Neurodegeneration, Myelin Loss and Glial Response in the Three-Vessel Global Ischemia Model in Rat

 , and
, and
Abstract
:1. Introduction
2. Results
2.1. Survival and Neurological Deficit
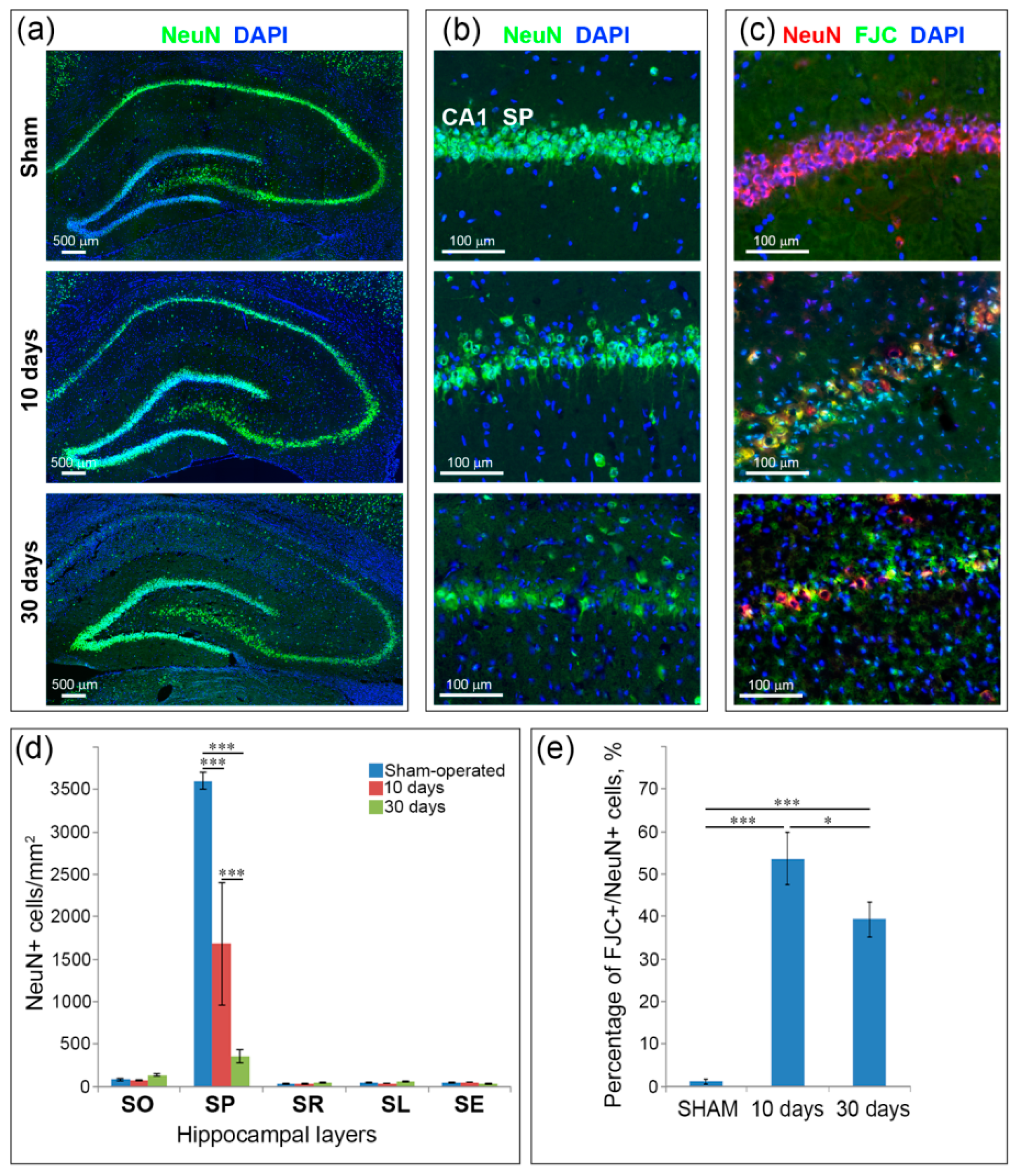
2.2. Neuronal Loss and Neurodegeneration in the Hippocampus, Neocortex and Thalamus
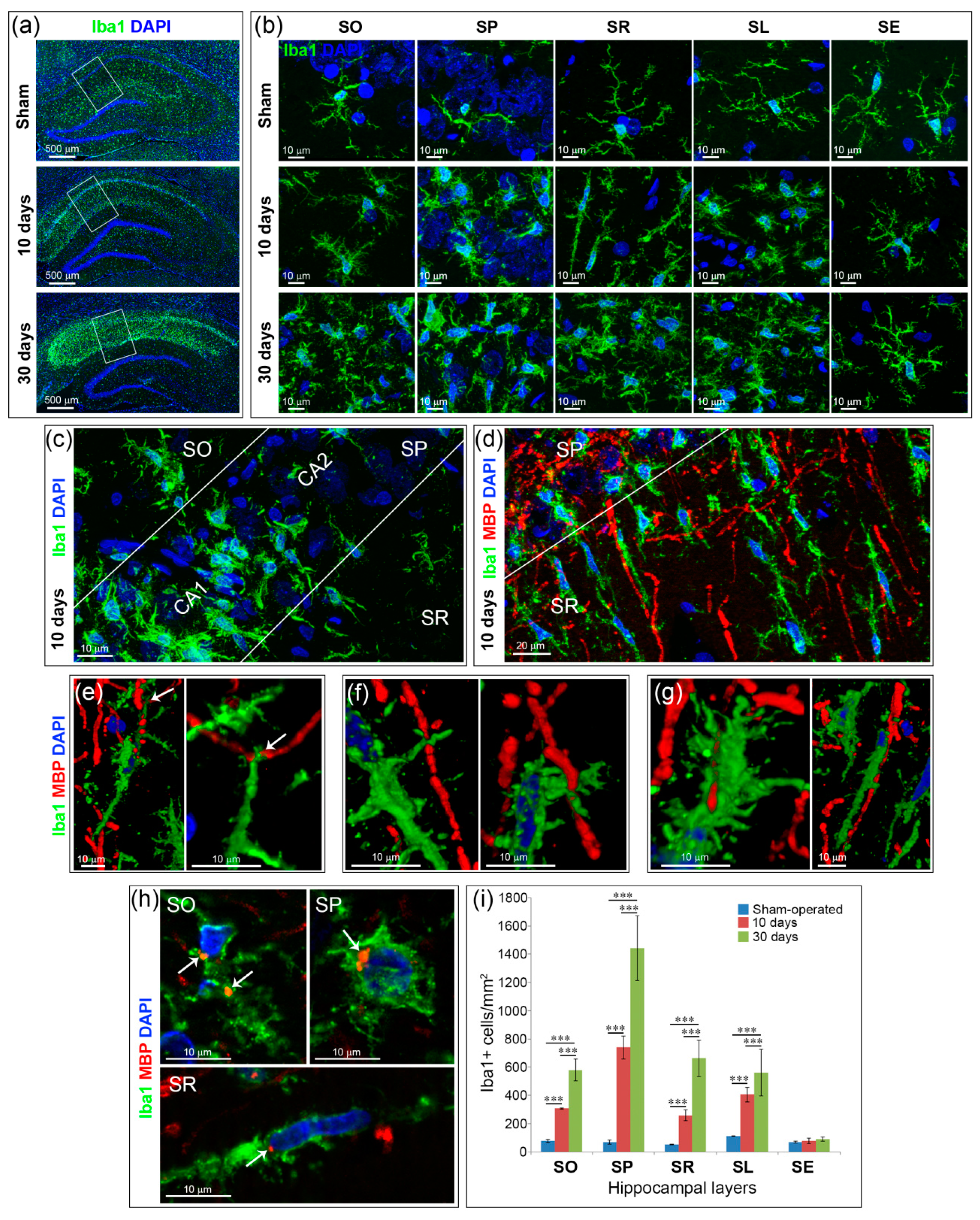
2.3. Inflammation and Specific Changes of Microglial Morphology after GCI
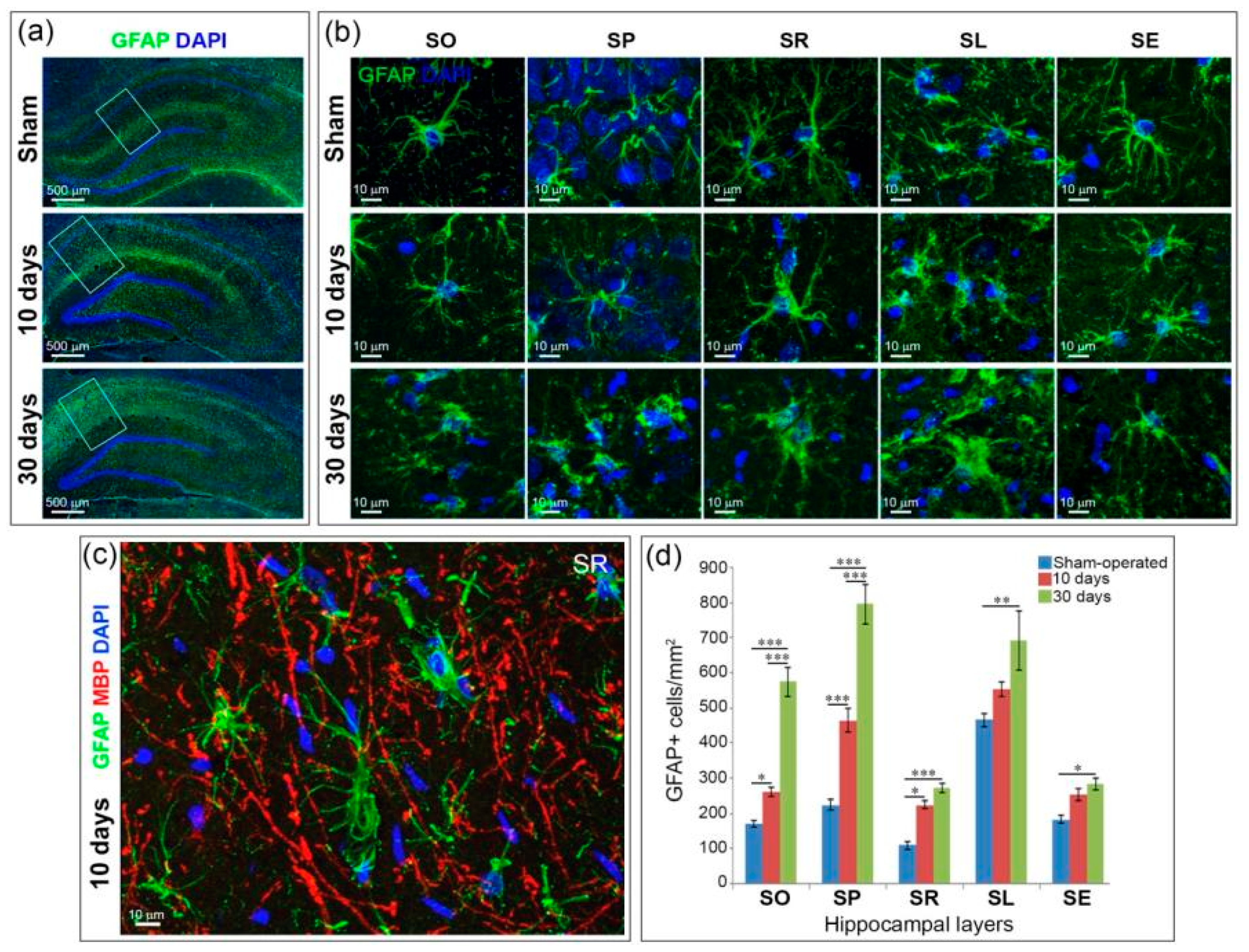
2.4. Astrogliosis
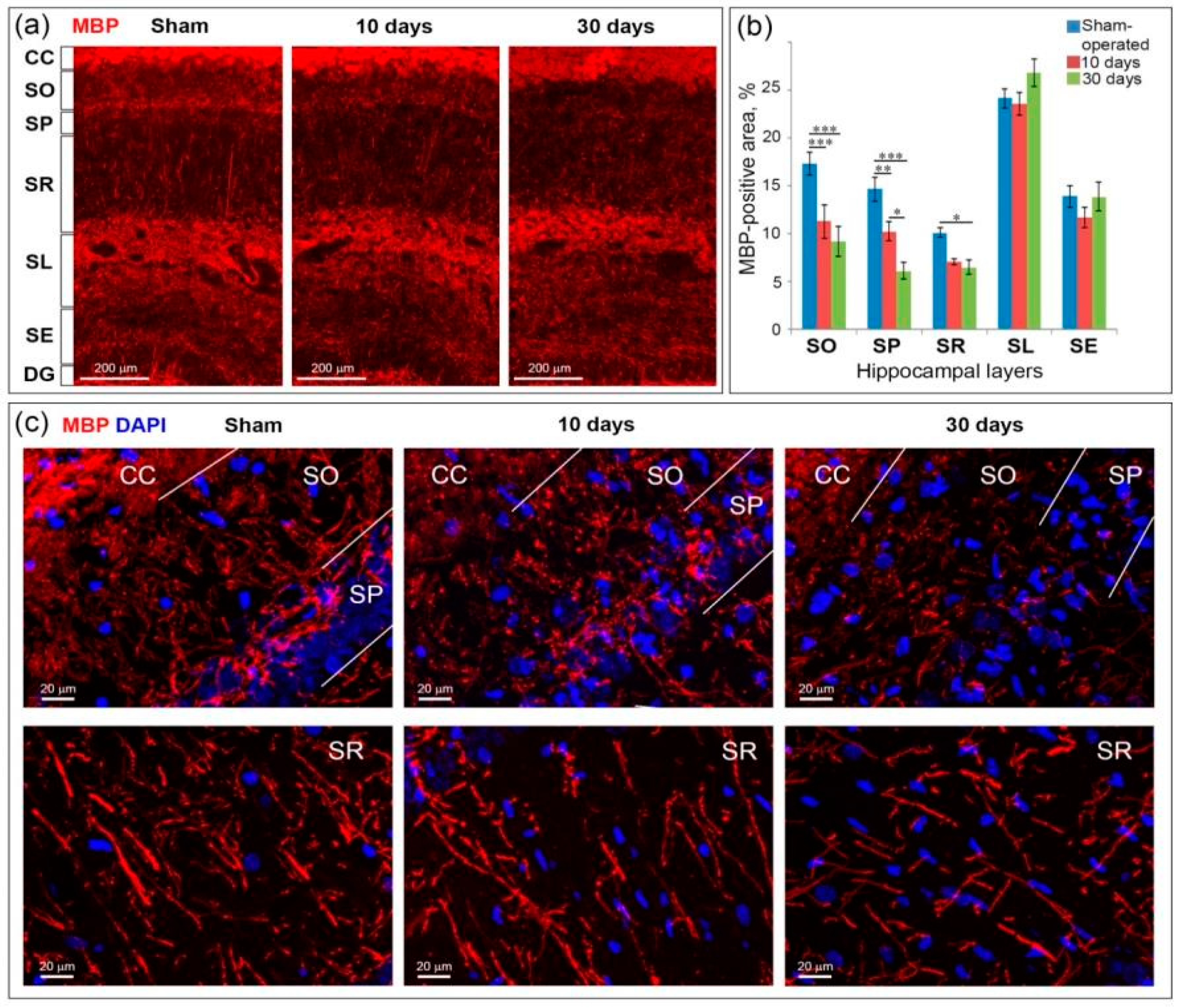
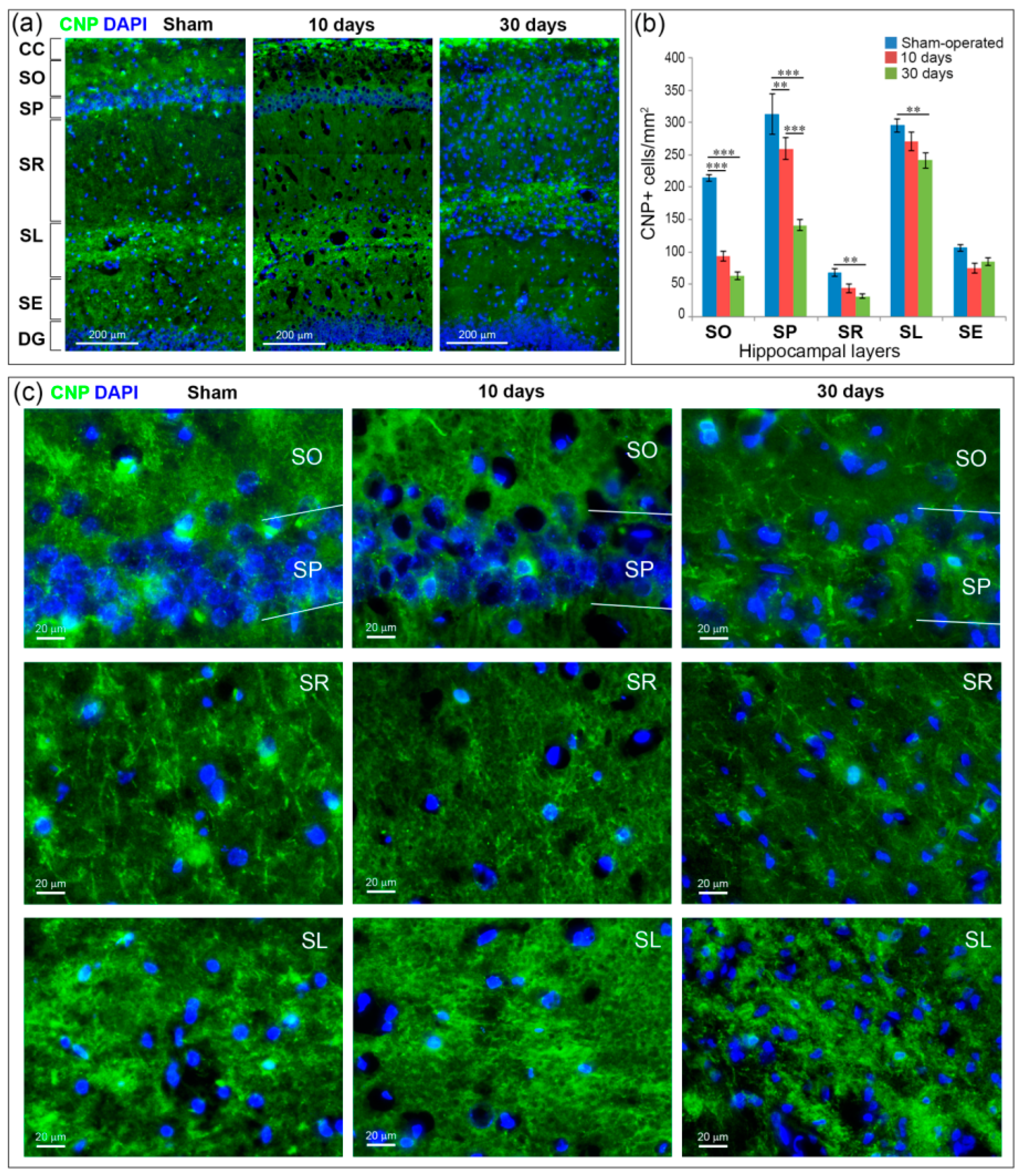
2.5. GCI Causes Myelin and Oligodendrocyte Loss in the SO, SP and SR the Hippocampus
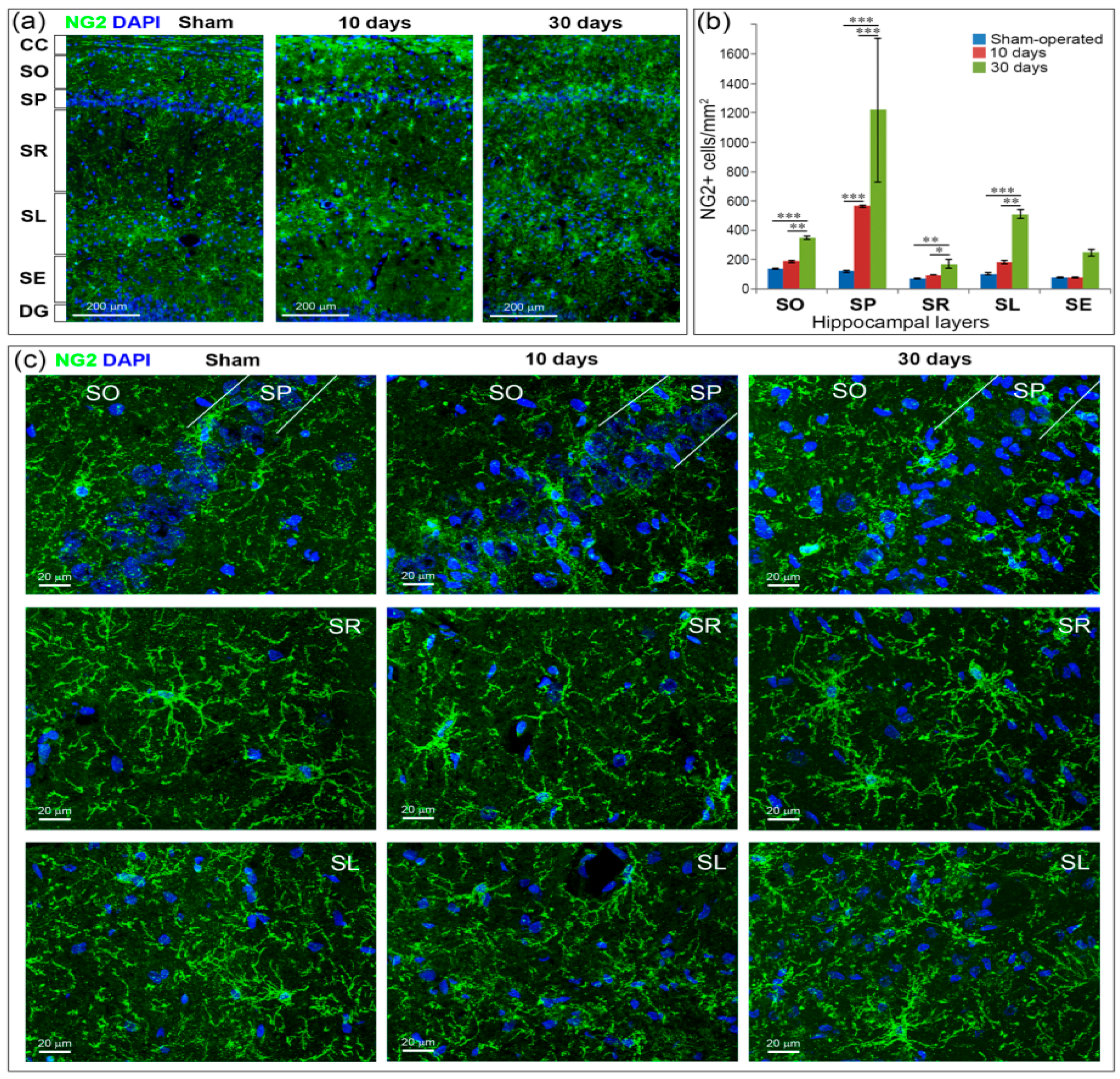
2.6. GCI Increases the Number of Immature Oligodendrocytes in the Hippocampus
3. Discussion
4. Materials and Methods
4.1. Animals and Housing
4.2. Experimental Design
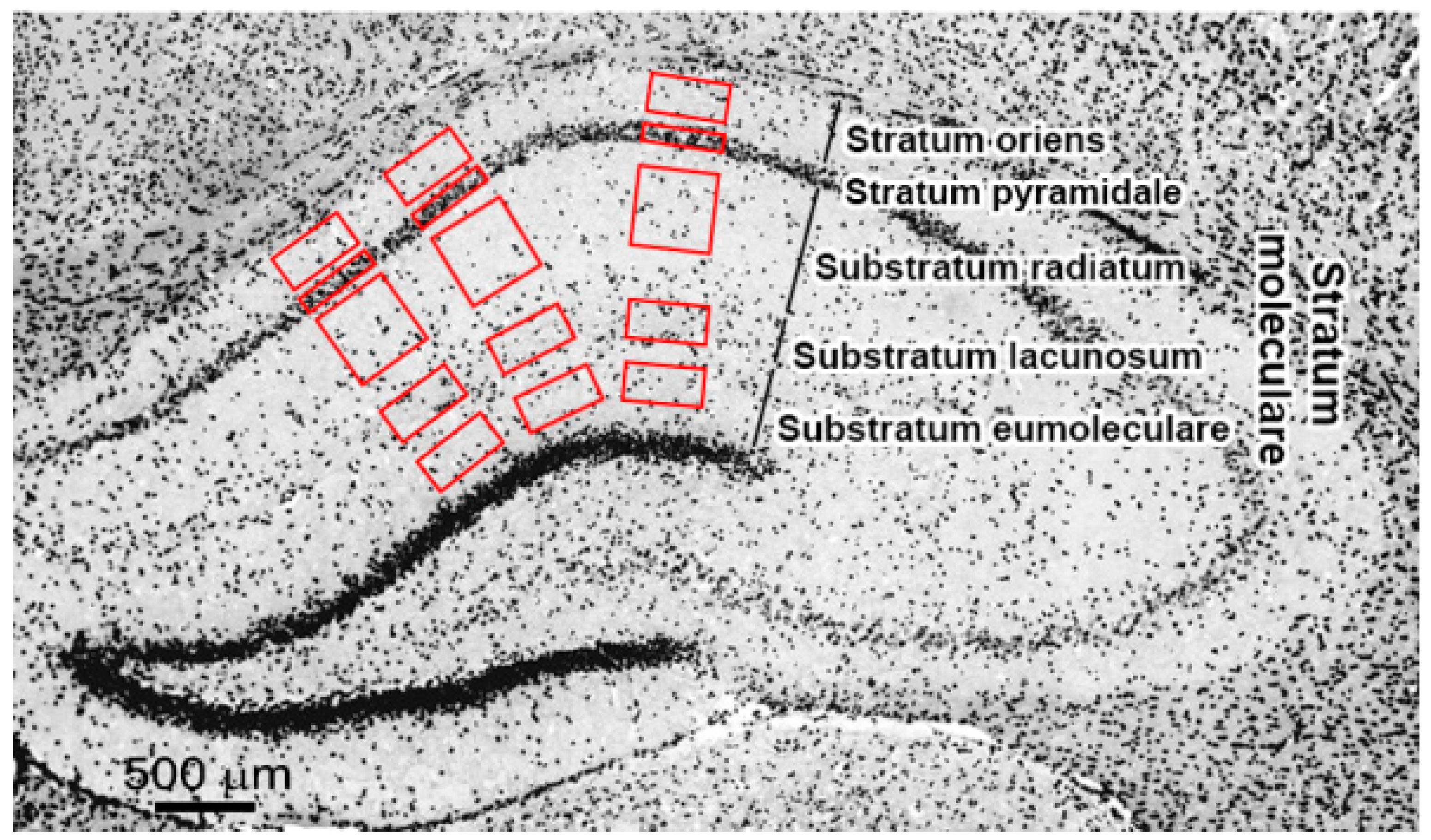
4.3. Immunochemistry and Microscopy
- rabbit-anti-NeuN (ABN78, Merck Millipore, Bedford, MA, USA) for detection of mature neurons;
- rabbit-anti-GFAP (ab7260, Abcam, Cambridge, MA, USA) for detection of astrocytes;
- rabbit-anti-Iba1 (019-19741, Wako Pure Chemical, Osaka, Japan) for detection of microglia/macrophages;
- rabbit-anti-NG2 (AB5320, Merck Millipore, Bedford, MA, USA) for detection of oligodendrocyte precursors (OPCs);
- mouse-anti-CNPase (MAB326, Merck Millipore, Bedford, MA, USA) for detection of myelinating oligodendrocytes;
- goat-anti-MBP (C-16, sc-13914, Santa Cruz Biotechnology, CA, USA) for myelin detection.
4.4. Image Processing
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| GCI | Global cerebral ischemia |
| CA1, 2, 3 | Subfields of the hippocampus |
| SO | Stratum oriens |
| SP | Stratum pyramidale |
| SR | Substratum radiatum |
| SL | Substratum lacunosum |
| SE | Substratum eumoleculare |
| NeuN | Neuronal nuclear antigen |
| FJC | Fluoro-Jade C |
| Iba1 | Ionized calcium-binding adapter molecule 1 |
| GFAP | Glial fibrillary acidic protein |
| MBP | Myelin basic protein |
| OPCs | Oligodendrocyte progenitor cells |
| CNP | 2′,3′-Cyclic nucleotide 3′-phosphodiesterase |
| NG2 | Neural/glial antigen 2 |
| MIP | Maximum intensity projection |
| LSD | Least significant difference |
References
- Etherton, M.R.; Wu, O.; Giese, A.; Lauer, A.; Boulouis, G.; Mills, B.; Cloonan, L.; Donahue, K.L.; Copen, W.; Schaefer, P.; et al. White Matter Integrity and Early Outcomes after Acute Ischemic Stroke. Transl. Stroke Res. 2019, 10, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Desmond, D.W. Cognition and White Matter Lesions. Cerebrovasc. Dis. 2002, 13, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Nave, K.A. Oligodendrocytes: Myelination and axonal support. Cold Spring Harb. Perspect. Biol. 2016, 8, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Khodanovich, M.Y.; Kisel, A.A.; Akulov, A.E.; Atochin, D.N.; Kudabaeva, M.S.; Glazacheva, V.Y.; Svetlik, M.V.; Medvednikova, Y.A.; Mustafina, L.R.; Yarnykh, V.L. Quantitative assessment of demyelination in ischemic stroke in vivo using macromolecular proton fraction mapping. J. Cereb. Blood Flow Metab. 2018, 38, 919–931. [Google Scholar] [CrossRef] [PubMed]
- Dewar, D.; Underhill, S.M.; Goldberg, M.P. Oligodendrocytes and ischemic brain injury. J. Cereb. Blood Flow Metab. 2003, 23, 263–274. [Google Scholar] [CrossRef] [Green Version]
- Pantoni, L.; Garcia, J.H.; Gutierrez, J.A. Cerebral white matter is highly vulnerable to ischemia. Stroke 1996, 27, 1641–1647. [Google Scholar] [CrossRef]
- Jing, L.; He, Q.; Zhang, J.Z.; Andy Li, P. Temporal profile of astrocytes and changes of oligodendrocyte-based myelin following middle cerebral artery occlusion in diabetic and non-diabetic rats. Int. J. Biol. Sci. 2013, 9, 190–199. [Google Scholar] [CrossRef]
- Han, C.W.; Lee, K.H.; Noh, M.G.; Kim, J.M.; Kim, H.S.; Kim, H.S.; Kim, R.G.; Cho, J.; Kim, H.I.; Lee, M.C. An experimental infarct targeting the internal capsule: Histopathological and ultrastructural changes. J. Pathol. Transl. Med. 2017, 51, 292–305. [Google Scholar] [CrossRef]
- McIver, S.R.; Muccigrosso, M.; Gonzales, E.R.; Lee, J.M.; Roberts, M.S.; Sands, M.S.; Goldberg, M.P. Oligodendrocyte degeneration and recovery after focal cerebral ischemia. Neuroscience 2010, 169, 1364–1375. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Hua, X.; Leak, R.; Shi, Y.; An, C.; Suenagac, J.; Chen, J.; Gao, Y. Demyelination as a Rational Therapeutic Target for Ischemic or Traumatic Brain Injury. Physiol. Behav. 2016, 176, 139–148. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Lee, M.; Hong, Y.; Won, J.; Lee, Y.; Kang, S.G.; Chang, K.T.; Hong, Y. Middle cerebral artery occlusion methods in rat versus mouse models of transient focal cerebral ischemic stroke. Neural Regen. Res. 2014, 9, 757–758. [Google Scholar] [CrossRef] [PubMed]
- Khodanovich, M.Y.; Kisel, A.A. Animal models of cerebral ischemia. AIP Conf. Proc. 2015, 1688, 1–8. [Google Scholar] [CrossRef]
- Kirino, T.; Tamura, A.; Sano, K. Delayed neuronal death in the rat hippocampus following transient forebrain ischemia. Acta Neuropathol. 1984, 64, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Kirino, T. Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res. 1982, 239, 57–69. [Google Scholar] [CrossRef]
- Nitatori, T.; Sato, N.; Waguri, S.; Karasawa, Y.; Araki, H.; Shibanai, K.; Kominami, E.; Uchiyama, Y. Delayed neuronal death in the CA1 pyramidal cell layer of the gerbil hippocampus following transient ischemia is apoptosis. J. Neurosci. 1995, 15, 1001–1011. [Google Scholar] [CrossRef] [Green Version]
- Atochin, D.N.; Chernysheva, G.A.; Aliev, O.I.; Smolyakova, V.I.; Osipenko, A.N.; Logvinov, S.V.; Zhdankina, A.A.; Plotnikova, T.M.; Plotnikov, M.B. An improved three-vessel occlusion model of global cerebral ischemia in rats. Brain Res. Bull. 2017, 132, 213–221. [Google Scholar] [CrossRef]
- Khodanovich, M.; Kisel, A.; Kudabaeva, M.; Chernysheva, G.; Smolyakova, V.; Krutenkova, E.; Wasserlauf, I.; Plotnikov, M.; Yarnykh, V. Effects of fluoxetine on hippocampal neurogenesis and neuroprotection in the model of global cerebral ischemia in rats. Int. J. Mol. Sci. 2018, 19, 162. [Google Scholar] [CrossRef] [Green Version]
- Khodanovich, M.Y.; Kisel’, A.A.; Chernysheva, G.A.; Smol’yakova, V.I.; Kudabaeva, M.S.; Krutenkova, E.P.; Tyumentseva, Y.; Plotnikov, M.B. p-Tyrosol Enhances the Production of New Neurons in the Hippocampal CA1 Field after Transient Global Cerebral Ischemia in Rats. Bull. Exp. Biol. Med. 2019, 168, 7–8. [Google Scholar] [CrossRef]
- Khodanovich, M.Y.; Kisel’, A.A.; Chernysheva, G.A.; Smol’yakova, V.I.; Savchenko, R.R.; Plotnikov, M.B. Effect of Fluoxetine on Neurogenesis in Hippocampal Dentate Gyrus after Global Transient Cerebral Ischemia in Rats. Bull. Exp. Biol. Med. 2016, 161, 351–354. [Google Scholar] [CrossRef]
- Martínez, N.S.; Machado, J.M.; Saad, H.P.; Antich, R.M.C.; Acosta, J.A.B.; Salgueiro, S.R.; Illera, G.G.; Alba, J.S.; del Barco Herrera, D.G. Global brain ischemia in Mongolian gerbils: Assessing the level of anastomosis in the cerebral circle of Willis. Acta Neurobiol. Exp. (Wars.) 2012, 72, 377–384. [Google Scholar]
- Squire, L.R.; Wixted, J.T. The Cognitive Neuroscience of Human Memory Since H.M. Annu. Rev. Neurosci. 2011, 34, 259–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Dede, A.J.O.; Hopkins, R.O.; Squire, L.R. Memory, scene construction, and the human hippocampus. Proc. Natl. Acad. Sci. USA 2015, 112, 4767–4772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Borst, G.; Thompson, W.L.; Hopkins, R.O.; Kosslyn, S.M.; Squire, L.R. Sparing of spatial mental imagery in patients with hippocampal lesions. Learn. Mem. 2013, 20, 657–663. [Google Scholar] [CrossRef] [Green Version]
- Zola-Morgan, S.; Squire, L.R.; Amaral, D.G. Human amnesia and the medial temporal region: Enduring memory impairment following a bilateral lesion limited to field CA1 of the hippocampus. J. Neurosci. 1986, 6, 2950–2967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rempel-Clower, N.L.; Zola, S.M.; Squire, L.R.; Amaral, D.G. Three cases of enduring memory impairment after bilateral damage limited to the hippocampal formation. J. Neurosci. 1996, 16, 5233–5255. [Google Scholar] [CrossRef] [PubMed]
- Ekstrom, D.K.J.; Diemer, N.H. Complete cerebral ischaemia in the rat: An ultrastructural and stereological analysis of the distal stratum radiatum in the hippocampal CA-1 region. Neuropathol. Appl. Neurol. 1982, 8, 197–215. [Google Scholar]
- Benveniste, H.; Jorgensen, M.B.; Sandberg, M.; Christensen, T.; Hagberg, H.; Diemer, N.H. Ischemic damage in hippocampal CA1 is dependent on glutamate release and intact innervation from CA3. J. Cereb. Blood Flow Metab. 1989, 9, 629–639. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, R.; Yamashiro, K.; Mochizuki, H.; Cho, N.; Onodera, M.; Mizuno, Y.; Urabe, T. Neurogenesis after transient global ischemia in the adult hippocampus visualized by improved retroviral vector. Stroke 2004, 35, 1454–1459. [Google Scholar] [CrossRef] [Green Version]
- Nakatomi, H.; Nakatomi, H.; Kuriu, T.; Kuriu, T.; Okabe, S.; Okabe, S.; Yamamoto, S.-I.; Yamamoto, S.-I.; Hatano, O.; Hatano, O.; et al. Regeneration of hippocampal pyramidal neurons afetr ischemic brain injury by recruitnt of endogenous neural progenitors. Cell 2002, 110, 429–441. [Google Scholar] [CrossRef] [Green Version]
- Kudabayeva, M.; Kisel, A.; Chernysheva, G.; Smol’Yakova, V.; Plotnikov, M.; Khodanovich, M. The increase in the number of astrocytes in the total cerebral ischemia model in rats. J. Phys. Conf. Ser. 2017, 886, 1–3. [Google Scholar] [CrossRef]
- Dohi, K.; Shioda, S.; Mizushima, H.; Homma, H.; Ozawa, H.; Nakai, Y.; Matsumoto, K. Delayed neuronal cell death and microglial cell reactivity in the CA1 region of the rat hippocampus in the cardiac arrest model. Med. Mol. Morphol. 1998, 31, 85–93. [Google Scholar] [CrossRef]
- Pforte, C.; Henrich-Noack, P.; Baldauf, K.; Reymann, K.G. Increase in proliferation and gliogenesis but decrease of early neurogenesis in the rat forebrain shortly after transient global ischemia. Neuroscience 2005. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Park, J.H.; Ahn, J.H.; Kim, I.H.; Cho, J.H.; Choi, J.H.; Yoo, K.; Lee, C.H.; Hwang, I.K.; Cho, J.H.; et al. New GABAergic Neurogenesis in the Hippocampal CA1 Region of a Gerbil Model of Long-Term Survival after Transient Cerebral Ischemic Injury. Brain Pathol. 2016, 581–592. [Google Scholar] [CrossRef]
- Johansen, F.F.; Balslev Jørgensen, M.; Diemer, N.H. Resistance of hippocampal CA-1 interneurons to 20 min of transient cerebral ischemia in the rat. Acta Neuropathol. 1983, 61, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Khodanovich, M.Y.; Pishchelko, A.O.; Glazacheva, V.Y.; Pan, E.S.; Krutenkova, E.P.; Trusov, V.B.; Yarnykh, V.L. Plant polyprenols reduce demyelination and recover impaired oligodendrogenesis and neurogenesis in the cuprizone murine model of multiple sclerosis. Phyther. Res. 2019, 33, 1363–1373. [Google Scholar] [CrossRef] [Green Version]
- Pulsinelli, W.A.; Brierley, J.B. A New Model of Bilateral Hemispheric Ischemia in the Unanesthetized Rat. Stroke 1979, 10, 267–272. [Google Scholar] [CrossRef] [Green Version]
- Radovsky, A.N.N.; Katz, L.; Ebmeyer, U.W.E.; Safar, P. Ischemic Neurons in Rat Brains After 6, 8, or 10 Minutes of Transient Hypoxic Ischemia. Toxicol. Pathol. 1997, 25, 500–505. [Google Scholar] [CrossRef]
- Böttiger, B.W.; Schmitz, B.; Wiessner, C.; Vogel, P.; Hossmann, K.A. Neuronal stress response and neuronal cell damage after cardiocirculatory arrest in rats. J. Cereb. Blood Flow Metab. 1998, 18, 1077–1087. [Google Scholar] [CrossRef] [Green Version]
- Shoykhet, M.; Simons, D.J.; Alexander, H.; Hosler, C.; Kochanek, P.M.; Clark, R.S.B. Thalamocortical dysfunction and thalamic injury after asphyxial cardiac arrest in developing rats. J. Neurosci. 2012, 32, 4972–4981. [Google Scholar] [CrossRef]
- Wahul, A.B.; Joshi, P.C.; Kumar, A.; Chakravarty, S. Transient global cerebral ischemia differentially affects cortex, striatum and hippocampus in Bilateral Common Carotid Arterial occlusion (BCCAo) mouse model. J. Chem. Neuroanat. 2018, 92, 1–15. [Google Scholar] [CrossRef]
- Paschen, W. Comparison of biochemical disturbances in hippocampal slices of gerbil and rat during and after in vitro ischemia. Neurosci. Lett. 1995, 199, 41–44. [Google Scholar] [CrossRef]
- Scotti, A.L.; Kalt, G.; Bollag, O.; Nitsch, C. Parvalbumin disappears from GABAergic CA1 neurons of the gerbil hippocampus with seizure onset while its presence persists in the perforant path. Brain Res. 1997, 760, 109–117. [Google Scholar] [CrossRef]
- Kang, T.C.; Park, S.K.; Bahn, J.H.; Chang, J.S.; Cho, S.W.; Choi, S.Y.; Won, M.H. Comparative studies on the GABA-transaminase immunoreactivity in rat and gerbil brains. Mol. Cells 2001, 11, 321–325. [Google Scholar] [PubMed]
- Butler, T.R.; Self, R.L.; Smith, K.J.; Sharrett-field, L.J.; Berry, J.N.; Littleton, J.M.; Pauly, J.R.; Mulholland, P.J.; Prendergast, A. Selective vulnerability of hippocampal cornu ammonis 1 pyramidal cells to excitotoxic insult is associated with the expression of polyamine-sensitive N-methyl- D-asparate-type glutamate receptors. Neuroscience 2010, 165, 525–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Megías, M.; Emri, Z.; Freund, T.F.; Gulyás, A.I. Total number and distribution of inhibitory and excitatory synapses on hippocampal CA1 pyramidal cells. Neuroscience 2001, 102, 527–540. [Google Scholar] [CrossRef]
- Deller, T.; Adelmann, G.; Nitsch, R.; Frotscher, M. The alvear pathway of the rat hippocampus. Cell Tissue Res. 1996, 286, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Migliore, R.; Lupascu, C.A.; Bologna, L.L.; Romani, A.; Courcol, J.D.; Antonel, S.; Van Geit, W.A.H.; Thomson, A.M.; Mercer, A.; Lange, S.; et al. The physiological variability of channel density in hippocampal CA1 pyramidal cells and interneurons explored using a unified data-driven modeling workflow. PLoS Comput. Biol. 2018, 14, 1–25. [Google Scholar] [CrossRef]
- Szilágyi, T.; Orbán-Kis, K.; Horváth, E.; Metz, J.; Pap, Z.; Pávai, Z. Morphological identification of neuron types in the rat hippocampus. Rom. J. Morphol. Embryol. 2011, 52, 15–20. [Google Scholar]
- Meier, S.; Bräuer, A.U.; Heimrich, B.; Nitsch, R.; Savaskan, N.E. Myelination in the hippocampus during development and following lesion. Cell. Mol. Life Sci. 2004, 61, 1082–1094. [Google Scholar] [CrossRef]
- Arszovszki, A.; Borhegyi, Z.; Klausberger, T. Three axonal projection routes of individual pyramidal cells in the ventral CA1 hippocampus. Front. Neuroanat. 2014, 8, 1–11. [Google Scholar] [CrossRef]
- Andersen, P.; Silfvenius, H.; Sundberg, S.H.; Sveen, O.; Wigstrom, H. Functional characteristics of unmyelinated fibres in the hippocampal cortex. Brain Res. 1978, 144, 19–29. [Google Scholar] [CrossRef]
- Jørgensen, M.B.; Finsen, B.R.; Jensen, M.B.; Castellano, B.; Diemer, N.H.; Zimmer, J. Microglial and Astroglial Reactions to Ischemic and Kainic Acid-Induced Lesions of the Adult Rat Hippocampus. Exp. Neurol. 1993, 120, 70–88. [Google Scholar] [CrossRef]
- Sugawara, T.; Leẃn, A.; Noshita, N.; Gasche, Y.; Chan, P.H. Effects of global ischemia duration on neuronal, astroglial, oligodendroglial, and microglial reactions in the vulnerable hippocampal CA1 subregion in rats. J. Neurotrauma 2002, 19, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Sadelli, K.; Stamegna, J.C.; Girard, S.D.; Baril, N.; Escoffier, G.; Brus, M.; Véron, A.D.; Khrestchatisky, M.; Roman, F.S. Global cerebral ischemia in rats leads to amnesia due to selective neuronal death followed by astroglial scar formation in the CA1 layer. Neurobiol. Learn. Mem. 2017, 141, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Holloway, O.G.; Canty, A.J.; King, A.E.; Ziebell, J.M. Rod microglia and their role in neurological diseases. Semin. Cell Dev. Biol. 2019, 94, 96–103. [Google Scholar] [CrossRef]
- Taylor, S.E.; Morganti-Kossmann, C.; Lifshitz, J.; Ziebell, J.M. Rod microglia: A morphological definition. PLoS ONE 2014, 9, e97096. [Google Scholar] [CrossRef]
- Au, N.P.B.; Ma, C.H.E. Recent advances in the study of bipolar/rod-shaped microglia and their roles in neurodegeneration. Front. Aging Neurosci. 2017, 9, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Bachstetter, A.D.; Ighodaro, E.T.; Hassoun, Y.; Aldeiri, D.; Neltner, J.H.; Patel, E.; Abner, E.L.; Nelson, P.T. Rod-shaped microglia morphology is associated with aging in 2 human autopsy series. Neurobiol. Aging 2017, 52, 98–105. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Wang, L.; Ju, F.; Ran, Y.; Wang, C.; Zhang, S. Transient global cerebral ischemia induces rapid and sustained reorganization of synaptic structures. J. Cereb. Blood Flow Metab. 2017, 37, 2756–2767. [Google Scholar] [CrossRef] [Green Version]
- Weinhard, L.; Di Bartolomei, G.; Bolasco, G.; Machado, P.; Schieber, N.L.; Neniskyte, U.; Exiga, M.; Vadisiute, A.; Raggioli, A.; Schertel, A.; et al. Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Henstridge, C.M.; Tzioras, M.; Paolicelli, R.C. Glial contribution to excitatory and inhibitory synapse loss in neurodegeneration. Front. Cell. Neurosci. 2019, 13, 1–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, K.; Akgul, G.; Wollmuth, L.P.; Tsirka, S.E. Microglia Actively Regulate the Number of Functional Synapses. PLoS ONE 2013, 8, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bechade, C.; Cantaut-belarif, Y.; Bessis, A. Microglial control of neuronal activity. Front. Cell. Neurosci. 2013, 7, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Du, X.; Liu, C.; Wen, Z.; Du, J. Reciprocal Regulation between Resting Microglial Dynamics and Neuronal Activity In Vivo. Dev. Cell 2012, 23, 1189–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef] [Green Version]
- Dawson, M.R.L.; Polito, A.; Levine, J.M.; Reynolds, R. NG2-expressing glial progenitor cells: An abundant and widespread population of cycling cells in the adult rat CNS. Mol. Cell. Neurosci. 2003, 24, 476–488. [Google Scholar] [CrossRef]
- Song, F.E.; Huang, J.L.; Lin, S.H.; Wang, S.; Ma, G.F.; Tong, X.P. Roles of NG2-glia in ischemic stroke. CNS Neurosci. Ther. 2017, 23, 547–553. [Google Scholar] [CrossRef]
- Zhao, B.; Zhao, C.Z.; Zhang, X.Y.; Huang, X.Q.; Shi, W.Z.; Fang, S.H.; Lu, Y.B.; Zhang, W.P.; Xia, Q.; Wei, E.Q. The new P2Y-like receptor G protein-coupled receptor 17 mediates acute neuronal injury and late microgliosis after focal cerebral ischemia in rats. Neuroscience 2012, 202, 42–57. [Google Scholar] [CrossRef]
- Tatsumi, K.; Takebayashi, H.; Manabe, T.; Tanaka, K.F.; Makinodan, M.; Yamauchi, T.; Makinodan, E.; Matsuyoshi, H.; Okuda, H.; Ikenaka, K.; et al. Genetic fate mapping of Olig2 progenitors in the injured adult cerebral cortex reveals preferential differentiation into astrocytes. J. Neurosci. Res. 2008, 86, 3494–3502. [Google Scholar] [CrossRef]
- Komitova, M.; Serwanski, D.R.; Richard Lu, Q.; Nishiyama, A. NG2 cells are not a major source of reactive astrocytes after neocortical stab wound injury. Glia 2011, 59, 800–809. [Google Scholar] [CrossRef] [Green Version]
- Heinrich, C.; Bergami, M.; Gascón, S.; Lepier, A.; Viganò, F.; Dimou, L.; Sutor, B.; Berninger, B.; Götz, M. Sox2-mediated conversion of NG2 glia into induced neurons in the injured adult cerebral cortex. Stem Cell Reports 2014, 3, 1000–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chernysheva, G.A.; Smol’yakova, V.I.; Osipenko, A.N.; Plotnikov, M.B. Evaluation of Survival and Neurological Deficit in Rats in the New Model of Global Transient Cerebral Ischemia. Bull. Exp. Biol. Med. 2014, 158, 197–199. [Google Scholar] [CrossRef] [PubMed]
- McGraw, C.P. Experimental Cerebral Infarction Effects of Pentobarbital in Mongolian Gerbils. Arch. Neurol. 1977, 34, 334–336. [Google Scholar] [CrossRef]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates; Academic Press: San Diego, CA, USA, 2007; ISBN 9780125476126. [Google Scholar]
- Otsu, N. A threshold selection method from gray-level histograms. IEEE Trans. Syst. Man Cybern. 1979, 9, 62–66. [Google Scholar] [CrossRef] [Green Version]
- Ercan, E.; Han, J.M.; Di Nardo, A.; Winden, K.; Han, M.-J.; Hoyo, L.; Saffari, A.; Leask, A.; Geschwind, D.H.; Sahin, M. Neuronal CTGF/CCN2 negatively regulates myelination in a mouse model of tuberous sclerosis complex. J. Exp. Med. 2017, 214, 681–697. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Total Number | Survival, % | Neurological Scores | ||
|---|---|---|---|---|---|
| 10 Days | 30 Days | 10 Days | 30 Days | ||
| Sham-operated | 10 | 100 | 100 | 0 | 0 |
| Ischemia | 19 | 52.6 | 47.4 | 6 (3–8) * | 4 (2–9) * |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anan’ina, T.; Kisel, A.; Kudabaeva, M.; Chernysheva, G.; Smolyakova, V.; Usov, K.; Krutenkova, E.; Plotnikov, M.; Khodanovich, M. Neurodegeneration, Myelin Loss and Glial Response in the Three-Vessel Global Ischemia Model in Rat. Int. J. Mol. Sci. 2020, 21, 6246. https://doi.org/10.3390/ijms21176246
Anan’ina T, Kisel A, Kudabaeva M, Chernysheva G, Smolyakova V, Usov K, Krutenkova E, Plotnikov M, Khodanovich M. Neurodegeneration, Myelin Loss and Glial Response in the Three-Vessel Global Ischemia Model in Rat. International Journal of Molecular Sciences. 2020; 21(17):6246. https://doi.org/10.3390/ijms21176246
Chicago/Turabian StyleAnan’ina, Tatiana, Alena Kisel, Marina Kudabaeva, Galina Chernysheva, Vera Smolyakova, Konstantin Usov, Elena Krutenkova, Mark Plotnikov, and Marina Khodanovich. 2020. "Neurodegeneration, Myelin Loss and Glial Response in the Three-Vessel Global Ischemia Model in Rat" International Journal of Molecular Sciences 21, no. 17: 6246. https://doi.org/10.3390/ijms21176246
APA StyleAnan’ina, T., Kisel, A., Kudabaeva, M., Chernysheva, G., Smolyakova, V., Usov, K., Krutenkova, E., Plotnikov, M., & Khodanovich, M. (2020). Neurodegeneration, Myelin Loss and Glial Response in the Three-Vessel Global Ischemia Model in Rat. International Journal of Molecular Sciences, 21(17), 6246. https://doi.org/10.3390/ijms21176246