointestinal tract. Gastroenterology 2000, 119, 1228–1235. [Google Scholar] [CrossRef]
- Kato, A.; Kitamura, T.; Takasaka, T.; Tominaga, T.; Ishikawa, A.; Zheng, H.-Y.; Yogo, Y. Detection of the archetypal regulatory region of JC virus from the tonsil tissue of patients with tonsillitis and tonsilar hypertrophy. J. Neurovirol. 2004, 10, 244–249. [Google Scholar] [CrossRef] [PubMed]
- Dörries, K.; Vogel, E.; Günther, S.; Czub, S. Infection of human polyomaviruses JC and BK in peripheral blood leukocytes from immunocompetent individuals. Virology 1994, 198, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Gallia, G.L.; Houff, S.A.; Major, E.O.; Khalili, K. JC virus infection of lymphocytes—Revisited. J. Infect. Dis. 1997, 176, 1603–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciappi, S.; Azzi, A.; De Santis, R.; Leoncini, F.; Sterrantino, G.; Mazzotta, F.; Mecocci, L. Archetypal and rearranged sequences of human polyomavirus JC transcription control region in peripheral blood leukocytes and in cerebrospinal fluid. J. Gen. Virol. 1999, 80, 1017–1023. [Google Scholar] [CrossRef]
- Chalkias, S.; Dang, X.; Bord, E.; Stein, M.C.; Kinkel, R.P.; Sloane, J.A.; Donnelly, M.; Ionete, C.; Houtchens, M.K.; Buckle, G.J.; et al. JC virus reactivation during prolonged natalizumab monotherapy for multiple sclerosis. Ann. Neurol. 2014, 75, 925–934. [Google Scholar] [CrossRef]
- Lafon, M.; Dutronc, H.; Dubois, V.; Pellegrin, I.; Barbeau, P.; Ragnaud, J.; Pellegrin, J.; Fleury, H.J.A.; Véronique, D. JC virus remains latent in peripheral blood B lymphocytes but replicates actively in urine from AIDS patients. J. Infect. Dis. 1998, 177, 1502–1505. [Google Scholar] [CrossRef]
- Chesters, P.M.; Heritage, J.; McCance, D.J. Persistence of DNA sequences of BK virus and JC virus in normal human tissues and in diseased tissues. J. Infect. Dis. 1983, 147, 676–684. [Google Scholar] [CrossRef]
- Monaco, M.C.G.; Major, E.O. Immune system involvement in the pathogenesis of JC virus induced PML: What is learned from studies of patients with underlying diseases and therapies as risk factors. Front. Immunol. 2015, 6, 159. [Google Scholar] [CrossRef] [Green Version]
- Wei, G.; Liu, C.K.; Atwood, W.J. JC virus binds to primary human glial cells, tonsillar stromal cells, and B-lymphocytes, but not to T lymphocytes. J. Neurovirol. 2000, 6, 127–136. [Google Scholar] [CrossRef]
- Chapagain, M.L.; Nerurkar, V.R. Human polyomavirus JC (JCV) infection of human B lymphocytes: A possible mechanism for JCV transmigration across the blood-brain barrier. J. Infect. Dis. 2010, 202, 184–191. [Google Scholar] [CrossRef] [Green Version]
- Dubois, V.; Dutronc, H.; Lafon, M.E.; Poinsot, V.; Pellegrin, J.L.; Ragnaud, J.M.; Ferrer, A.M.; Fleury, H.J. Latency and reactivation of JC virus in peripheral blood of human immunodeficiency virus type 1-infected patients. J. Clin. Microbiol. 1997, 35, 2288–2292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Liz, G.; Del Valle, L.; Gentilella, A.; Croul, S.; Khalili, K. Detection of JC virus DNA fragments but not proteins in normal brain tissue. Ann. Neurol. 2008, 64, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Wüthrich, C.; Cheng, Y.M.; Joseph, J.T.; Kesari, S.; Beckwith, C.; Stopa, E.; Bell, J.E.; Koralnik, I.J. Frequent infection of cerebellar granule cell neurons by polyomavirus JC in progressive multifocal leukoencephalopathy. J. Neuropathol. Exp. Neurol. 2009, 68, 15–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayliss, J.; Karasoulos, T.; Bowden, S.; Glogowski, I.; McLean, C.A. Immunosuppression increases latent infection of brain by JC polyomavirus. Pathology 2011, 43, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.N.; Major, E.O. Viral variant nucleotide sequences help expose leukocytic positioning in the JC virus pathway to the CNS. J. Leukoc. Boil. 1999, 65, 428–438. [Google Scholar] [CrossRef] [PubMed]
- DeCaprio, J.A.; Garcea, R.L. A cornucopia of human polyomaviruses. Nat. Rev. Genet. 2013, 11, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Atwood, W.J.; Shah, K.V. Polyomaviruses. In Fields Virology, 3rd ed.; Lippincott-Raven Publishers: Philadelphia, PA, USA, 1996; ISBN 0781702534. [Google Scholar]
- Frisque, R.J.; Bream, G.L.; Cannella, M.T. Human polyomavirus JC virus genome. J. Virol. 1984, 51, 458–469. [Google Scholar] [CrossRef] [Green Version]
- Ranganathan, P.N.; Khalili, K. The transcriptional enhancer element kappaB, regulates promoter activity of the human neurotropic virus, JCV, in cells derived from the CNS. Nucleic Acids Res. 1993, 21, 1959–1964. [Google Scholar] [CrossRef]
- Romagnoli, L.; Wollebo, H.S.; Deshmane, S.L.; Mukerjee, R.; Del Valle, L.; Safak, M.; Khalili, K.; White, M.K. Modulation of JC virus transcription by C/EBPβ. Virus Res. 2009, 146, 97–106. [Google Scholar] [CrossRef] [Green Version]
- Wollebo, H.S.; Melis, S.; Khalili, K.; Safak, M.; White, M.K. Cooperative roles of NF-κB and NFAT4 in polyomavirus JC regulation at the KB control element. Virology 2012, 432, 146–154. [Google Scholar] [CrossRef] [Green Version]
- White, M.K.; Kaminski, R.; Khalili, K.; Wollebo, H.S. Rad51 activates polyomavirus JC early transcription. PLoS ONE 2014, 9, e110122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ault, G.S.; Stoner, G.L. Human polyomavirus JC promoter/enhancer rearrangement patterns from progressive multifocal leukoencephalopathy brain are unique derivatives of a single archetypal structure. J. Gen. Virol. 1993, 74, 1499–1507. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.D.; King, D.M.; Slauch, J.M.; Frisque, R.J. Differences in regulatory sequences of naturally occurring JC virus variants. J. Virol. 1985, 53, 306–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenney, S.; Natarajan, V.; Strike, D.; Khoury, G.; Salzman, N.P. JC virus enhancer-promoter active in human brain cells. Science 1984, 226, 1337–1339. [Google Scholar] [CrossRef] [PubMed]
- Tada, H.; Lashgari, M.; Rappaport, J.; Khalili, K. Cell type-specific expression of JC virus early promoter is determined by positive and negative regulation. J. Virol. 1989, 63, 463–466. [Google Scholar] [CrossRef] [Green Version]
- Safak, M.; Barrucco, R.; Darbinyan, A.; Okada, Y.; Nagashima, K.; Khalili, K. Interaction of JC virus agno protein with T antigen modulates transcription and replication of the viral genome in glial cells. J. Virol. 2001, 75, 1476–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saribas, A.S.; Datta, P.K.; Safak, M. A comprehensive proteomics analysis of JC virus Agnoprotein-interacting proteins: Agnoprotein primarily targets the host proteins with coiled-coil motifs. Virology 2019, 540, 104–118. [Google Scholar] [CrossRef] [PubMed]
- Darbinyan, A.; Darbinian, N.; Safak, M.; Radhakrishnan, S.; Giordano, A.; Khalili, K. Evidence for dysregulation of cell cycle by human polyomavirus, JCV, late auxiliary protein. Oncogene 2002, 21, 5574–5581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, T.; Orba, Y.; Okada, Y.; Sunden, Y.; Kimura, T.; Tanaka, S.; Nagashima, K.; Hall, W.W.; Sawa, H. The human polyoma JC virus agnoprotein acts as a viroporin. PLoS Pathog. 2010, 6, e1000801. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Orba, Y.; Makino, Y.; Okada, Y.; Sunden, Y.; Hasegawa, H.; Hall, W.W.; Sawa, H. Viroporin activity of the JC polyomavirus is regulated by interactions with the adaptor protein complex 3. Proc. Natl. Acad. Sci. USA 2013, 110, 18668–18673. [Google Scholar] [CrossRef] [Green Version]
- Okada, S.E.Y.; Endo, S.; Takahashi, H.; Sawa, H.; Umemura, T.; Nagashima, K.; Okada, Y. Distribution and function of JCV agnoprotein. J. Neurovirol. 2001, 7, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Saribas, A.S.; White, M.K.; Safak, M. JC virus agnoprotein enhances large T antigen binding to the origin of viral DNA replication: Evidence for its involvement in viral DNA replication. Virology 2012, 433, 12–26. [Google Scholar] [CrossRef] [Green Version]
- Jay, G.; Nomura, S.; Anderson, C.W.; Khoury, G. Identification of the SV40 agnogene product: A DNA binding protein. Nature 1981, 291, 346–349. [Google Scholar] [CrossRef] [PubMed]
- Buch, M.H.; Liaci, A.M.; O’Hara, S.D.; Garcea, R.L.; Neu, U.; Stehle, T. Structural and functional analysis of murine Polyomavirus capsid proteins establish the determinants of kigand recognition and pathogenicity. PLoS Pathog. 2015, 11, e1005104. [Google Scholar] [CrossRef] [PubMed]
- Trowbridge, P.W.; Frisque, R.J. Identification of three new JC virus proteins generated by alternative splicing of the early viral mRNA. J. Neurovirol. 1995, 1, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Maginnis, M.S.; Atwood, W.J. JC virus: An oncogenic virus in animals and humans? Semin. Cancer Boil. 2009, 19, 261–269. [Google Scholar] [CrossRef] [Green Version]
- Lashgari, M.S.; Tada, H.; Amini, S.; Khalili, K. Regulation of JCVL promoter function: Transactivation of JCVL promoter by JCV and SV40 early proteins. Virology 1989, 170, 292–295. [Google Scholar] [CrossRef]
- Khalili, K.; Feigenbaum, L.; Khoury, G. Evidence for a shift in 5′-termini of early viral RNA during the lytic cycle of JC virus. Virology 1987, 158, 469–472. [Google Scholar] [CrossRef]
- Waga, S.; Bauer, G.; Stillman, B. Reconstitution of complete SV40 DNA replication with purified replication factors. J. Boil. Chem. 1994, 269, 10923–10934. [Google Scholar]
- Prins, C.; Prins, C.; Frisque, R.J. JC virus T′ proteins encoded by alternatively spliced early mRNAs enhance T antigen-mediated viral DNA replication in human cells. J. Neurovirol. 2001, 7, 250–264. [Google Scholar] [CrossRef]
- An, P.; Robles, M.T.S.; Pipas, J.M. Large T antigens of polyomaviruses: Amazing molecular machines. Annu. Rev. Microbiol. 2012, 66, 213–236. [Google Scholar] [CrossRef]
- White, M.K.; Gordon, J.; Reiss, K.; Del Valle, L.; Croul, S.; Giordano, A.; Darbinyan, A.; Khalili, K. Human polyomaviruses and brain tumors. Brain Res. Rev. 2005, 50, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Dyson, N.; Bernards, R.; Friend, S.H.; Gooding, L.R.; Hassell, J.A.; Major, E.O.; Pipas, J.M.; VanDyke, T.; Harlow, E. Large T antigens of many polyomaviruses are able to form complexes with the retinoblastoma protein. J. Virol. 1990, 64, 1353–1356. [Google Scholar] [CrossRef] [Green Version]
- Kao, C.; Huang, J.; Wu, S.-Q.; Hauser, P.; Reznikoff, C.A. Role of SV40 T antigen binding to pRB and p53 in multistep transformation In Vitro of human uroepithelial cells. Carcinogenesis 1993, 14, 2297–2302. [Google Scholar] [CrossRef] [PubMed]
- Caracciolo, V.; Reiss, K.; Crozier-Fitzgerald, C.; De Pascali, F.; Macaluso, M.; Khalili, K.; Giordano, A. Interplay between the retinoblastoma related pRb2/p130 and E2F-4 and -5 in relation to JCV-TAg. J. Cell. Physiol. 2007, 212, 96–104. [Google Scholar] [CrossRef]
- Cress, W.D.; Nevins, J.R. Use of the E2F transcription factor by dna tumor virus regulatory proteins. Inducible Lymphoid Organs 1996, 208, 63–78. [Google Scholar] [CrossRef]
- Krynska, B.; Gordon, J.; Otte, J.; Franks, R.; Knobler, R.; DeLuca, A.; Giordano, A.; Khalili, K. Role of cell cycle regulators in tumor formation in transgenic mice expressing the human neurotropic virus, JCV, early protein. J. Cell. Biochem. 1997, 67, 223–230. [Google Scholar] [CrossRef]
- Reich, N.C.; Levine, A.J. Specific interaction of the SV40 T antigen-cellular p53 protein complex with SV40 DNA. Virology 1982, 117, 286–290. [Google Scholar] [CrossRef]
- Tan, T.H.; Wallis, J.; Levine, A.J. Identification of the p53 protein domain involved in formation of the simian virus 40 large T-antigen-p53 protein complex. J. Virol. 1986, 59, 574–583. [Google Scholar] [CrossRef] [Green Version]
- Uppal, S.; Coatesworth, A.P. Neurofibromatosis type 2. Int. J. Clin. Pract. 2003, 57, 698–703. [Google Scholar]
- Shollar, D.; Del Valle, L.; Khalili, K.; Otte, J.; Gordon, J. JCV T-antigen interacts with the neurofibromatosis type 2 gene product in a transgenic mouse model of malignant peripheral nerve sheath tumors. Oncogene 2004, 23, 5459–5467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Kwak, N.-J.; Lee, J.Y.; Choi, B.H.; Lim, Y.; Ko, Y.J.; Kim, Y.-H.; Huh, P.-W.; Lee, K.-H.; Rha, H.K.; et al. Merlin neutralizes the inhibitory effect of Mdm2 on p53. J. Boil. Chem. 2003, 279, 7812–7818. [Google Scholar] [CrossRef] [Green Version]
- Gan, D.-D.; Khalili, K. Interaction between JCV large T-antigen and β-catenin. Oncogene 2004, 23, 483–490. [Google Scholar] [CrossRef] [Green Version]
- Lassak, A.; Peruzzi, F.; Enam, S.; Croul, S.; Khalili, K.; Reiss, K.; Del Valle, L.; Wang, J.Y. Insulin receptor substrate 1 translocation to the nucleus by the human JC virus T-antigen. J. Boil. Chem. 2002, 277, 17231–17238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piña-Oviedo, S.; Urbanska, K.; Radhakrishnan, S.; Sweet, T.; Reiss, K.; Khalili, K.; Del Valle, L. Effects of JC virus infection on anti-apoptotic protein survivin in progressive multifocal leukoencephalopathy. Am. J. Pathol. 2007, 170, 1291–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ripple, M.J.; Struckhoff, A.P.; Trillo-Tinoco, J.; Li, L.; Margolin, D.A.; McGoey, R.; Del Valle, L. Activation of c-Myc and cyclin D1 by JCV T-antigen and β-catenin in colon cancer. PLoS ONE 2014, 9, e106257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coyle-Rink, J.; Del Valle, L.; Sweet, T.; Khalili, K.; Amini, S. Developmental expression of Wnt signaling factors in mouse brain. Cancer Boil. Ther. 2003, 1, 640–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Valle, L.; White, M.K.; Enam, S.; Bromer, M.Q.; Thomas, R.M.; Parkman, H.P.; Khalili, K. Detection of JC virus DNA sequences and expression of viral T antigen and agnoprotein in esophageal carcinoma. Cancer 2005, 103, 516–527. [Google Scholar] [CrossRef] [PubMed]
- Gan, D.-D.; Reiss, K.; Carrill, T.; Del Valle, L.; Croul, S.; Giordano, A.; Fishman, P.; Khalili, K. Involvement of Wnt signaling pathway in murine medulloblastoma induced by human neurotropic JC virus. Oncogene 2001, 20, 4864–4870. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharyya, R.; Noch, E.K.; Khalili, K. A novel role of Rac1 GTPase in JCV T-antigen-mediated β-catenin stabilization. Oncogene 2007, 26, 7628–7636. [Google Scholar] [CrossRef]
- Trojanek, J.B.; Croul, S.; Ho, T.; Wang, J.Y.; Darbinyan, A.; Nowicki, M.; Del Valle, L.; Skorski, T.; Khalili, K.; Reiss, K. T-antigen of the human polyomavirus JC attenuates faithful DNA repair by forcing nuclear interaction between IRS-1 and Rad51. J. Cell. Physiol. 2005, 206, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Gualco, E.; Urbanska, K.; Perez-Liz, G.; Sweet, T.; Peruzzi, F.; Reiss, K.; Del Valle, L. IGF-IR-dependent expression of Survivin is required for T-antigen-mediated protection from apoptosis and proliferation of neural progenitors. Cell Death Differ. 2009, 17, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Darbinyan, A.; White, M.K.; Akan, S.; Radhakrishnan, S.; Del Valle, L.; Amini, S.; Khalili, K. Alterations of DNA damage repair pathways resulting from JCV infection. Virology 2007, 364, 73–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazutka, J.R.; Neel, J.; Major, E.; Dedonyt, V.; Mierauskin, J.; Slapšyt, G.; Kesminien, A. High titers of antibodies to two human polyomaviruses, JCV and BKV, correlate with increased frequency of chromosomal damage in human lymphocytes. Cancer Lett. 1996, 109, 177–183. [Google Scholar] [CrossRef]
- Kenan, D.J.; Mieczkowski, P.A.; Burger-Calderon, R.; Singh, H.K.; Nickeleit, V. The oncogenic potential of BK-polyomavirus is linked to viral integration into the human genome. J. Pathol. 2015, 237, 379–389. [Google Scholar] [CrossRef]
- White, M.K.; Bellizzi, A.; Ibba, G.; Pietropaolo, V.; Palamara, A.T.; Wollebo, H.S. The DNA damage response promotes polyomavirus JC infection by nucleus to cytoplasm NF- kappaB activation. Virol. J. 2017, 14, 31. [Google Scholar] [CrossRef] [Green Version]
- Erickson, K.D.; Garcea, R.L. Viral replication centers and the DNA damage response in JC virus-infected cells. Virology 2019, 528, 198–206. [Google Scholar] [CrossRef]
- Tahseen, D.; Rady, P.L.; Tyring, S.K. Human polyomavirus modulation of the host DNA damage response. Virus Genes 2020, 56, 128–135. [Google Scholar] [CrossRef]
- Justice, J.L.; Verhalen, B.; Jiang, M. Polyomavirus interaction with the DNA damage response. Virol. Sin. 2015, 30, 122–129. [Google Scholar] [CrossRef]
- Starrett, G.J.; Buck, C.B. The case for BK polyomavirus as a cause of bladder cancer. Curr. Opin. Virol. 2019, 39, 8–15. [Google Scholar] [CrossRef]
- Verhalen, B.; Justice, J.L.; Imperiale, M.J.; Jiang, M. Viral DNA replication-dependent DNA damage response activation during BK polyomavirus infection. J. Virol. 2015, 89, 5032–5039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Justice, J.L.; Needham, J.M.; Thompson, S.R. BK polyomavirus activates the DNA damage response to prolong s phase. J. Virol. 2019, 93, 00130–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Wang, X.; Diaz, J.; Tsang, S.H.; Buck, C.B.; You, J. Merkel cell polyomavirus large T antigen disrupts host genomic integrity and inhibits cellular proliferation. J. Virol. 2013, 87, 9173–9188. [Google Scholar] [CrossRef] [Green Version]
- Verhaegen, M.E.; Mangelberger, R.; Harms, P.W.; Vozheiko, T.D.; Weick, J.W.; Wilbert, D.M.; Saunders, T.L.; Ermilov, A.N.; Bichakjian, C.K.; Johnson, T.M.; et al. Merkel cell polyomavirus small T antigen is oncogenic in transgenic mice. J. Investig. Dermatol. 2014, 135, 1415–1424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bollag, B.; Hofstetter, C.A.; Reviriego-Mendoza, M.M.; Frisque, R.J. JC virus small t antigen binds phosphatase PP2A and Rb family proteins and is required for efficient viral DNA replication activity. PLoS ONE 2010, 5, e10606. [Google Scholar] [CrossRef]
- Sariyer, I.K.; Khalili, K.; Safak, M. Dephosphorylation of JC virus agnoprotein by protein phosphatase 2A: Inhibition by small t antigen. Virology 2008, 375, 464–479. [Google Scholar] [CrossRef] [Green Version]
- Pallas, D.C.; Shahrik, L.K.; Martin, B.L.; Jaspers, S.; Miller, T.B.; Brautigan, D.L.; Roberts, T.M. Polyoma small and middle T antigens and SV40 small t antigen form stable complexes with protein phosphatase 2A. Cell 1990, 60, 167–176. [Google Scholar] [CrossRef]
- Huang, J.-L.; Lin, C.-S.; Chang, C.-C.; Lu, Y.-N.; Hsu, Y.-L.; Wong, T.-Y.; Wang, Y.-F. Human JC virus small tumour antigen inhibits nucleotide excision repair and sensitises cells to DNA-damaging agents. Mutagenesis 2015, 30, 475–485. [Google Scholar] [CrossRef]
- Nunbhakdi-Craig, V.; Craig, L.; Machleidt, T.; Sontag, E. Simian virus 40 small tumor antigen induces deregulation of the actin cytoskeleton and tight junctions in kidney epithelial cells. J. Virol. 2003, 77, 2807–2818. [Google Scholar] [CrossRef] [Green Version]
- Shuda, M.; Kwun, H.J.; Feng, H.; Chang, Y.; Moore, P. Human Merkel cell polyomavirus small T antigen is an oncoprotein targeting the 4E-BP1 translation regulator. J. Clin. Investig. 2011, 121, 3623–3634. [Google Scholar] [CrossRef]
- Wendzicki, J.A.; Moore, P.; Chang, Y. Large T and small T antigens of Merkel cell polyomavirus. Curr. Opin. Virol. 2015, 11, 38–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shuda, M.; Chang, Y.; Moore, P. Merkel cell polyomavirus-positive Merkel cell carcinoma requires viral small T-antigen for cell proliferation. J. Investig. Dermatol. 2013, 134, 1479–1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhaegen, M.E.; Mangelberger, R.; Weick, J.W.; Vozheiko, T.D.; Harms, P.W.; Nash, K.T.; Quintana, E.; Baciu, P.; Johnson, T.M.; Bichakjian, C.K.; et al. Merkel cell carcinoma dependence on Bcl-2 family members for survival. J. Investig. Dermatol. 2014, 134, 2241–2250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darbinyan, A.; Siddiqui, K.M.; Slonina, D.; Darbinian, N.; Amini, S.; White, M.K.; Khalili, K. Role of JC virus agnoprotein in DNA repair. J. Virol. 2004, 78, 8593–8600. [Google Scholar] [CrossRef] [Green Version]
- Padgett, B.L.; Walker, D.L.; ZuRhein, G.M.; Varakis, J.N. Differential neurooncogenicity of strains of JC virus, a human polyoma virus, in newborn Syrian hamsters. Cancer Res. 1977, 37, 718–720. [Google Scholar]
- ZuRhein, G.R.; Varakis, J.N. Perinatal induction of medulloblastomas in Syrian golden hamsters by a human polyomavirus (JC). Natl. Cancer Inst. Monogr. 1979, 51, 205–208. [Google Scholar]
- Walker, D.L.; Padgett, B.L.; ZuRhein, G.M.; Albert, A.E.; Marsh, R.F. Human Papovavirus (JC): Induction of brain tumors in hamsters. Science 1973, 181, 674–676. [Google Scholar] [CrossRef]
- Varakis, J.; ZuRhein, G.M.; Padgett, B.L.; Walker, D.L. Induction of peripheral neuroblastomasin Syrian hamsters after injection as neonates with JC virus, a humanpolyoma virus. Cancer Res. 1978, 38, 1718–1722. [Google Scholar]
- Zu Rhein, G.M. Studies of JC virus-induced nervous system tumors in the Syrian hamster: A review. Prog. Clin. Boil. Res. 1983, 105, 205–221. [Google Scholar]
- London, W.; Houff, S.; Madden, D.; Fuccillo, D.; Gravell, M.; Wallen, W.; Palmer, A.; Sever, J.; Padgett, B.; Walker, D.; et al. Brain tumors in owl monkeys inoculated with a human polyomavirus (JC virus). Science 1978, 201, 1246–1249. [Google Scholar] [CrossRef]
- Eddy, B.E.; Borman, G.S.; Grubbs, G.E.; Young, R.D. Identification of the oncogenic substance in rhesus monkey kidney cell cultures as simian virus 40. Virology 1962, 17, 65–75. [Google Scholar] [CrossRef]
- Khalili, K.; Croul, S.; Del Valle, L.; Krynska, B.; Gordon, J. Oncogenic potential of human neurotropic virus: Laboratory and clinical observations. Isr. Med. Assoc. J. IMAJ 2001, 3, 210–215. [Google Scholar] [PubMed]
- Khalili, K.; Del Valle, L.; Otte, J.; Weaver, M.; Gordon, J. Human neurotropic polyomavirus, JCV, and its role in carcinogenesis. Oncogene 2003, 22, 5181–5191. [Google Scholar] [CrossRef] [Green Version]
- London, W.T.; Houff, S.A.; McKeever, P.E.; Wallen, W.C.; Sever, J.L.; Padgett, B.L.; Walker, D.L. Viral-induced astrocytomas in squirrel monkeys. Prog. Clin. Boil. Res. 1983, 105, 227–237. [Google Scholar]
- Major, E.O.; Mourrain, P.; Cummins, C. JC virus-induced owl monkey glioblastoma cells in culture: Biological properties associated with the viral early gene product. Virology 1984, 136, 359–367. [Google Scholar] [CrossRef]
- Nagashima, K.; Yasui, K.; Kimura, J.; Washizu, M.; Yamaguchi, K.; Mori, W. Induction of brain tumors by a newly isolated JC virus (Tokyo-1 strain). Am. J. Pathol. 1984, 116, 455–463. [Google Scholar]
- Ohsumi, S.; Ikehara, I.; Motoi, M.; Ogawa, K.; Nagashima, K.; Yasui, K. Induction of undifferentiated brain tumors in rats by a human polyomavirus (JC virus). Jpn. J. Cancer Res. 1985, 76, 429–431. [Google Scholar]
- Ohsumi, S.; Motoi, M.; Ogawa, K. Induction of undifferentiated tumors by JC virus in the cerebrum of rats. Pathol. Int. 2008, 36, 815–825. [Google Scholar] [CrossRef]
- Krynska, B.; Otte, J.; Franks, R.; Khalili, K.; Croul, S. Human ubiquitous JCV(CY) T-antigen gene induces brain tumors in experimental animals. Oncogene 1999, 18, 39–46. [Google Scholar] [CrossRef] [Green Version]
- Gordon, J.; Del Valle, L.; Otte, J.; Khalili, K. Pituitary neoplasia induced by expression of human neurotropic polyomavirus, JCV, early genome in transgenic mice. Oncogene 2000, 19, 4840–4846. [Google Scholar] [CrossRef] [Green Version]
- Small, J.A.; Scangos, G.A.; Cork, L.; Jay, G.; Khoury, G. The early region of human papovavirus JC induces dysmyelination in transgenic mice. Cell 1986, 46, 13–18. [Google Scholar] [CrossRef]
- Franks, R.R.; Rencic, A.; Gordon, J.; Zoltick, P.W.; Curtis, M.; Knobler, R.L.; Khalili, K. Formation of undifferentiated mesenteric tumors in transgenic mice expressing human neurotropic polymavirus early protein. Oncogene 1996, 12, 2573–2578. [Google Scholar] [PubMed]
- Richardson, E.P. Progressive multifocal leukoencephalopathy. N. Engl. J. Med. 1961, 265, 815–823. [Google Scholar] [CrossRef] [PubMed]
- Castaigne, P.; Rondot, P.; Escourolle, R.; Dumas, J.L.R.; Cathala, F.; Hauw, J.J. Progressive multifocal leukoencephalopathy and multiple gliomas. Rev. Neurol. 1974, 130, 379–392. [Google Scholar] [PubMed]
- Sima, A.A.; Finkelstein, S.D.; McLachlan, D.R. Multiple malignant astrocytomas in a patient with spontaneous progressive multifocal leukoencephalopathy. Ann. Neurol. 1983, 14, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Shintaku, M.; Matsumoto, R.; Sawa, H.; Nagashima, K. Infection with JC virus and possible dysplastic ganglion-like transformation of the cerebral cortical neurons in a case of progressive multifocal leukoencephalopathy. J. Neuropathol. Exp. Neurol. 2000, 59, 921–929. [Google Scholar] [CrossRef] [Green Version]
- Barbanti-Brodano, G.; Martini, F.; De Mattei, M.; Lazzarin, L.; Corallini, A.; Tognon, M. BK and JC human polyomaviruses and simian virus 40: Natural history of infection in humans, experimental oncogenicity, and association with human tumors. Adv. Appl. Microbiol. 1998, 50, 69–99. [Google Scholar] [CrossRef]
- Northcott, P.A.; Robinson, G.W.; Kratz, C.P.; Mabbott, D.J.; Pomeroy, S.L.; Clifford, S.C.; Rutkowski, S.; Ellison, D.W.; Malkin, D.; Taylor, M.; et al. Medulloblastoma. Nat. Rev. Dis. Prim. 2019, 5, 11. [Google Scholar] [CrossRef]
- Del Valle, L.; Enam, S.; Lara, C.; Miklossy, J.; Khalili, K.; Gordon, J. Primary central nervous system lymphoma expressing the human neurotropic polyomavirus, JC virus, genome. J. Virol. 2004, 78, 3462–3469. [Google Scholar] [CrossRef] [Green Version]
- Egan, J.D.; Ring, B.L.; Reding, M.J.; Wells, I.C.; Shuman, R.M. Reticulum cell sarcoma and progressive multifocal leukoencephalopathy following renal transplantation. Transplantation 1980, 29, 84–85. [Google Scholar] [CrossRef]
- Gallia, G.L.; Del Valle, L.; Laine, C.; Curtis, M.; Khalili, K. Concomitant progressive multifocal leucoencephalopathy and primary central nervous system lymphoma expressing JC virus oncogenic protein, large T antigen. Mol. Pathol. 2001, 54, 354–359. [Google Scholar] [CrossRef] [Green Version]
- Major, E.O. Progressive multifocal leukoencephalopathy in patients on immunomodulatory therapies. Annu. Rev. Med. 2010, 61, 35–47. [Google Scholar] [CrossRef] [Green Version]
- Houff, S.A.; Major, E.O.; Katz, D.A.; Kufta, C.V.; Sever, J.L.; Pittaluga, S.; Roberts, J.R.; Gitt, J.; Saini, N.; Lux, W. Involvement of JC virus–infected mononuclear cells from the bone marrow and spleen in the pathogenesis of progressive multifocal leukoencephalopathy. N. Engl. J. Med. 1988, 318, 301–305. [Google Scholar] [CrossRef]
- Sabath, B.F.; Major, E.O. Traffic of JC virus from sites of initial infection to the brain: The path to progressive multifocal leukoencephalopathy. J. Infect. Dis. 2002, 186, S180–S186. [Google Scholar] [CrossRef] [Green Version]
- Velásquez, C.; Amako, Y.; Harold, A.; Toptan, T.; Chang, Y.; Shuda, M. Characterization of a Merkel cell polyomavirus-positive merkel cell carcinoma cell line CVG-1. Front. Microbiol. 2018, 9, 713. [Google Scholar] [CrossRef]
- Hayashi, H.; Endo, S.; Suzuki, S.; Tanaka, K.; Sawa, H.; Ozaki, Y.; Sawamura, Y.; Nagashima, K. JC virus large T protein transforms rodent cells but is not involved in human medulloblastoma. Neuropathology 2001, 21, 129–137. [Google Scholar] [CrossRef]
- Arthur, R.R.; Grossman, S.A.; Ronnett, B.M.; Bigner, S.H.; Vogelstein, B.; Shah, K.V. Lack of association of human polyomaviruses with human brain tumors. J. Neuro-Oncol. 1994, 20, 55–58. [Google Scholar] [CrossRef]
- Munoz-Marmol, A.M.; Mola, G.; Ruiz-Larroya, T.; Fernández-Vasalo, A.; Vela, E.; Mate, J.L.; Ariza, A. Rarity of JC virus DNA sequences and early proteins in human gliomas and medulloblastomas: The controversial role of JC virus in human neurooncogenesis. Neuropathol. Appl. Neurobiol. 2006, 32, 131–140. [Google Scholar] [CrossRef]
- Blumberg, B.S.; London, W.T. Hepatitis B virus and the prevention of primary cancer of the liver3. J. Natl. Cancer Inst. 1985, 74, 267–273. [Google Scholar] [CrossRef]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P. Clonal integration of a polyomavirus in human merkel cell carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef] [Green Version]
- Young, L.S.; Rickinson, A.B. Epstein–Barr virus: 40 years on. Nat. Rev. Cancer 2004, 4, 757–768. [Google Scholar] [CrossRef]
- Lauer, G.M.; Walker, B.D. Hepatitis C virus infection. N. Engl. J. Med. 2001, 345, 41–52. [Google Scholar] [CrossRef]
- Chang, Y.; Cesarman, E.; Pessin, M.S.; Lee, F.; Culpepper, J.; Knowles, D.M.; Moore, P. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 1994, 266, 1865–1869. [Google Scholar] [CrossRef] [Green Version]
- Wong, M.; Pagano, J.S.; Schiller, J.T.; Tevethia, S.S.; Raab-Traub, N.; Gruber, J. New Associations of human papillomavirus, Simian sirus 40, and Epstein-Barr virus with human cancer. J. Natl. Cancer Inst. 2002, 94, 1832–1836. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, M.; Miyoshi, I.; Hinuma, Y. Isolation and characterization of retrovirus from cell lines of human adult T-cell leukemia and its implication in the disease. Proc. Natl. Acad. Sci. USA 1982, 79, 2031–2035. [Google Scholar] [CrossRef] [Green Version]


| Brain Tumor | JCV Factor | Cellular Factor | Assay | References |
|---|---|---|---|---|
| Glioblastoma | VP1, NCCR | - | PCR and sequencing (Mad-4 NCCR and genotype1 VP1) | Boldorini R et al. 2003 [12]; Delbue S et al. 2005 [13] |
| LTAg | p53 | IHC (p53 and LTAg), PCR (LTAg) and SB (LTAg) | Del Valle et al. 2000 [14], Del Valle et al. 2001a [15] | |
| LTAg, VP1, Agno, NCCR | p53 | IHC (p53 and LTAg–VP1 not detected), PCR (LTAg, VP1, Agno, NCCR), SB (LTAg, VP1, Agno, NCCR), sequencing (Mad-1NCCR) and LCM LTAg positive cells | Piña-Oviedo S et al. 2006 (case report) [16]; | |
| LTAg, VP1, Agno, NCCR | p53 | IHC (p53 and LTAg–VP1 not detected), PCR (LTAg, VP1, Agno, NCCR), SB (LTAg, VP1, Agno), sequencing (Mad-4 NCCR) | Del Valle L et al. 2002b (case report) [23] | |
| Astrocytoma | LTAg | p53 | IHC (p53 and LTAg), PCR (LTAg) and SB (LTAg) | Del Valle et al. 2001a [15] |
| LTAg, NCCR | - | IHC (LTAg), PCR (LTAg and NCCR) and sequencing (Mad-4 NCCR) | Caldarelli-Stefano R et al. 2000 [17] | |
| Oligoastrocytoma | LTAg | p53 | IHC (p53 and LTAg), PCR (LTAg) and SB (LTAg) | Del Valle et al. 2001a [15] |
| LTAg, NCCR | Ki67 | IHC (Ki67 proliferation marker and LTAg), PCR (LTAg and NCCR), SB (LTAg), primer extension (LTAg), IP (LTAg) and sequencing (Mad-4 NCCR) | Rencic A et al. 1996 [24] | |
| Oligodendroglioma | LTAg | p53 | IHC (p53 and LTAg), PCR (LTAg) and SB (LTAg) | Del Valle et al. 2001a [15] |
| LTAg, VP1, Agno, NCCR | p53 | IHC (p53, LTAg, Agno–Vp1 not detected), PCR (LTAg, VP1, Agno, NCCR), SB (LTAg, VP1 and Agno), sequencing (Mad-4 and Archetype NCCR) | Del Valle et al. 2002c [25] | |
| Ependymoma | LTAg | p53 | IHC (p53 and LTAg), PCR (LTAg) and SB (LTAg) | Del Valle et al. 2001a [15] |
| Medulloblastoma | LTAg, VP1 | - | IHC (LTAg–VP1 not detected), PCR (LTAg, VP1), SB (LTAg, VP1) | Krynska B et al. 1999a [20] |
| LTAg, VP1 | p53, pRb (p107, pRb2/p130) | IHC (p53, pRb, LTAg), PCR (LTAg, VP1) | Del Valle et al. 2001c [21] | |
| LTAg, Agno | p53 | IHC (p53, LTAg and Agno), PCR (LTAg, Agno), SB (LTAg, Agno) | Del Valle et al. 2002a [22] | |
| Primary CNS lymphoma | LTAg, Agno, VP1 | p53 | IHC (p53 and LTAg–VP1 not detected), PCR (LTAg, VP1, Agno), SB (LTAg, VP1, Agno), LCM LTAg positive cells | Del Valle et al. 2004 [161] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahye, N.; Bellizzi, A.; May, D.; Wollebo, H.S. The Role of the JC Virus in Central Nervous System Tumorigenesis. Int. J. Mol. Sci. 2020, 21, 6236. https://doi.org/10.3390/ijms21176236
Ahye N, Bellizzi A, May D, Wollebo HS. The Role of the JC Virus in Central Nervous System Tumorigenesis. International Journal of Molecular Sciences. 2020; 21(17):6236. https://doi.org/10.3390/ijms21176236
Chicago/Turabian StyleAhye, Nicholas, Anna Bellizzi, Dana May, and Hassen S. Wollebo. 2020. "The Role of the JC Virus in Central Nervous System Tumorigenesis" International Journal of Molecular Sciences 21, no. 17: 6236. https://doi.org/10.3390/ijms21176236
APA StyleAhye, N., Bellizzi, A., May, D., & Wollebo, H. S. (2020). The Role of the JC Virus in Central Nervous System Tumorigenesis. International Journal of Molecular Sciences, 21(17), 6236. https://doi.org/10.3390/ijms21176236