Fanconi–Bickel Syndrome: A Review of the Mechanisms That Lead to Dysglycaemia



Abstract
:1. Introduction
2. Physiological Roles of GLUT2
2.1. Overview
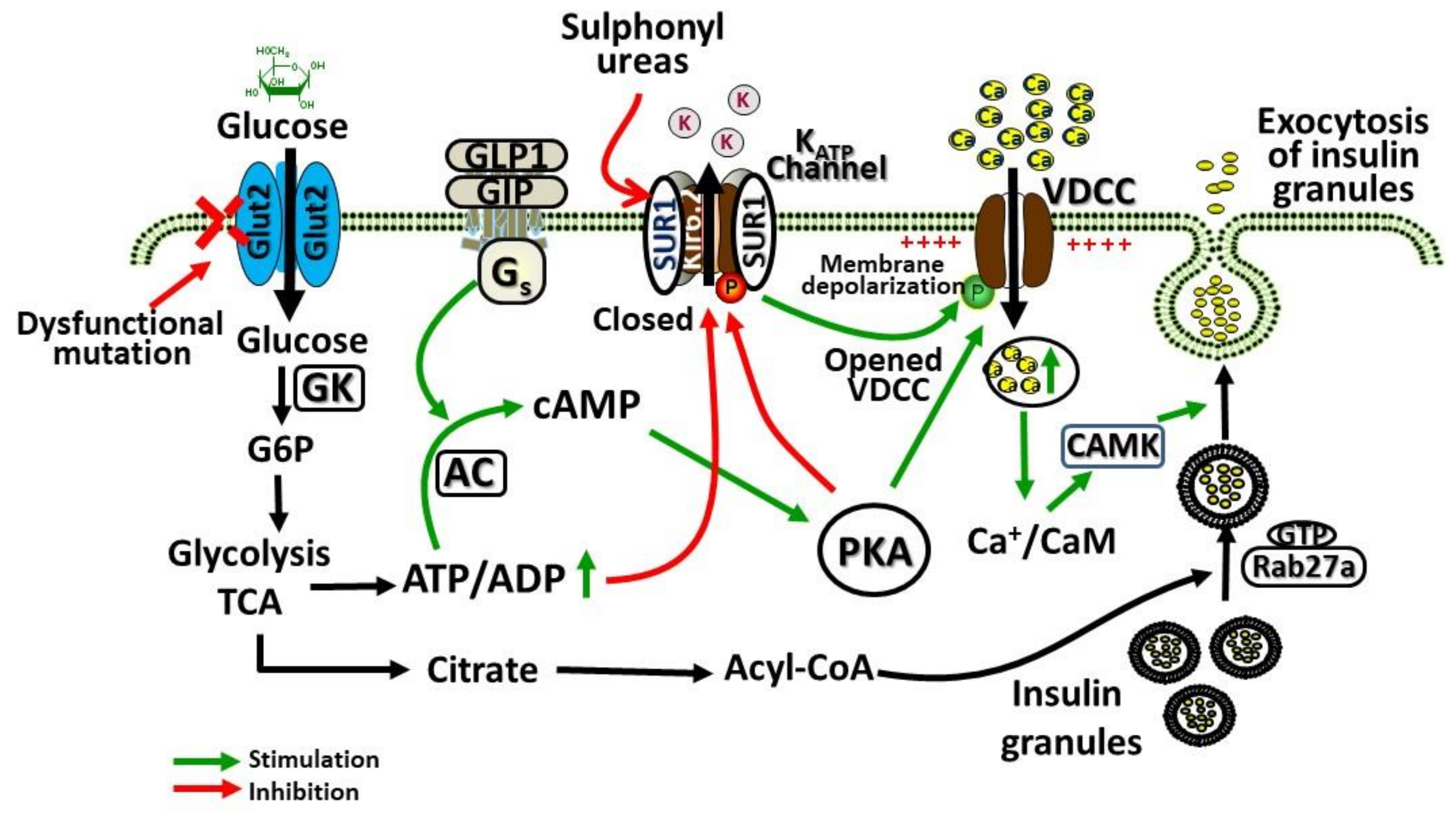
2.2. Role of GLUT2 in β-Cells
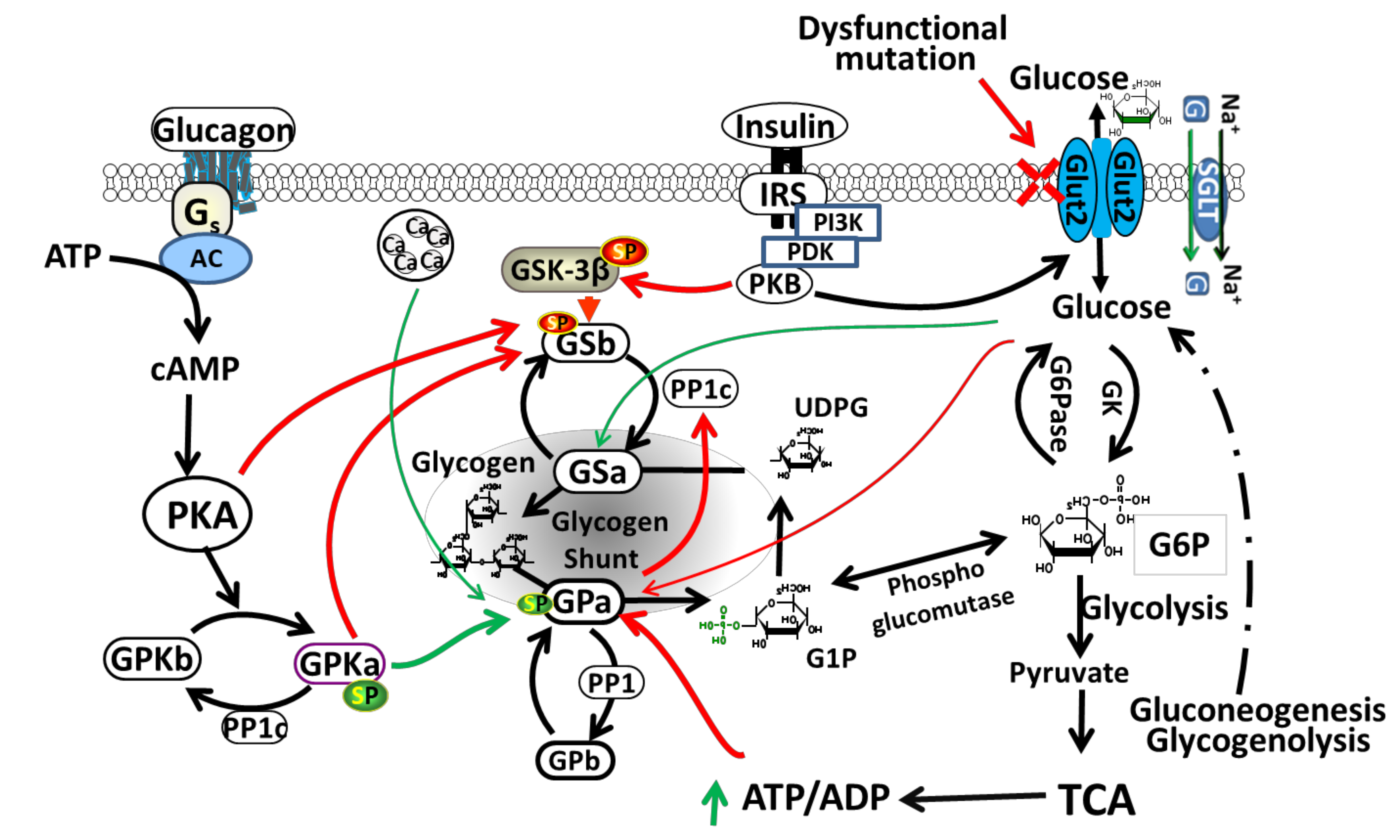
2.3. Role of GLUT2 in Liver
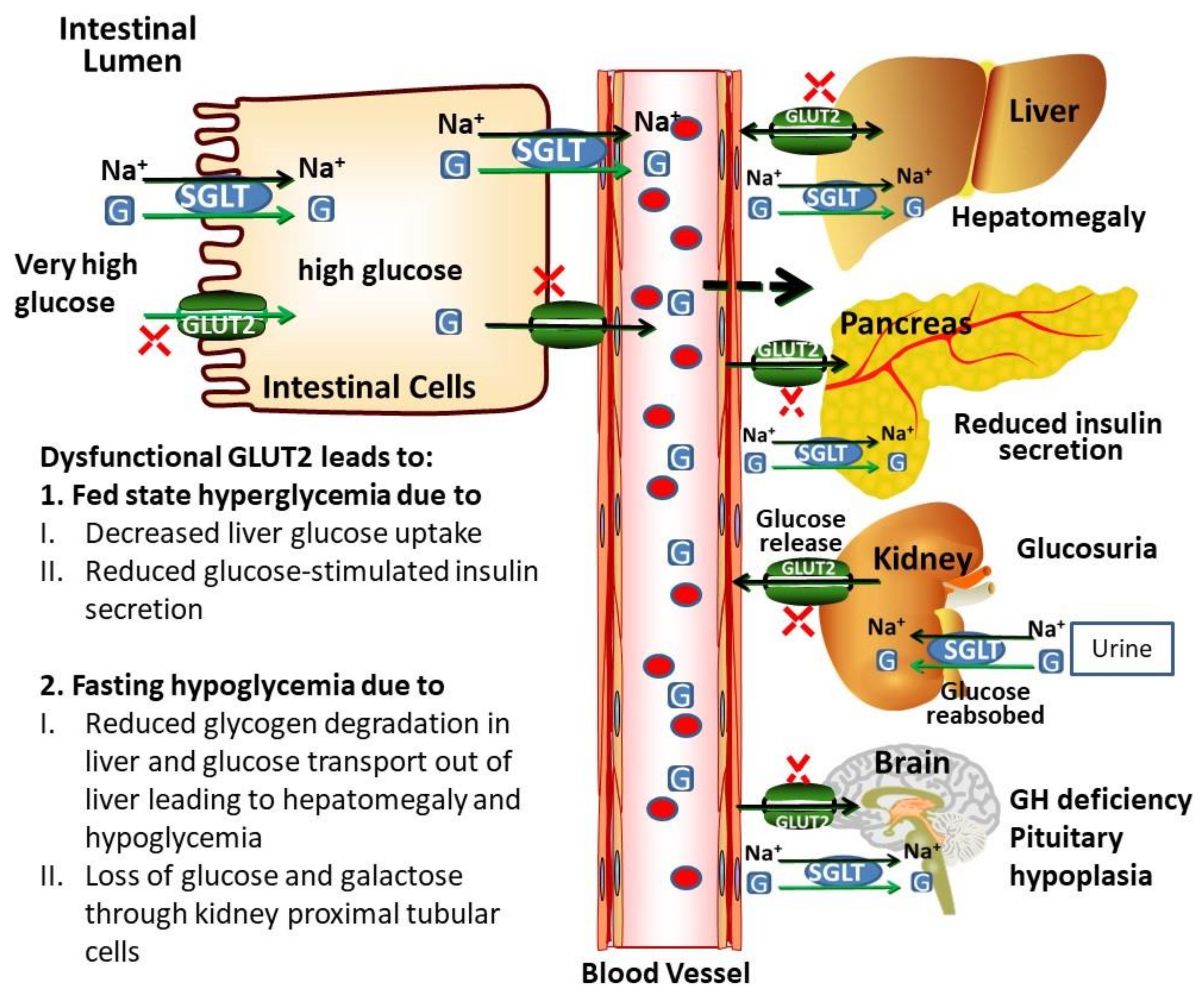
2.4. Role of GLUT2 in Kidney
2.5. Role of GLUT2 in Intestine
2.6. Role of GLUT2 in Brain
3. SLC2A2 (GLUT2) Mutations and Patterns of Dysglycaemia
3.1. Potential Biochemical Mechanisms Leading to Dysglycaemia in Patients with FBS
3.2. Birth Weight in FBS
3.3. Neonatal Diabetes in FBS
3.4. Frank Diabetes in FBS
3.5. Glycogen Storage in FBS
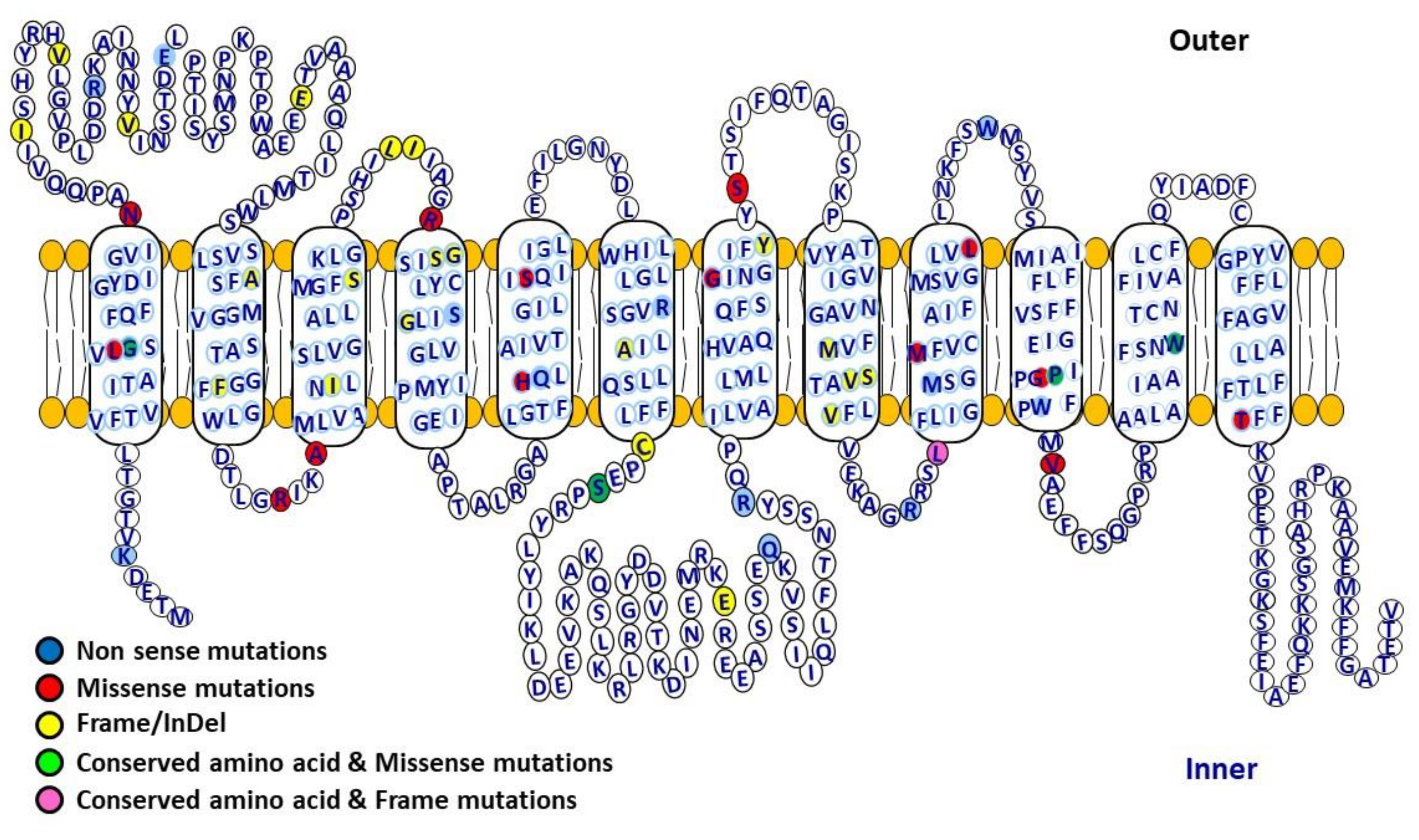
3.6. Structure Function Relationship of GLUT2 in FBS
4. Conclusions and Future Perspective
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| GLUT2 | Glucose transporter 2 |
| cAMP | Cyclic adenosine monophosphate |
| FBS | Fanconi–Bickel Syndrome |
| HbA1c | Haemoglobin-A1c |
| GP | Glycogen phosphorylase |
| PKA | Protein kinase A |
| G6P | Glucose-6-phosphate |
| GK | Glucokinase |
| G | Glucose |
| NR | Not Reported |
| aa | amino acid |
References
- Fanconi, G.; Bickel, H. [Chronic aminoaciduria (amino acid diabetes or nephrotic-glucosuric dwarfism) in glycogen storage and cystine disease]. Helv. Paediatr. Acta 1949, 4, 359–396. [Google Scholar] [PubMed]
- Manz, F.; Bickel, H.; Brodehl, J.; Feist, D.; Gellissen, K.; Gescholl-Bauer, B.; Gilli, G.; Harms, E.; Helwig, H.; Nutzenadel, W.; et al. Fanconi–Bickel syndrome. Pediatr. Nephrol. 1987, 1, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Permutt, M.A.; Kornyi, L.; Keller, K.; Lacy, P.E.; Scharp, D.W.; Mueckler, M. Cloning and functional expression of a human pancreatic islet glucose-transporter cDNA. Proc. Natl. Acad. Sci. USA 1989, 86, 8688–8692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueckler, M.; Kruse, M.; Strube, M.; Riggs, A.C.; Chiu, K.C.; Permutt, M.A. A mutation in the Glut2 glucose transporter gene of a diabetic patient abolishes transport activity. J. Biol. Chem. 1994, 269, 17765–17767. [Google Scholar]
- Tanizawa, Y.; Riggs, A.C.; Chiu, K.C.; Janssen, R.C.; Bell, D.S.; Go, R.P.; Roseman, J.M.; Acton, R.T.; Permutt, M.A. Variability of the pancreatic islet beta cell/liver (GLUT 2) glucose transporter gene in NIDDM patients. Diabetologia 1994, 37, 420–427. [Google Scholar] [CrossRef]
- Santer, R.; Schneppenheim, R.; Dombrowski, A.; Gotze, H.; Steinmann, B.; Schaub, J. Mutations in GLUT2, the gene for the liver-type glucose transporter, in patients with Fanconi–Bickel syndrome. Nat. Genet. 1997, 17, 324–326. [Google Scholar] [CrossRef]
- Mohandas Nair, K.; Sakamoto, O.; Jagadeesh, S.; Nampoothiri, S. Fanconi–Bickel syndrome. Indian J. Pediatr. 2012, 79, 112–114. [Google Scholar] [CrossRef]
- Al-Haggar, M. Fanconi–Bickel syndrome as an example of marked allelic heterogeneity. World J. Nephrol. 2012, 1, 63–68. [Google Scholar] [CrossRef]
- Batool, H.; Zubaida, B.; Hashmi, M.A.; Naeem, M. Genetic testing of two Pakistani patients affected with rare autosomal recessive Fanconi–Bickel syndrome and identification of a novel SLC2A2 splice site variant. J. Pediatr. Endocrinol. Metab. 2019, 32, 1229–1233. [Google Scholar] [CrossRef]
- Kehar, M.; Bijarnia, S.; Ellard, S.; Houghton, J.; Saxena, R.; Verma, I.C.; Wadhwa, N. Fanconi–Bickel syndrome–mutation in SLC2A2 gene. Indian J. Pediatr. 2014, 81, 1237–1239. [Google Scholar] [CrossRef] [Green Version]
- Fukumoto, H.; Seino, S.; Imura, H.; Seino, Y.; Eddy, R.L.; Fukushima, Y.; Byers, M.G.; Shows, T.B.; Bell, G.I. Sequence, tissue distribution, and chromosomal localization of mRNA encoding a human glucose transporter-like protein. Proc. Natl. Acad. Sci. USA 1988, 85, 5434–5438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, J.; Kayano, T.; Fukomoto, H.; Bell, G.I. Organization of the human GLUT2 (pancreatic beta-cell and hepatocyte) glucose transporter gene. Diabetes 1993, 42, 773–777. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Fritz, J.D.; Powers, A.C. Different functional domains of GLUT2 glucose transporter are required for glucose affinity and substrate specificity. Endocrinology 1998, 139, 4205–4212. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, J.; Benito, C.; Gomis, R. Pancreatic islet GLUT2 glucose transporter mRNA and protein expression in humans with and without NIDDM. Diabetes 1995, 44, 1369–1374. [Google Scholar] [CrossRef]
- McCulloch, L.J.; van de Bunt, M.; Braun, M.; Frayn, K.N.; Clark, A.; Gloyn, A.L. GLUT2 (SLC2A2) is not the principal glucose transporter in human pancreatic beta cells: Implications for understanding genetic association signals at this locus. Mol. Genet. Metab. 2011, 104, 648–653. [Google Scholar] [CrossRef]
- Kellett, G.L.; Brot-Laroche, E. Apical GLUT2: A major pathway of intestinal sugar absorption. Diabetes 2005, 54, 3056–3062. [Google Scholar] [CrossRef] [Green Version]
- Freitas, H.S.; Schaan, B.D.; Seraphim, P.M.; Nunes, M.T.; Machado, U.F. Acute and short-term insulin-induced molecular adaptations of GLUT2 gene expression in the renal cortex of diabetic rats. Mol. Cell Endocrinol. 2005, 237, 49–57. [Google Scholar] [CrossRef]
- Thorens, B.; Cheng, Z.Q.; Brown, D.; Lodish, H.F. Liver glucose transporter: A basolateral protein in hepatocytes and intestine and kidney cells. Am. J. Physiol. 1990, 259, C279–C285. [Google Scholar] [CrossRef]
- Leturque, A.; Brot-Laroche, E.; Le Gall, M.; Stolarczyk, E.; Tobin, V. The role of GLUT2 in dietary sugar handling. J. Physiol. Biochem. 2005, 61, 529–537. [Google Scholar] [CrossRef]
- Mounien, L.; Marty, N.; Tarussio, D.; Metref, S.; Genoux, D.; Preitner, F.; Foretz, M.; Thorens, B. Glut2-dependent glucose-sensing controls thermoregulation by enhancing the leptin sensitivity of NPY and POMC neurons. FASEB J. 2010, 24, 1747–1758. [Google Scholar] [CrossRef]
- Garcia, M.; Millan, C.; Balmaceda-Aguilera, C.; Castro, T.; Pastor, P.; Montecinos, H.; Reinicke, K.; Zuniga, F.; Vera, J.C.; Onate, S.A.; et al. Hypothalamic ependymal-glial cells express the glucose transporter GLUT2, a protein involved in glucose sensing. J. Neurochem. 2003, 86, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Thorens, B. Molecular and cellular physiology of GLUT-2, a high-Km facilitated diffusion glucose transporter. Int. Rev. Cytol. 1992, 137, 209–238. [Google Scholar] [PubMed]
- Berger, C.; Zdzieblo, D. Glucose transporters in pancreatic islets. Pflug. Arch. 2020, 1–24. [Google Scholar] [CrossRef]
- Thorens, B. GLUT2, glucose sensing and glucose homeostasis. Diabetologia 2015, 58, 221–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, F.Q.; Keating, A.F. Functional properties and genomics of glucose transporters. Curr. Genom. 2007, 8, 113–128. [Google Scholar] [CrossRef]
- Johnson, J.H.; Newgard, C.B.; Milburn, J.L.; Lodish, H.F.; Thorens, B. The high Km glucose transporter of islets of Langerhans is functionally similar to the low affinity transporter of liver and has an identical primary sequence. J. Biol. Chem. 1990, 265, 6548–6551. [Google Scholar]
- De Vos, A.; Heimberg, H.; Quartier, E.; Huypens, P.; Bouwens, L.; Pipeleers, D.; Schuit, F. Human and rat beta cells differ in glucose transporter but not in glucokinase gene expression. J. Clin. Investig. 1995, 96, 2489–2495. [Google Scholar] [CrossRef] [Green Version]
- Richardson, C.C.; Hussain, K.; Jones, P.M.; Persaud, S.; Löbner, K.; Boehm, A.; Clark, A.; Christie, M.R. Low levels of glucose transporters and K+ATP channels in human pancreatic beta cells early in development. Diabetologia 2007, 50, 1000–1005. [Google Scholar] [CrossRef] [Green Version]
- Mally, M.I.; Otonkoski, T.; Lopez, A.D.; Hayek, A. Developmental gene expression in the human fetal pancreas. Pediatr. Res. 1994, 36, 537–544. [Google Scholar] [CrossRef] [Green Version]
- Guillemain, G.; Loizeau, M.; Pinçon-Raymond, M.; Girard, J.; Leturque, A. The large intracytoplasmic loop of the glucose transporter GLUT2 is involved in glucose signaling in hepatic cells. J. Cell Sci. 2000, 113 Pt 5, 841–847. [Google Scholar]
- Stolarczyk, E.; Le Gall, M.; Even, P.; Houllier, A.; Serradas, P.; Brot-Laroche, E.; Leturque, A. Loss of sugar detection by GLUT2 affects glucose homeostasis in mice. PLoS ONE 2007, 2, e1288. [Google Scholar] [CrossRef] [Green Version]
- Gould, G.W.; Thomas, H.M.; Jess, T.J.; Bell, G.I. Expression of human glucose transporters in Xenopus oocytes: Kinetic characterization and substrate specificities of the erythrocyte, liver, and brain isoforms. Biochemistry 1991, 30, 5139–5145. [Google Scholar] [CrossRef] [PubMed]
- Thorens, B.; Sarkar, H.K.; Kaback, H.R.; Lodish, H.F. Cloning and functional expression in bacteria of a novel glucose transporter present in liver, intestine, kidney, and beta-pancreatic islet cells. Cell 1988, 55, 281–290. [Google Scholar] [CrossRef]
- Leturque, A.; Brot-Laroche, E.; Le Gall, M. GLUT2 mutations, translocation, and receptor function in diet sugar managing. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E985–E992. [Google Scholar] [CrossRef] [PubMed]
- Carabaza, A.; Ciudad, C.J.; Baque, S.; Guinovart, J.J. Glucose has to be phosphorylated to activate glycogen synthase, but not to inactivate glycogen phosphorylase in hepatocytes. FEBS Lett. 1992, 296, 211–214. [Google Scholar] [CrossRef] [Green Version]
- Seyer, P.; Vallois, D.; Poitry-Yamate, C.; Schütz, F.; Metref, S.; Tarussio, D.; Maechler, P.; Staels, B.; Lanz, B.; Grueter, R.; et al. Hepatic glucose sensing is required to preserve β cell glucose competence. J. Clin. Investig. 2013, 123, 1662–1676. [Google Scholar] [CrossRef] [Green Version]
- Burcelin, R.; del Carmen Muñoz, M.; Guillam, M.T.; Thorens, B. Liver hyperplasia and paradoxical regulation of glycogen metabolism and glucose-sensitive gene expression in GLUT2-null hepatocytes. Further evidence for the existence of a membrane-based glucose release pathway. J. Biol. Chem. 2000, 275, 10930–10936. [Google Scholar] [CrossRef] [Green Version]
- Guillam, M.T.; Burcelin, R.; Thorens, B. Normal hepatic glucose production in the absence of GLUT2 reveals an alternative pathway for glucose release from hepatocytes. Proc. Natl. Acad. Sci. USA 1998, 95, 12317–12321. [Google Scholar] [CrossRef] [Green Version]
- Hosokawa, M.; Thorens, B. Glucose release from GLUT2-null hepatocytes: Characterization of a major and a minor pathway. Am. J. Physiol. Endocrinol. Metab. 2002, 282, E794–E801. [Google Scholar] [CrossRef]
- Weinstein, S.P.; O’Boyle, E.; Fisher, M.; Haber, R.S. Regulation of GLUT2 glucose transporter expression in liver by thyroid hormone: Evidence for hormonal regulation of the hepatic glucose transport system. Endocrinology 1994, 135, 649–654. [Google Scholar] [CrossRef]
- Eisenberg, M.L.; Maker, A.V.; Slezak, L.A.; Nathan, J.D.; Sritharan, K.C.; Jena, B.P.; Geibel, J.P.; Andersen, D.K. Insulin receptor (IR) and glucose transporter 2 (GLUT2) proteins form a complex on the rat hepatocyte membrane. Cell Physiol. Biochem. 2005, 15, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rodriguez, A.; Nevado, C.; Escriva, F.; Sesti, G.; Rondinone, C.M.; Benito, M.; Valverde, A.M. PTP1B deficiency increases glucose uptake in neonatal hepatocytes: Involvement of IRA/GLUT2 complexes. Am. J. Physiol. Gastrointest Liver Physiol. 2008, 295, G338–G347. [Google Scholar] [CrossRef] [Green Version]
- Ghezzi, C.; Loo, D.D.F.; Wright, E.M. Physiology of renal glucose handling via SGLT1, SGLT2 and GLUT2. Diabetologia 2018, 61, 2087–2097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, M.A.; Forbes, J.M. Glucose and glycogen in the diabetic kidney: Heroes or villains? EBioMedicine 2019, 47, 590–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannstadt, M.; Magen, D.; Segawa, H.; Stanley, T.; Sharma, A.; Sasaki, S.; Bergwitz, C.; Mounien, L.; Boepple, P.; Thorens, B.; et al. Fanconi–Bickel syndrome and autosomal recessive proximal tubulopathy with hypercalciuria (ARPTH) are allelic variants caused by GLUT2 mutations. J. Clin. Endocrinol. Metab. 2012, 97, E1978–E1986. [Google Scholar] [CrossRef]
- Santer, R.; Schneppenheim, R.; Dombrowski, A.; Gotze, H.; Steinmann, B.; Schaub, J. Fanconi–Bickel syndrome--a congenital defect of the liver-type facilitative glucose transporter. SSIEM Award. Society for the Study of Inborn Errors of Metabolism. J. Inherit. Metab. Dis. 1998, 21, 191–194. [Google Scholar] [CrossRef]
- Santer, R.; Steinmann, B.; Schaub, J. Fanconi–Bickel syndrome--a congenital defect of facilitative glucose transport. Curr. Mol. Med. 2002, 2, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V. Glucose transporters in the kidney in health and disease. Pflugers Arch. 2020, 1–26. [Google Scholar] [CrossRef]
- Kellett, G.L.; Brot-Laroche, E.; Mace, O.J.; Leturque, A. Sugar absorption in the intestine: The role of GLUT2. Annu. Rev. Nutr. 2008, 28, 35–54. [Google Scholar] [CrossRef]
- Mace, O.J.; Affleck, J.; Patel, N.; Kellett, G.L. Sweet taste receptors in rat small intestine stimulate glucose absorption through apical GLUT2. J. Physiol. 2007, 582, 379–392. [Google Scholar] [CrossRef]
- Uldry, M.; Ibberson, M.; Hosokawa, M.; Thorens, B. GLUT2 is a high affinity glucosamine transporter. FEBS Lett. 2002, 524, 199–203. [Google Scholar] [CrossRef]
- Bady, I.; Marty, N.; Dallaporta, M.; Emery, M.; Gyger, J.; Tarussio, D.; Foretz, M.; Thorens, B. Evidence from glut2-null mice that glucose is a critical physiological regulator of feeding. Diabetes 2006, 55, 988–995. [Google Scholar] [CrossRef] [PubMed]
- Tarussio, D.; Metref, S.; Seyer, P.; Mounien, L.; Vallois, D.; Magnan, C.; Foretz, M.; Thorens, B. Nervous glucose sensing regulates postnatal β cell proliferation and glucose homeostasis. J. Clin. Investig. 2014, 124, 413–424. [Google Scholar] [CrossRef]
- Gözmen, S.K.; Çelik, K.; Çalkavur, S.; Serdaroğlu, E. A Novel Mutation in Fanconi Bickel Syndrome Diagnosed in the Neonatal Period. J. Pediatr. Res. 2019, 6, 155–157. [Google Scholar] [CrossRef]
- Khandelwal, P.; Sinha, A.; Jain, V.; Houghton, J.; Hari, P.; Bagga, A. Fanconi syndrome and neonatal diabetes: Phenotypic heterogeneity in patients with GLUT2 defects. CEN Case Rep. 2018, 7, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Pogoriler, J.; O’Neill, A.F.; Voss, S.D.; Shamberger, R.C.; Perez-Atayde, A.R. Hepatocellular Carcinoma in Fanconi–Bickel Syndrome. Pediatr. Dev. Pathol. 2018, 21, 84–90. [Google Scholar] [CrossRef] [Green Version]
- Tastemel-Ozturk, T.; Bilginer-Gurbuz, B.; Teksam, O.; Sivri, S. A Fanconi–Bickel syndrome patient with a novel mutation and accompanying situs inversus totalis. Turk. J. Pediatr. 2017, 59, 693–695. [Google Scholar] [CrossRef]
- Seker-Yilmaz, B.; Kor, D.; Bulut, F.D.; Yuksel, B.; Karabay-Bayazit, A.; Topaloglu, A.K.; Ceylaner, G.; Onenli-Mungan, N. Impaired glucose tolerance in Fanconi–Bickel syndrome: Eight patients with two novel mutations. Turk. J. Pediatr. 2017, 59, 434–441. [Google Scholar] [CrossRef] [Green Version]
- Shafeghati, Y.; Sarkheil, P.; Baghdadi, T.; Hadipour, F.; Hadipour, Z.; Noruzinia, M. Osteogenesis Imperfecta or Fanconi–Bickel Syndrome? (Report of a Very Rare Disease Due to New Mutation on GLUT2 Gene). Sarem J. Reprod. Med. 2017, 1, 73–76. [Google Scholar] [CrossRef] [Green Version]
- In, N.S.; Amaral., V.; Tzun., R.C.; Marques, J.S. Fanconi Syndrome: Report of 2 Cases. EC Pediatrics 2017, 4.6, 165–169. [Google Scholar]
- Amita, M.; Srivastava, P.; Mandal, K.; De, S.; Phadke, S.R. Fanconi–Bickel Syndrome: Another Novel Mutation in SLC2A2. Indian J. Pediatr. 2017, 84, 236–237. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, J.; Ahmad, I.; Ahmed, A.; Ahangar, A.A. Fanconi Bickel Syndrome: A Rare Entity. J. Clin. Case Rep. 2016, 6, 7. [Google Scholar] [CrossRef]
- Garg, M.; Gupta, A.; Mathur, P.; Sharma, M.; Kumar, R.; Gupta, V.; Manjunath, M. A rare case of Glycogen storage disease type XI Fanconi–Bickel Syndrome. J. Pediatric Crit. Care 2016, 3, 66–68. [Google Scholar] [CrossRef]
- Shah, R.; Rao, S.; Parikh, R.; Sophia, T.; Khalid, H. Fanconi Bickel Syndrome with Hypercalciuria due to GLUT 2 Mutation. Indian Pediatr. 2016, 53, 829–830. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Nambam, B.; Weinstein, D.A.; Shoemaker, L.R. Late Diagnosis of Fanconi–Bickel Syndrome: Challenges With the Diagnosis and Literature Review. J. Inborn Errors Metab. Screen. 2016, 4, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Dweikat, I.M.; Alawneh, I.S.; Bahar, S.F.; Sultan, M.I. Fanconi–Bickel syndrome in two Palestinian children: Marked phenotypic variability with identical mutation. BMC Res. Notes 2016, 9, 387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afroze, B.; Chen, M. Fanconi–Bickel Syndrome: Two Pakistani Patients Presenting with Hypophosphatemic Rickets. J. Pediatr. Genet 2016, 5, 161–166. [Google Scholar]
- Wang, W.; Wei, M.; Song, H.M.; Qiu, Z.Q.; Zhang, L.J.; Li, Z.; Tang, X.Y. SLC2A2 gene analysis in three Chinese children with Fanconi–Bickel syndrome. Zhongguo Dang Dai Er Ke Za Zhi 2015, 17, 362–366. [Google Scholar]
- Abbasi, F.; Azizi, F.; Javaheri, M.; Mosallanejad, A.; Ebrahim-Habibi, A.; Ghafouri-Fard, S. Segregation of a novel homozygous 6 nucleotide deletion in GLUT2 gene in a Fanconi–Bickel syndrome family. Gene 2015, 557, 103–105. [Google Scholar] [CrossRef]
- Mihout, F.; Devuyst, O.; Bensman, A.; Brocheriou, I.; Ridel, C.; Wagner, C.A.; Mohebbi, N.; Boffa, J.J.; Plaisier, E.; Ronco, P. Acute metabolic acidosis in a GLUT2-deficient patient with Fanconi–Bickel syndrome: New pathophysiology insights. Nephrol. Dial. Transpl. 2014, 29 (Suppl. 4), iv113–iv116. [Google Scholar] [CrossRef]
- Hadipour, F.; Sarkheil, P.; Noruzinia, M.; Hadipour, Z.; Baghdadi, T.; Shafeghati, Y. Fanconi–Bickel syndrome versus osteogenesis imperfeeta: An Iranian case with a novel mutation in glucose transporter 2 gene, and review of literature. Indian J. Hum. Genet 2013, 19, 84–86. [Google Scholar] [PubMed] [Green Version]
- Jahnavi, S.; Poovazhagi, V.; Mohan, V.; Bodhini, D.; Raghupathy, P.; Amutha, A.; Suresh Kumar, P.; Adhikari, P.; Shriraam, M.; Kaur, T.; et al. Clinical and molecular characterization of neonatal diabetes and monogenic syndromic diabetes in Asian Indian children. Clin. Genet 2013, 83, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Dayal, D.; Dekate, P.; Sharda, S.; Das, A.; Attri, S. An Indian girl with Fanconi–Bickel syndrome without SLC2A2 gene mutation. J. Pediatr. Genet 2013, 2, 109–112. [Google Scholar]
- Sansbury, F.H.; Flanagan, S.E.; Houghton, J.A.; Shuixian Shen, F.L.; Al-Senani, A.M.; Habeb, A.M.; Abdullah, M.; Kariminejad, A.; Ellard, S.; Hattersley, A.T. SLC2A2 mutations can cause neonatal diabetes, suggesting GLUT2 may have a role in human insulin secretion. Diabetologia 2012, 55, 2381–2385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Setoodeh, A.; Rabbani, A. Transient neonatal diabetes as a presentation of Fanconi- Bickel Syndrome. Acta Med. Iran 2012, 50, 836–838. [Google Scholar] [PubMed]
- Al-Haggar, M.; Sakamoto, O.; Shaltout, A.; Al-Hawari, A.; Wahba, Y.; Abdel-Hadi, D. Mutation analysis of the GLUT2 gene in three unrelated Egyptian families with Fanconi–Bickel syndrome: Revisited gene atlas for renumbering. Clin. Exp. Nephrol. 2012, 16, 604–610. [Google Scholar] [CrossRef]
- Al-Haggar, M.; Sakamoto, O.; Shaltout, A.; El-Hawary, A.; Wahba, Y.; Abdel-Hadi, D. Fanconi Bickel Syndrome: Novel Mutations in GLUT 2 Gene Causing a Distinguished Form of Renal Tubular Acidosis in Two Unrelated Egyptian Families. Case Rep. Nephrol. 2011, 2011, 754369. [Google Scholar] [CrossRef]
- Pena, L.; Charrow, J. Fanconi–Bickel syndrome: Report of life history and successful pregnancy in an affected patient. Am. J. Med. Genet. A 2011, 155a, 415–417. [Google Scholar] [CrossRef]
- Simsek, E.; Savas-Erdeve, S.; Sakamoto, O.; Doganci, T.; Dallar, Y. A novel mutation of the GLUT2 gene in a Turkish patient with Fanconi–Bickel syndrome. Turk. J. Pediatr. 2009, 51, 166–168. [Google Scholar]
- Hoffman, T.L.; Blanco, E.; Lane, A.; Galvin-Parton, P.; Gadi, I.; Santer, R.; DeLeon, D.; Stanley, C.; Wilson, T.A. Glucose metabolism and insulin secretion in a patient with ABCC8 mutation and Fanconi–Bickel syndrome caused by maternal isodisomy of chromosome 3. Clin. Genet. 2007, 71, 551–557. [Google Scholar] [CrossRef]
- Saltik-Temizel, I.N.; Coskun, T.; Yuce, A.; Kocak, N. Fanconi–Bickel syndrome in three Turkish patients with different homozygous mutations. Turk. J. Pediatr. 2005, 47, 167–169. [Google Scholar] [PubMed]
- Peduto, A.; Spada, M.; Alluto, A.; La Dolcetta, M.; Ponzone, A.; Santer, R. A novel mutation in the GLUT2 gene in a patient with Fanconi–Bickel syndrome detected by neonatal screening for galactosaemia. J. Inherit. Metab. Dis. 2004, 27, 279–280. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.W.; Shin, Y.L.; Seo, E.J.; Kim, G.H. Identification of a novel mutation in the GLUT2 gene in a patient with Fanconi–Bickel syndrome presenting with neonatal diabetes mellitus and galactosaemia. Eur. J. Pediatr. 2002, 161, 351–353. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, T.; Tamura, T.; Chinen, Y.; Ohta, T. A novel mutation (N32K) of GLUT2 gene in a Japanese patient with Fanconi–Bickel syndrome. Clin. Genet 2002, 62, 255–256. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, M.; Kitasawa, E.; Ida, H.; Eto, Y.; Owada, M. A newly recognized missense mutation in the GLUT2 gene in a patient with Fanconi–Bickel syndrome. Eur. J. Pediatr. 2000, 159, 867. [Google Scholar] [CrossRef]
- Sakamoto, O.; Ogawa, E.; Ohura, T.; Igarashi, Y.; Matsubara, Y.; Narisawa, K.; Iinuma, K. Mutation analysis of the GLUT2 gene in patients with Fanconi–Bickel syndrome. Pediatr. Res. 2000, 48, 586–589. [Google Scholar] [CrossRef] [Green Version]
- Akagi, M.; Inui, K.; Nakajima, S.; Shima, M.; Nishigaki, T.; Muramatsu, T.; Kokubu, C.; Tsukamoto, H.; Sakai, N.; Okada, S. Mutation analysis of two Japanese patients with Fanconi–Bickel syndrome. J. Hum. Genet 2000, 45, 60–62. [Google Scholar] [CrossRef]
- Burwinkel, B.; Sanjad, S.A.; Al-Sabban, E.; Al-Abbad, A.; Kilimann, M.W. A mutation in GLUT2, not in phosphorylase kinase subunits, in hepato-renal glycogenosis with Fanconi syndrome and low phosphorylase kinase activity. Hum. Genet 1999, 105, 240–243. [Google Scholar] [CrossRef]
- Santer, R.; Schneppenheim, R.; Suter, D.; Schaub, J.; Steinmann, B. Fanconi–Bickel syndrome--the original patient and his natural history, historical steps leading to the primary defect, and a review of the literature. Eur. J. Pediatr. 1998, 157, 783–797. [Google Scholar] [CrossRef]
- Müller, D.; Santer, R.; Krawinkel, M.; Christiansen, B.; Schaub, J. Fanconi–Bickel syndrome presenting in neonatal screening for galactosaemia. J. Inherit. Metab. Dis. 1997, 20, 607–608. [Google Scholar] [CrossRef]
- Lee, P.J.; Van’t Hoff, W.G.; Leonard, J.V. Catch-up growth in Fanconi–Bickel syndrome with uncooked cornstarch. J. Inherit. Metab. Dis. 1995, 18, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Karamizadeh, Z.; Saki, F.; Imanieh, M.H.; Zahmatkeshan, M.; Fardaee, M. A new mutation of Fanconi–Bickel syndrome with liver failure and pseudotumour cerebri. J. Genet. 2012, 91, 359–361. [Google Scholar] [CrossRef] [PubMed]
- Grunert, S.C.; Schwab, K.O.; Pohl, M.; Sass, J.O.; Santer, R. Fanconi–Bickel syndrome: GLUT2 mutations associated with a mild phenotype. Mol. Genet. Metab. 2012, 105, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Du, M.L.; Chen, H.S.; Chen, Q.L.; Yu, C.S.; Mal, H.M. Two cases of Fanconi–Bickel syndrome: First report from China with novel mutations of SLC2A2 gene. J. Pediatr. Endocrinol. Metab. 2011, 24, 749–753. [Google Scholar] [CrossRef]
- Roy, M.; Bose, K.; Paul, D.K.; Anand, P. Hypophosphatemic rickets: Presenting features of Fanconi–Bickel syndrome. Case Rep. Pathol. 2011, 2011, 314696. [Google Scholar] [CrossRef] [Green Version]
- Karande, S.; Kumbhare, N.; Kulkarni, M. Fanconi–Bickel syndrome. Indian Pediatr. 2007, 44, 223–225. [Google Scholar]
- Riva, S.; Ghisalberti, C.; Parini, R.; Furlan, F.; Bettinelli, A.; Somaschini, M. The Fanconi–Bickel syndrome: A case of neonatal onset. J. Perinatol. 2004, 24, 322–323. [Google Scholar] [CrossRef] [Green Version]
- Ozer, E.A.; Aksu, N.; Uclar, E.; Erdogan, H.; Bakiler, A.R.; Tsuda, M.; Kitasawa, E.; Coker, M.; Ozer, E. No mutation in the SLC2A2 ( GLUT2) gene in a Turkish infant with Fanconi–Bickel syndrome. Pediatr. Nephrol. 2003, 18, 397–398. [Google Scholar] [CrossRef]
- Santer, R.; Groth, S.; Kinner, M.; Dombrowski, A.; Berry, G.T.; Brodehl, J.; Leonard, J.V.; Moses, S.; Norgren, S.; Skovby, F.; et al. The mutation spectrum of the facilitative glucose transporter gene SLC2A2 (GLUT2) in patients with Fanconi–Bickel syndrome. Hum. Genet. 2002, 110, 21–29. [Google Scholar] [CrossRef]
- Muraoka, A.; Hashiramoto, M.; Clark, A.E.; Edwards, L.C.; Sakura, H.; Kadowaki, T.; Holman, G.D.; Kasuga, M. Analysis of the structural features of the C-terminus of GLUT1 that are required for transport catalytic activity. Biochem. J. 1995, 311, 699–704. [Google Scholar] [CrossRef] [Green Version]
- Taha, D.; Al-Harbi, N.; Al-Sabban, E. Hyperglycemia and hypoinsulinemia in patients with Fanconi–Bickel syndrome. J. Pediatr. Endocrinol. Metab. 2008, 21, 581–586. [Google Scholar] [PubMed]
- Ganesh, R.; Arvindkumar, R.; Vasanthi, T. Infantile-onset diabetes mellitus: A 1-year follow-up study. Clin. Pediatr. 2009, 48, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Berry, G.T.; Baker, L.; Kaplan, F.S.; Witzleben, C.L. Diabetes-like renal glomerular disease in Fanconi–Bickel syndrome. Pediatr. Nephrol. 1995, 9, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Garty, R.; Cooper, M.; Tabachnik, E. The Fanconi syndrome associated with hepatic glycogenosis and abnormal metabolism of galactose. J. Pediatr. 1974, 85, 821–823. [Google Scholar] [CrossRef]
- Guillam, M.T.; Hummler, E.; Schaerer, E.; Yeh, J.I.; Birnbaum, M.J.; Beermann, F.; Schmidt, A.; Deriaz, N.; Thorens, B. Early diabetes and abnormal postnatal pancreatic islet development in mice lacking Glut-2. Nat. Genet. 1997, 17, 327–330. [Google Scholar] [CrossRef]
- Lemelman, M.B.; Letourneau, L.; Greeley, S.A.W. Neonatal Diabetes Mellitus: An Update on Diagnosis and Management. Clin. Perinatol. 2018, 45, 41–59. [Google Scholar] [CrossRef]
- Saponaro, C.; Mühlemann, M.; Acosta-Montalvo, A.; Piron, A.; Gmyr, V.; Delalleau, N.; Moerman, E.; Thévenet, J.; Pasquetti, G.; Coddeville, A.; et al. Interindividual Heterogeneity of SGLT2 Expression and Function in Human Pancreatic Islets. Diabetes 2020, 69, 902–914. [Google Scholar] [CrossRef]
- Matsutani, A.; Koranyi, L.; Cox, N.; Permutt, M.A. Polymorphisms of GLUT2 and GLUT4 genes. Use in evaluation of genetic susceptibility to NIDDM in blacks. Diabetes 1990, 39, 1534–1542. [Google Scholar] [CrossRef]
- Lesage, S.; Zouali, H.; Vionnet, N.; Philippi, A.; Velho, G.; Serradas, P.; Passa, P.; Demenais, F.; Froguel, P. Genetic analyses of glucose transporter genes in French non-insulin-dependent diabetic families. Diabetes Metab. 1997, 23, 137–142. [Google Scholar]
- Dupuis, J.; Langenberg, C.; Prokopenko, I.; Saxena, R.; Soranzo, N.; Jackson, A.U.; Wheeler, E.; Glazer, N.L.; Bouatia-Naji, N.; Gloyn, A.L.; et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat. Genet 2010, 42, 105–116. [Google Scholar] [CrossRef]
- Laukkanen, O.; Lindström, J.; Eriksson, J.; Valle, T.T.; Hämäläinen, H.; Ilanne-Parikka, P.; Keinänen-Kiukaanniemi, S.; Tuomilehto, J.; Uusitupa, M.; Laakso, M. Polymorphisms in the SLC2A2 (GLUT2) gene are associated with the conversion from impaired glucose tolerance to type 2 diabetes: The Finnish Diabetes Prevention Study. Diabetes 2005, 54, 2256–2260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemachandra, A.H.; Richard, M.C. Neonatal Hyperglycemia. Pediatrics Rev. 1999, 20, e16–e24. [Google Scholar] [CrossRef]
- Santer, R.; Hillebrand, G.; Steinmann, B.; Schaub, J. Intestinal glucose transport: Evidence for a membrane traffic-based pathway in humans. Gastroenterology 2003, 124, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Stümpel, F.; Burcelin, R.; Jungermann, K.; Thorens, B. Normal kinetics of intestinal glucose absorption in the absence of GLUT2: Evidence for a transport pathway requiring glucose phosphorylation and transfer into the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2001, 98, 11330–11335. [Google Scholar] [CrossRef] [Green Version]
- Michau, A.; Guillemain, G.; Grosfeld, A.; Vuillaumier-Barrot, S.; Grand, T.; Keck, M.; L’Hoste, S.; Chateau, D.; Serradas, P.; Teulon, J.; et al. Mutations in SLC2A2 gene reveal hGLUT2 function in pancreatic beta cell development. J. Biol. Chem. 2013, 288, 31080–31092. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Number | Reference | Sex | Origin | Mutation | Amino Acid Change |
|---|---|---|---|---|---|
| 1 | [9] | F | Pakistani | c.497-2A > T | p.(Gly166_Ser169del)] |
| 2 | [9] | M | Pakistani | c.497-2A > T | p.(Gly166_Ser169del)] |
| 3 | [54] | F | Turkish | IVS8g.24401-24406del6 | NR |
| 4 | [55] | F | India | c.952G>A | p.Gly318Arg |
| 5 | [55] | M | India | c.952G>A | p.Gly318Arg |
| 6 | [55] | F | India | c.952G>A | p.Gly318Arg |
| 7 | [56] | M | Kuwait | c.474A>C | p. Arg158Ser |
| 8 | [57] | M | Turkish | c.108+5G>A | NR |
| 9 | [58] | M | Turkish | c.1069delGinsAATAA | p. Val357AsnfsTer37 |
| 10 | [58] | F | Turkish | c.482_483insC | p. Gly162ArgfsTer17 |
| 11 | [58] | NR | Turkish | c.482_483insC | p. Gly162ArgfsTer17 |
| 12 | [58] | M | Turkish | c.482_483insC | p. Gly162ArgfsTer17 |
| 13 | [58] | NR | Turkish | c.482_483insC | p. Gly162ArgfsTer17 |
| 14 | [58] | M | Turkish | c.482_483insC | p. Gly162ArgfsTer17 |
| 15 | [58] | M | Turkish | c.482_483insC | p. Gly162ArgfsTer17 |
| 16 | [58] | M | Turkish | c.575A>G | p. His192Arg |
| 17 | [59] | M | Iran | C.685_701del GCCATCCTTCAGTCTCT ins CAGAAAA | p.A229 QfsX19 |
| 18 | [60] | M | NR | NR | E85fsX177 and G189D |
| 19 | [61] | F | Indian | c.1246G>A | p. Gly416Ser |
| 20 | [60] | M | NR | NR | NR |
| 21 | [62] | M | Kashmir | NR | NR |
| 22 | [63] | F | Indian | NR | NR |
| 23 | [64] | M | Indian? | c.16-1G>A or IVS 1-1G>A | NR |
| 24 | [65] | F | African American | c.670 | Cys224Del |
| 25 | [66] | F | Palestinian | c.901C>T | p. Arg301Ter |
| 26 | [66] | M | Palestinian | c.901C>T | p. Arg301Ter |
| 27 | [67] | F | Pakistani | c.339delC | p. Phe114LeufsTer16 |
| 28 | [67] | M | Pakistani | c.339delC | p. Phe114LeufsTer16 |
| 29 | [68] | NR | Chinese | c.380C>A and c.970dupT | p.Ala127Asp and p.324TyrfsX392 |
| 30 | [68] | NR | Chinese | c.1068+5G>C | IVS8+5G>C |
| 31 | [68] | NR | Chinese | c.1194T>A | p.Tyr398X |
| 32 | [69] | F | Iranian | c.1061_1066del6 | p.V355_S356del2 |
| 33 | [69] | M | Iranian | c.1061_1066del6 | p.V355_S356del2 |
| 34 | [70] | M | Algerian | IVS 3+2T>C/IVS 3+2T>C | NR |
| 35 | [10] | M | Indian | c.1330T>C | p. W444R |
| 36 | [71] | F | Iranian | c. 685_701del GCCATCCTTCAGTCTCTins CAGAAAA | P.A229QFsX19 |
| 37 | [72] | F | Indian | c.56T>C | p. Leu19Pro |
| 38 | [73] | F | Indian | NR | NR |
| 39 | [74] | F | Chinese | c.609T>A | p.Ser203Arg |
| 40 | [74] | M | Oman | c.1127T>G | p.Met376Arg |
| 41 | [74,75] | F | Iran | c.963+1G>A | NR |
| 42 | [74] | M | Sudanese | c.157C>T | p.Arg53X |
| 43 | [74] | M | Saudi Arabia | c.339del | p.Phe114LeufsX16 |
| 44 | [76] | F | Egyptian | c.1250C>T | p. P417L |
| 45 | [76] | M | Egyptian | c.253_254del GA | p. Glu85fs |
| 46 | [77] | M | Egyptian | c.776-1G>C | NR |
| 47 | [78] | F | Caucasian | c.1439C>G and c.1469delA | T480R and L490SfsX24 |
| 48 | [79] | M | Turkish | c.835_836delGA | p.E279KfsX6 |
| 49 | [80] | F | NR | c.1213C>T | NR |
| 50 | [81] | M | Turkish | 783del17 | NR |
| 51 | [81] | F | Turkish | c.818C>G | NR |
| 52 | [81] | F | Turkish | IVS5+1 G>T | NR |
| 53 | [82] | M | Italian | 425_7/delTAA | NR |
| 54 | [83] | F | Korean | c.322A>T | K5X |
| 55 | [84] | F | Japanese | c.96T>G | N32K |
| 56 | [85] | M | Japanese | c.1093 C>T and c.1642 T>C | p. Arg365Ter and p. Trp444Arg |
| 57 | [86] | F | Japanese | nt 1580T>A | V423E |
| 58 | [86] | M | Japanese | IVS2–2A>G | NR |
| 59 | [86] | M | Japanese | c.1171C>T and c.1478T>C | Q287X and L389P |
| 60 | [87] | M | Japanese | c.1159G>A | W420X |
| 61 | [87] | F | Japanese | NR | NR |
| 62 | [88] | M | Saudi-Arabian | c.1250 C>T | Pro417Leu |
| 63 | NR | Arabian | c.1250C>T | NR | |
| 64 | NR | Arabian | c.1250 C>T | NR | |
| 65 | NR | Arabian | c.1250 C>T | NR | |
| 66 | NR | Arabian | c.1250 C>T | NR | |
| 67 | NR | Arabian | c.1250 C>T | NR | |
| 68 | [6,89] | M | Swiss | c.1251C>T or 1213 C>T | R301X |
| 69 | [6] | F | NR | ΔT446-449 | |
| 70 | [6] | M | NR | ΔT446-449 | |
| 71 | [6] | M | NR | c.1405C>T | R365X |
| 72 | [90] | M | Japanese | c.1171C>T and c.1478 T>C | NR |
| 73 | [91] | M | Caucasian | NR | NR |
| 74 | [91] | M | Caucasian | NR | NR |
| 75 | [4] | NR | Japanese | c.1571G>A | NR |
| 76 | [2] | F | Arabian | c.1562C>T | NR |
| 77 | M | Arabian | c.1562C>T | NR | |
| 78 | M | Arabian | c.1562C>T | NR | |
| 79 | NR | Arabian | c.1562C>T | NR | |
| 80 | NR | Arabian | c.1562C>T | NR | |
| 81 | NR | Arabian | c.1562C>T | NR | |
| 82 | NR | Arabian | c.1562C>T | NR | |
| 83 | NR | Arabian | c.1562C>T | NR | |
| 84 | NR | Arabian | c.1562C>T | NR | |
| 85 | [92] | M | Iran | IVS8+1 G>T | NR |
| 86 | [93] | M | Caucasian | c.457_462delCTTATA and c.1250C>G | p.153_4delLI and p.P417R |
| 87 | [93] | F | Caucasian | c.457_462delCTTATA and c.1250C>G | p.153_4delLI and p.P417R |
| 88 | [45] | M | Dominican Republic | IVS 4-2A>G | p. Gln166AspfsTer4 |
| 89 | [45] | M | Dominican Republic | IVS 4-2A>G | p. Gln166AspfsTer4 |
| 90 | [45] | F | Israeli | c.372A>C | p. Arg124Ser |
| 91 | [45] | M | Israeli | c.372A>C | p. Arg124Ser |
| 92 | [45] | F | Israeli | c.372A>C | p. Arg124Ser |
| 93 | [94] | F | Chinese Han | c.682C>T and c.1185 G>A | p. Arg228X and p. Trp395X |
| 94 | [94] | M | Chinese Han/Yao | c.196G>T and c.1117delA | p. Glu66X and p. Met373X |
| 95 | [95] | M | NR | NR | NR |
| 96 | [80] | F | NR | c.1213C>T | NR |
| 97 | [96] | F | Indian | NR | NR |
| 98 | [97] | M | Arab | c.1213C>T | p.Phe405>Leu |
| 99 | [98] | F | Turkish | NR | NR |
| 100 | [99] | F | German | NR | NR |
| 101 | M | German | 627 delAGTTGGTGins GT | NR | |
| 102 | M | Turkish-Assyrian | 793–4 ins C | NR | |
| 103 | F | Turkish-Assyrian | 793–4 ins C | NR | |
| 104 | M | German | NR | NR | |
| 105 | M | German | NR | NR | |
| 106 | M | Italian (?) | c.1213C>T | NR | |
| 107 | F | Italian (?) | c.889C>T | NR | |
| 108 | M | English | 1363 del G and 1405C>T | NR | |
| 109 | M | English | 1364 del G and 1405C>T | NR | |
| 110 | M | Caucasian | 1264 G>A and 469C>T | NR | |
| 111 | M | Turkish | 449 del T | NR | |
| 112 | F | Turkish | 450 del T | NR | |
| 113 | M | Turkish | c.1405C>T | NR | |
| 114 | M | Caucasian | c.1405C>T and 1008 ins A | NR | |
| 115 | M | Arabian | c.1213C>T | NR | |
| 116 | M | Polish (?) | NR | NR | |
| 117 | F | Polish (?) | c.469C>T | NR | |
| 118 | F | Polish (?) | NR | NR | |
| 119 | M | Algerian | IVS 6 +1 G>C | NR | |
| 120 | M | Moroccan | 1288–9TC>AA | NR | |
| 121 | F | NR | c.1562C>T and IVS 8 + | NR | |
| 122 | 1 G>A | NR | |||
| 123 | M | Algerian | IVS 5 +5 G>C | NR | |
| 124 | M | Algerian | IVS 5 +5 G>C | NR | |
| 125 | F | French (?) | 1573 ins GT and 1751C>G | NR | |
| 126 | M | French (?) | 1574 ins GT and 1751C>G | NR | |
| 127 | F | Italian | 1264G>A | NR | |
| 128 | F | Italian | 371G>A and 1751C>G | NR | |
| 129 | M | Turkish | 1562C>T | NR | |
| 130 | F | NR | IVS 6 +1 g>a | NR | |
| 131 | M | NR | NR | NR | |
| 132 | M | NR | NR | NR | |
| 133 | F | French-Canadian | 494 ins 26 and 1751C>G | NR | |
| 134 | F | Eskimo | 1415–6 del TC | NR | |
| 135 | M | Arabian | 1213C>T | NR | |
| 136 | F | NR | IVS 8 +1 G>A | NR | |
| 137 | F | NR | NR | NR | |
| 138 | F | Turkish-Assyrian | 793–4 ins C | NR | |
| 139 | F | Greek | 712–3 del CT | NR | |
| 140 | M | Algerian | IVS 3 +2 T>C | NR | |
| 141 | M | NR | 1092C>A | NR | |
| 142 | M | Turkish | 738 del 17 | NR | |
| 143 | F | Turkish | IVS 5 +1 G>T | NR | |
| 144 | [100] | M | Japanese | c.1405C>T and c.1642T>C | NR |
| Missense Mutations | Nonsense Mutations | fs/indel Mutations | Intronic Mutations | Compound Heterozygous Mutations |
|---|---|---|---|---|
| G20E | K5X | I39 | c.497-2A>T | p.Ala127Asp and p.324TyrfsX392 |
| N32K | R53X | L153_I154 | c.108+5G>A | p. Arg365Ter and p. Trp444Arg |
| R158S | E66X | C239 | IVS 3+2T>C | Q287X and L389P |
| S203R | S169X | V355_S356 | c.963+1G>A | Gly20Glu and T480R |
| S242R | Q193X | V45 | c.776-1G>C | T480R and L490SfsX24 |
| G318R | R228X | V60 | IVS5+1 G>T | p.153_4delLI and p.P417R |
| S326K | Q287X | A105 | IVS 2 - 2 A>G | E85fsX177 and G189D |
| M376R | R301X | I133 | IVS4+1G>A | p. Arg228X and p. Trp395X |
| L389P | R365X | S145 | IVS 8+1 G>T | p. Glu66X and p. Met373X |
| G416S | W420X | S161 | IVS 5-1 G>A | |
| P417L | M350 | c.16-1G>A or IVS 1-1G>A | ||
| P417R | L368 | IVS8g.24401-24406del6 | ||
| V423E | W420 | c.1068+5 G>C | ||
| W444R | Glu85fs | |||
| T480R | E279KfsX6 | |||
| His192Arg | Val357AsnfsTer37 | |||
| Arg124Ser | Gly162ArgfsTer17 | |||
| Leu19Pro ** | A229QFsX19 | |||
| p.Phe405>Leu | Phe114LeufsX16 | |||
| Gln166AspfsTer4 | ||||
| Cys224Del |
| Type of Dysglycaemia | Mutation | Amino Acid Change | Reference |
|---|---|---|---|
| Transient neonatal diabetes | c.952G>A | p.Gly318Arg | [55] |
| c.609 T>A | p.Ser203Arg | [74] | |
| c.1127 T>G | p.Met376Arg | [74] | |
| c.963+1G>A | NR | [74] | |
| c.157C>T | p.Arg53X | [74] | |
| c.339del | p.Phe114LeufsX16 | [74] | |
| 322 A>T | K5X | [83] | |
| Glucose intolerance/diabetes mellitus | c.482_483insC | p. Gly162ArgfsTer17 | [58] |
| c.575A>G | p. His192Arg | [58] | |
| c.56 T>C | p. Leu19Pro ** | [72] | |
| Gestational diabetes | c.1439C>G and c.1469delA | T480R and L490SfsX24 | [78] |
| NR | valine 197 to isoleucine | [4] | |
| Other: 2 patients [101], and 1 patient [102] | |||
| Fasting hypoglycemia | c.108+5G>A | NR | [57] |
| 783del17 | NR | [81] | |
| 818C>G | NR | [81] | |
| IVS5+1 G>T | NR | [81] | |
| c.1580T>A | V423E | [86] | |
| IVS 2 - 2 A>G | NR | [86] | |
| c.952G>A | p.Gly318Arg | [55] | |
| NR | E85fsX177 and G189D | [60] | |
| NR | NR | [62] | |
| NR | NR | [91] | |
| NR | NR | [103] | |
| c.1246 G>A | p. Gly416Ser | [61] | |
| c.339delC | p. Phe114LeufsTer16 | [67] | |
| c.339delC | p. Phe114LeufsTer16 | [67] | |
| Post-prandial hyperglycemia | c.1061_1066del6 | p.V355_S356del2 | [69] |
| IVS8g.24401-24406del6 | NR | [54] | |
| NR | NR | [73] | |
| NR | NR | [96] | |
| Fasting hypoglycemia and postprandial hyperglycemia | c.1069delGinsAATAA | p. Val357AsnfsTer37 | [58] |
| c.482_483insC | p. Gly162ArgfsTer17 | [58] | |
| c.482_483insC | p. Gly162ArgfsTer17 | [58] | |
| c.482_483insC | p. Gly162ArgfsTer17 | [58] | |
| c.482_483insC | p. Gly162ArgfsTer17 | [58] | |
| c.482_483insC | p. Gly162ArgfsTer17 | [58] | |
| c.901C > T | p. Arg301Ter | [66] | |
| c.16-1G>A or IVS 1-1G>A | [64] | ||
| c.380C>A and c.970dupT | p.Ala127Asp and p.324TyrfsX392 | [68] | |
| c.1068+5 G>C | IVS8+5G>C | [68] | |
| c.1194T>A | p.Tyr398X | [68] | |
| c.1250C>T | p. P417L | [76] | |
| IVS 3+2t>c/IVS 3+2t>c) | NR | [70] | |
| c. 685_70l del GCCATCCTTCAGTCTCTins CAGAAAA | P.A229QFsX19 | [71] | |
| c.253_254delGA | p. Glu85fs | [77] | |
| c.776-1G>C | NR | [77] | |
| c.835_836delGA | p.E279KfsX6 | [79] | |
| C1213T | NR | [80] | |
| 96T>G | N32K | [84] | |
| c.1171C>T and c.1478T>C | Q287X and L389P | [86] | |
| c.1213C>T | R301X | [89] | |
| ΔT446-449 | [6] | ||
| ΔT446-449 | [6] | ||
| C1405T | R365X | [6] | |
| c.1213 C>T | p.Phe405>Leu | [97] | |
| C.685_701del GCCATCCTTCAGTCTCT ins CAGAAAA | p.A229 QfsX19 | [59] | |
| c.670 | Cys224Del | [65] | |
| c.682C>T and c.1185 G>A | p. Arg228X and p. Trp395X | [94] | |
| c.196G>T and c.1117delA | p. Glu66X and p. Met373X | [94] | |
| c.1330 T>C | p. W444R | [10] |
| Mutation | Birth Weight (kg) | References | |
|---|---|---|---|
| DNA | Protein | ||
| Missense | |||
| c.609T>A | p.Ser203Arg | 1.85 | [74] |
| c.1127T>G | p.Met376Arg | 2.5 | [74] |
| c.952G>A | p.Gly318>Arg | 2.4 | [55] |
| c.952G>A | p.Gly318>Arg | 2.8 | [55] |
| c.952G>A | p.Gly318>Arg | 2.3 | [55] |
| c.1213C>T | p.Phe405>Leu | 3.23 | [97] |
| Non-sense | |||
| c.901C>T | p.Arg301X | 2.8 | [66] |
| c.901C>T | p.Arg301X | 2.2 | [66] |
| fs/indel | |||
| c.322A>T | p.Lys5>X | 2 | [83] |
| c.339delC | p.Phe114LeufsX16 | 2.5 | [74] |
| c. 685_701 del GCCATCCTTCAGTCTCT ins CAGAAAA | P.A229QFsX19 | 2.6 | [71] |
| c.1069delGinsAATAA | p. Val357AsnfsTer37 | 3 | [58] |
| C.685_701del GCCATCCTTCAGTCTCT ins CAGAAAA | p.A229 QfsX19 | 2.6 | [59] |
| c.783del17 | 2.6 | [81] | |
| c.670 | Cys224Del | 2.09 | [65] |
| Intronic | |||
| c.963+1G>A | NR | 2 | [74] |
| c.16-1G>A or IVS 1- 1G>A | NR | 2.5 | [64] |
| IVS8 g.24401-24406del6 | NR | 2.6 | [54] |
| c.963+1 G>A | NR | 2.05 | [75] |
| (IVS2+5G>A[c.108+5G>A]) | NR | 3.25 | [57] |
| Compound Heterozygous | |||
| c.457_462delCTTATA in Exon 4 and c.1250C>G in Exon 10 | (p.153_4delLI) and (p.Pro417Arg) | 3.773.97 | [93] |
| E85fsX177 and G189D | 3.0 | [60] | |
| Undefined | |||
| 2.8 | [62] | ||
| 2.5 | [63] | ||
| 2.9 | [73] | ||
| 2.1 | [96] | ||
| 2.5 | [95] | ||
| 2.8 | [104] | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharari, S.; Abou-Alloul, M.; Hussain, K.; Ahmad Khan, F. Fanconi–Bickel Syndrome: A Review of the Mechanisms That Lead to Dysglycaemia. Int. J. Mol. Sci. 2020, 21, 6286. https://doi.org/10.3390/ijms21176286
Sharari S, Abou-Alloul M, Hussain K, Ahmad Khan F. Fanconi–Bickel Syndrome: A Review of the Mechanisms That Lead to Dysglycaemia. International Journal of Molecular Sciences. 2020; 21(17):6286. https://doi.org/10.3390/ijms21176286
Chicago/Turabian StyleSharari, Sanaa, Mohamad Abou-Alloul, Khalid Hussain, and Faiyaz Ahmad Khan. 2020. "Fanconi–Bickel Syndrome: A Review of the Mechanisms That Lead to Dysglycaemia" International Journal of Molecular Sciences 21, no. 17: 6286. https://doi.org/10.3390/ijms21176286
APA StyleSharari, S., Abou-Alloul, M., Hussain, K., & Ahmad Khan, F. (2020). Fanconi–Bickel Syndrome: A Review of the Mechanisms That Lead to Dysglycaemia. International Journal of Molecular Sciences, 21(17), 6286. https://doi.org/10.3390/ijms21176286