Repurposing Quinacrine for Treatment of Malignant Mesothelioma: In-Vitro Therapeutic and Mechanistic Evaluation

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Evaluation of Therapeutic Efficacy of Quinacrine (QA) in Treatment of Malignant Mesothelioma (MPM)
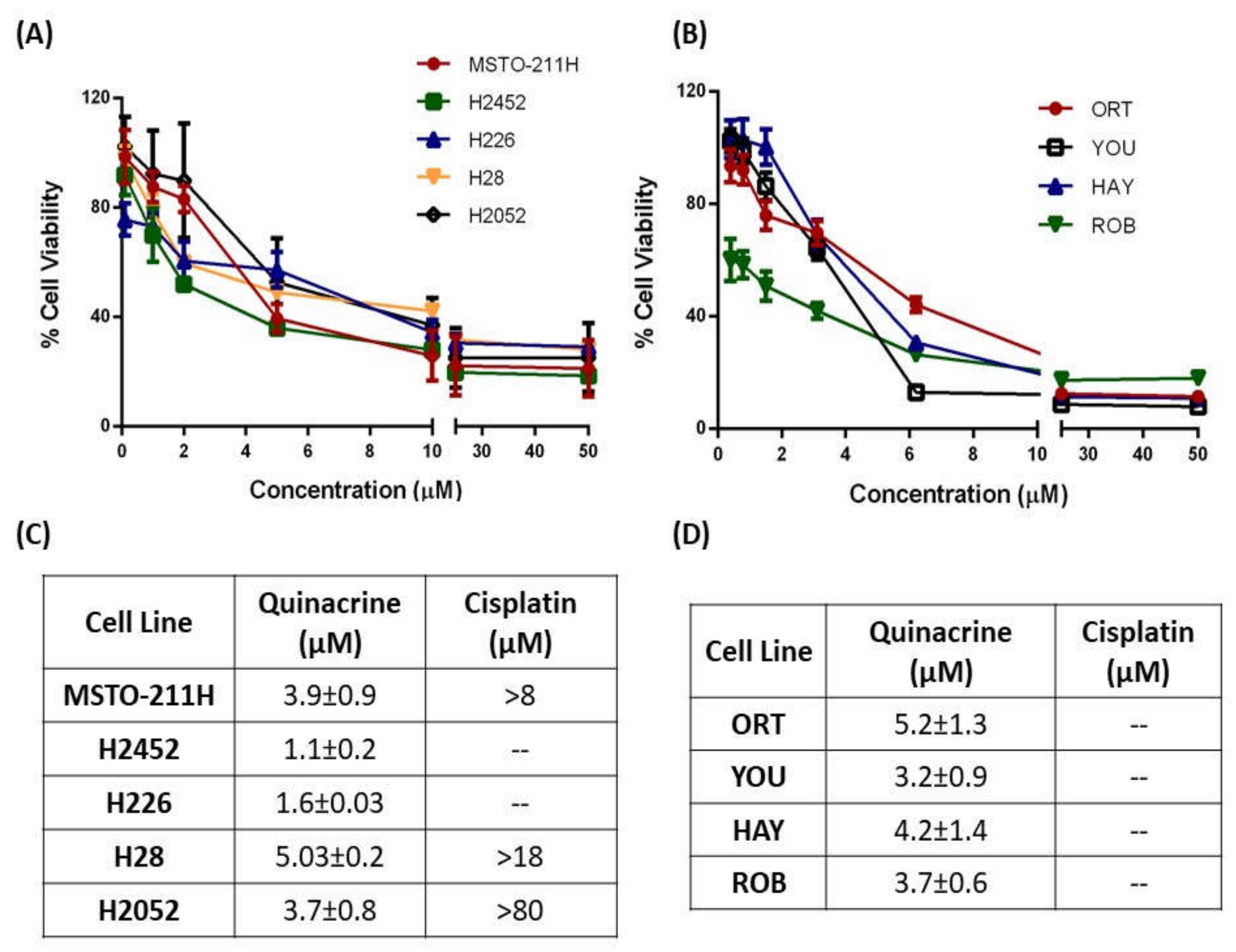
2.1.1. QA Demonstrates Enhanced Cytotoxic Ability as Compared to CS in Immortalized and Patient-Derived MPM Cells
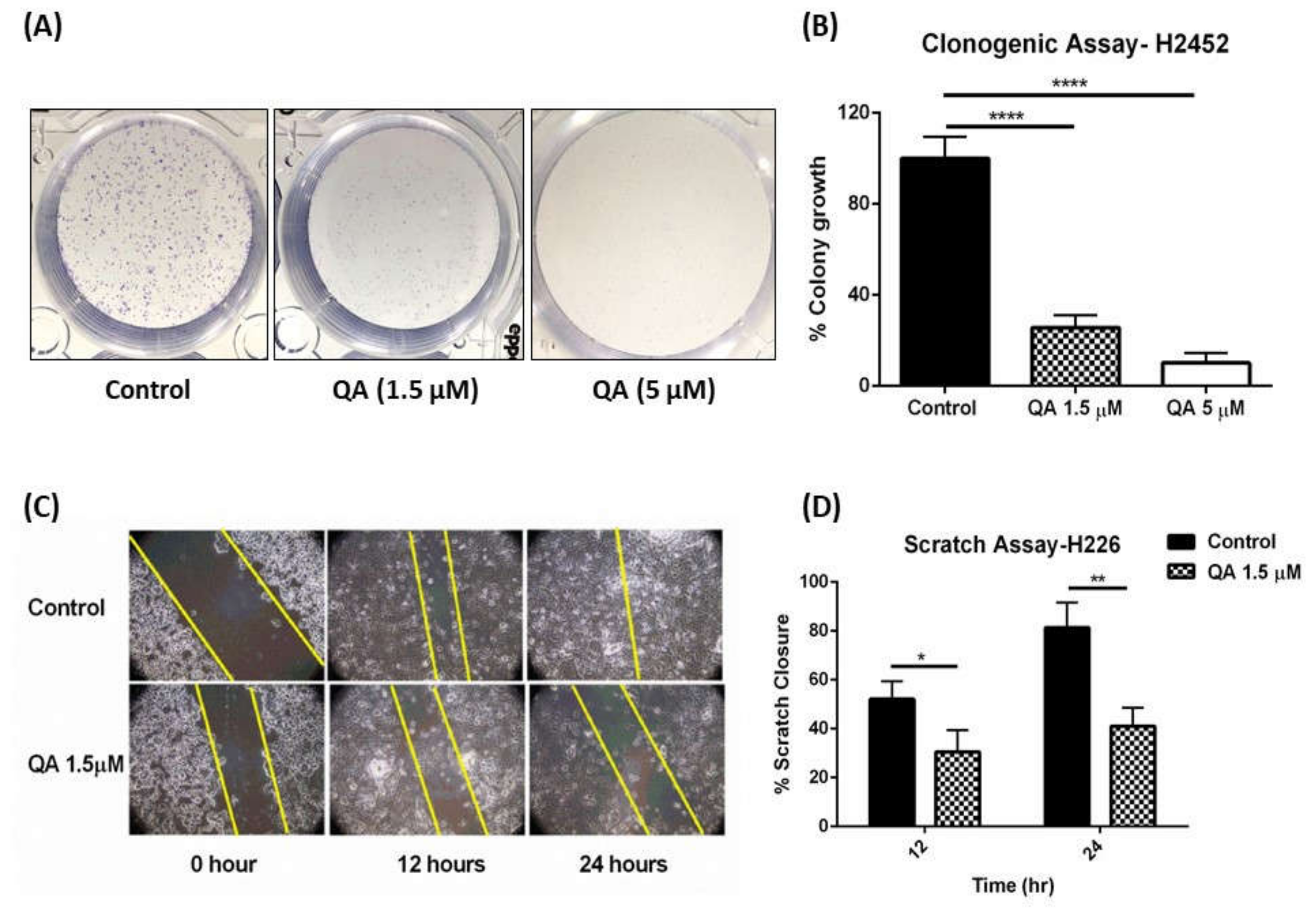
2.1.2. QA Attenuates Colony Formation in MPM Cells: Clonogenic Assay
2.1.3. QA Therapy Results in Inhibition of Cellular Migration in MPM Cells: Scratch Assay
2.1.4. QA Successfully Traverses across Cell Membrane and Localizes around the Nucleus of MPM Cells
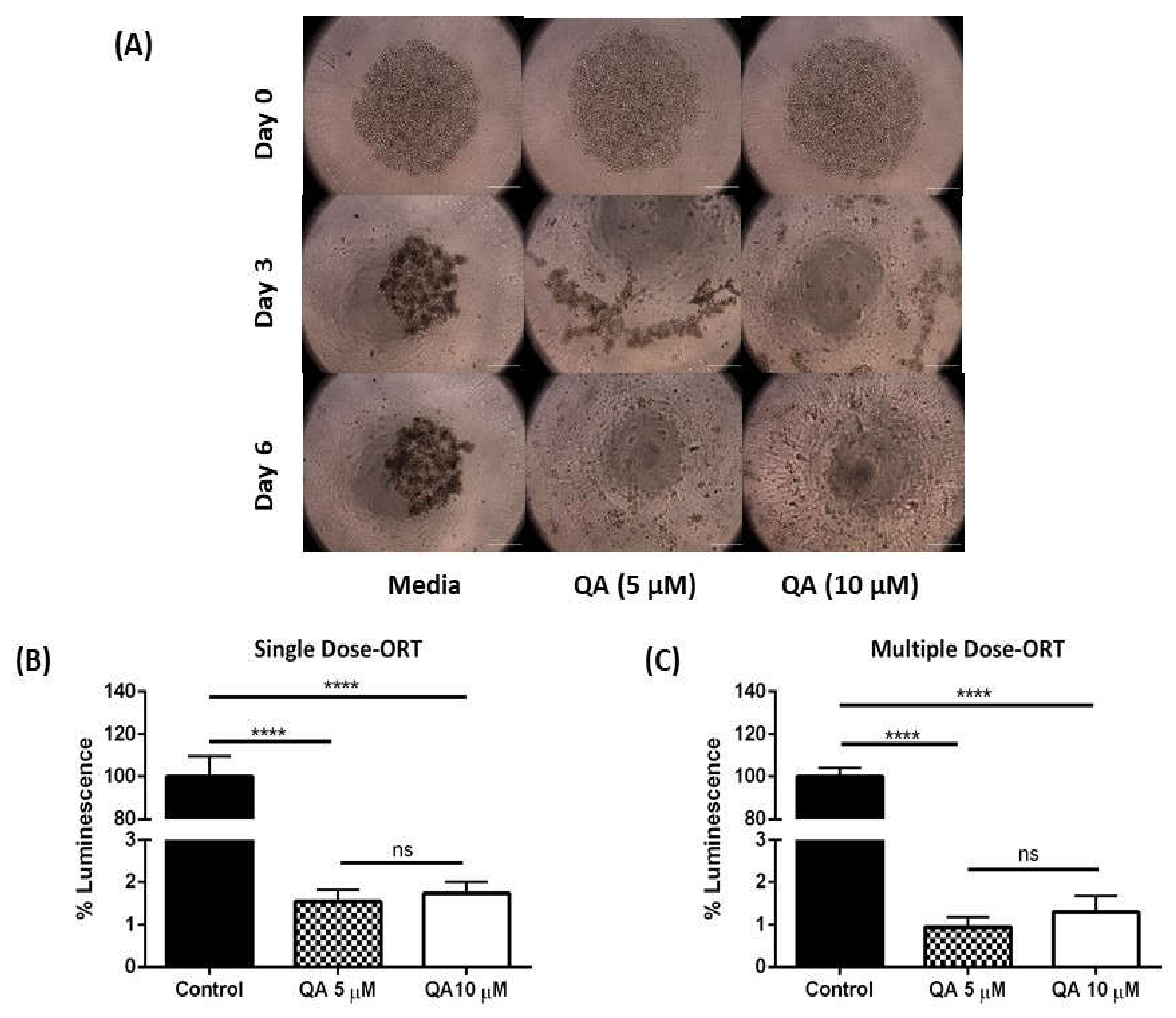
2.1.5. QA Exhibits Excellent Efficacy in an In-Vitro 3D Spheroid Model of MPM Cells Mimicking Physiological Tumors
Visual Estimation—MSTO-211H Spheroids
Live/Dead Cell Assay—MSTO-211H Spheroids
Viability Analysis—MSTO-211H Spheroids
Visual Estimation—ORT Spheroids
Viability Analysis—ORT Spheroids
2.2. Evaluation of Potential Mechanism of Action for QA Therapy in MPM Treatment
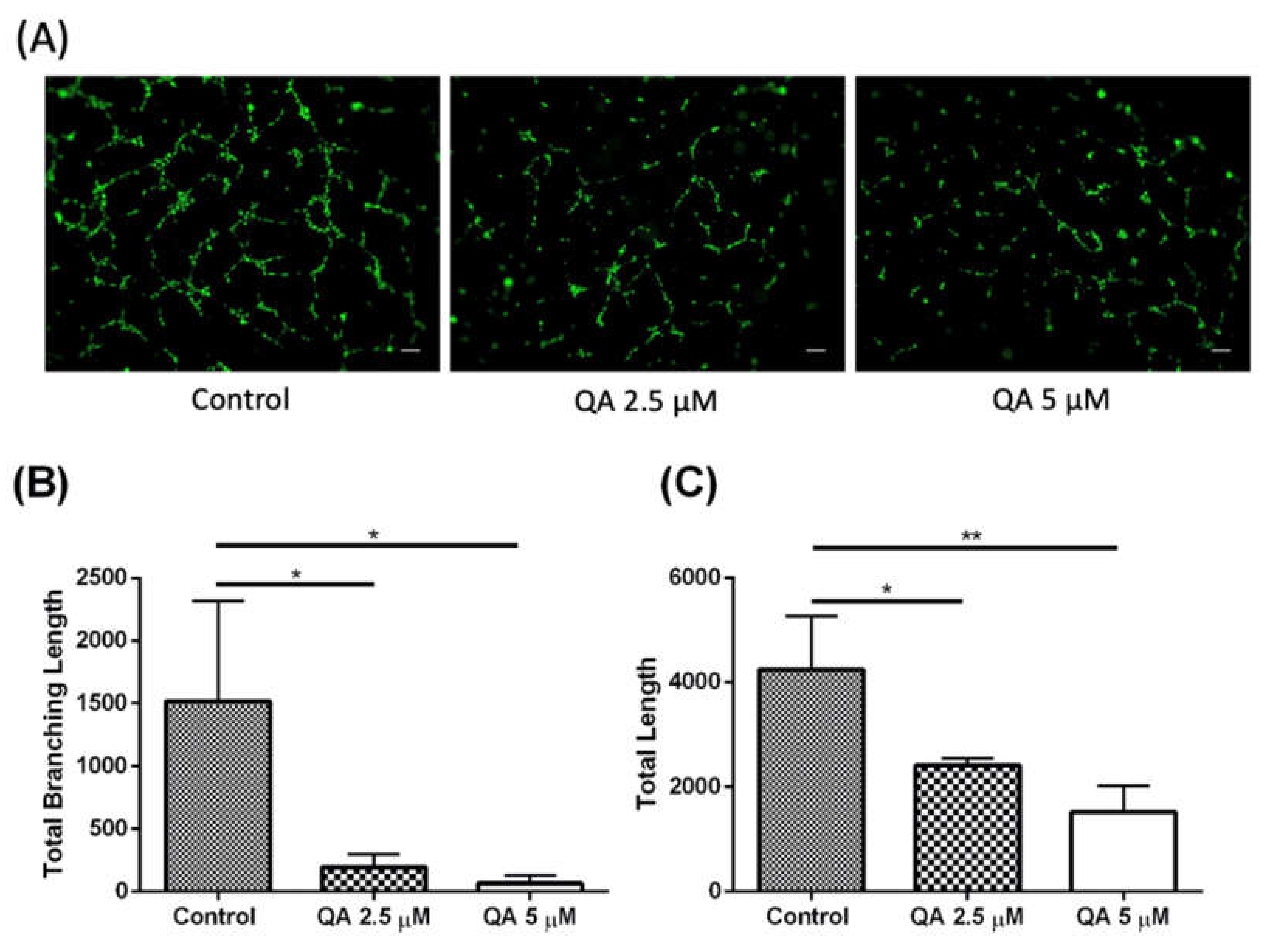
QA Successfully Demonstrates Anti-Angiogenic Traits in MPM Cells
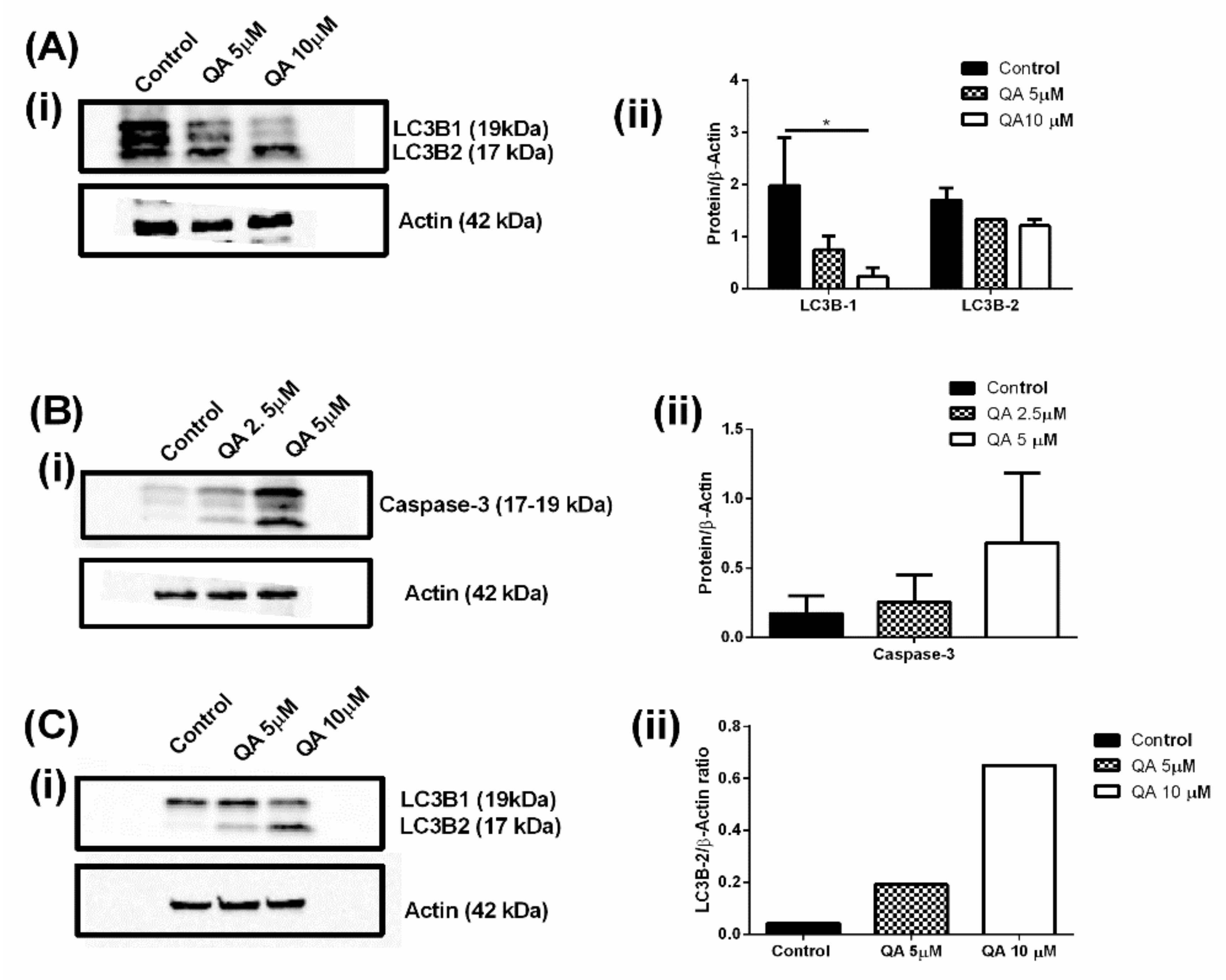
2.3. QA Inhibits the Autophagy Process Thereby Potentially Starving Cells of Essential Nutrients and ATP
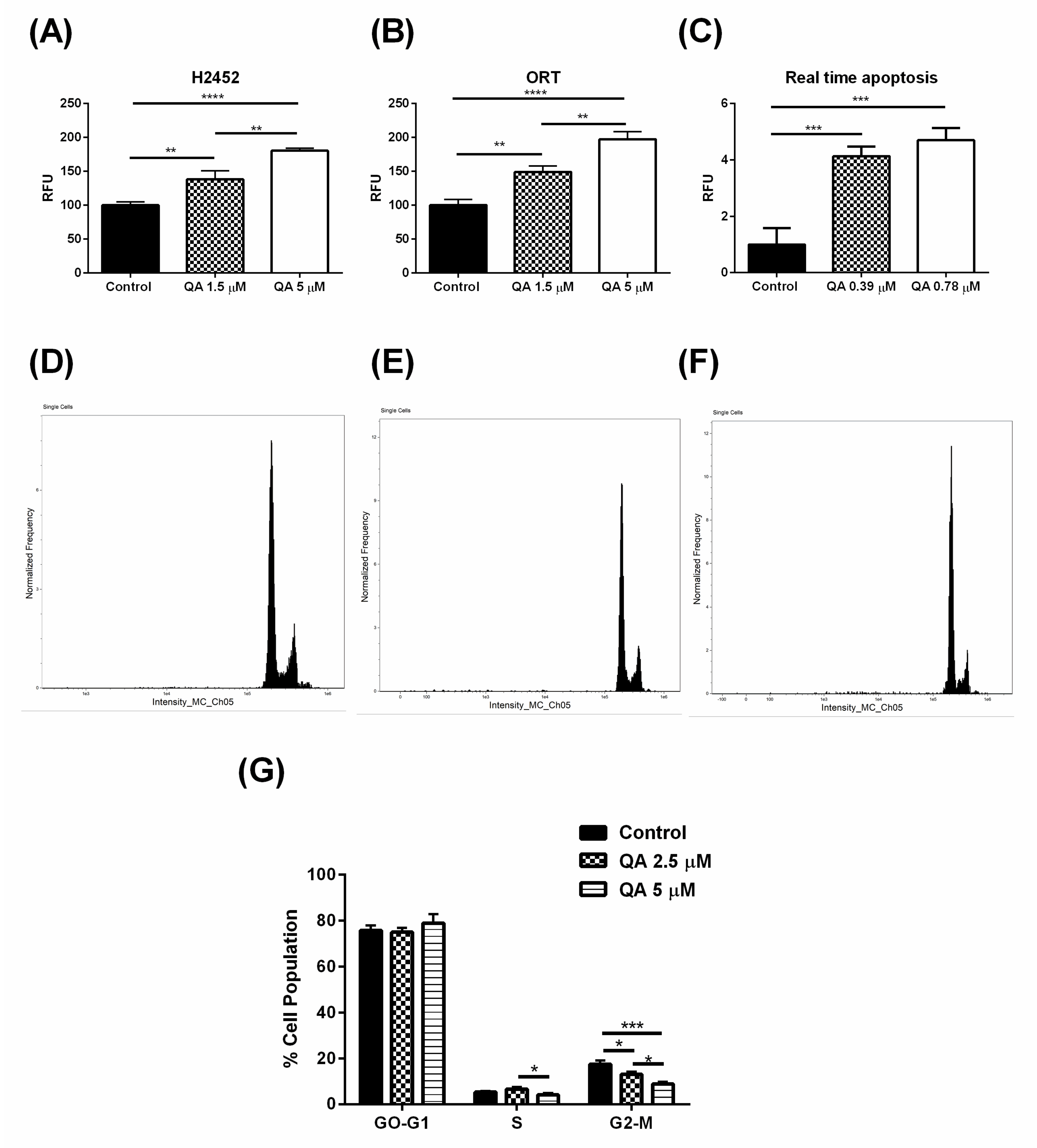
2.4. QA Therapy Leads to Induction of Apoptosis in MPM Cells
2.5. QA Arrests MPM Cells in G2-M Phase of Cell Cycle, Indicated by a Lower Cell Population
2.6. QA Affects Autophagic and Apoptotic Markers in MPM Cells
3. Materials and Methods
3.1. Materials
3.2. Cell Lines Used and Culture Method
3.3. In-Vitro Phenotypic Efficacy of QA in Mesothelioma Cell Lines
3.3.1. Cell Viability Studies
3.3.2. Evaluation of Colony formation: Clonogenic Assay
3.3.3. Evaluation of Cellular Migration: Scratch Assay
3.3.4. Evaluation of Cellular Internalization of QA in MPM Cells
3.3.5. QA Efficacy in In-Vitro 3D-Spheroids
3.3.6. Live/Dead Cellular Assay
3.4. Mechanistic Evaluation of QA’s Anti-Mesothelioma Efficacy
3.4.1. Evaluation of QA as an Angiogenesis Inhibitor
3.4.2. Effect of QA on Cellular Autophagy
3.4.3. Cell Cycle and Apoptosis Analysis by Flow Cytometry
3.4.4. RealTime-Glo™ Annexin V Apoptosis and Necrosis Assay
3.4.5. Western Blotting for Assessing QA’s Impact on MPM Molecular Markers
3.5. Statistical Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cinausero, M.; Rihawi, K.; Sperandi, F.; Melotti, B.; Ardizzoni, A. Chemotherapy treatment in malignant pleural mesothelioma: A difficult history. J. Thorac. Dis. 2018, 10, S304–S310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mott, F.E. Mesothelioma: A review. Ochsner J. 2012, 12, 70–79. [Google Scholar] [PubMed]
- Berzenji, L.; Van Schil, P. Multimodality treatment of malignant pleural mesothelioma. F1000Research 2018, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turaga, K.K.; Deraco, M.; Alexander, H.R. Current management strategies for peritoneal mesothelioma. Int. J. Hyperth. 2017, 33, 579–581. [Google Scholar] [CrossRef]
- Webb, J.; Yiu, Y.W.; Giastefani, S.; Carr-White, G. Pericardial mesothelioma. QJM 2016, 109, 631–632. [Google Scholar] [CrossRef] [Green Version]
- Key Statistics about Malignant Mesothelioma. Available online: https://www.cancer.org/cancer/malignant-mesothelioma/about/key-statistics.html (accessed on 7 July 2020).
- Clayson, H.; Seymour, J.; Noble, B. Mesothelioma from the patient’s perspective. Hematol. Oncol. Clin. N. Am. 2005, 19, 1175–1190. [Google Scholar] [CrossRef]
- Dragani, T.A.; Colombo, F.; Pavlisko, E.N.; Roggli, V.L. Malignant mesothelioma diagnosed at a younger age is associated with heavier asbestos exposure. Carcinogenesis 2018, 39, 1151–1156. [Google Scholar] [CrossRef] [Green Version]
- Shukla, S.K.; Chan, A.; Parvathaneni, V.; Gupta, V. Metformin-loaded chitosomes for treatment of malignant pleural mesothelioma—A rare thoracic cancer. Int. J. Biol. Macromol. 2020, 160, 128–141. [Google Scholar] [CrossRef]
- Kondola, S.; Manners, D.; Nowak, A.K. Malignant pleural mesothelioma: An update on diagnosis and treatment options. Ther. Adv. Respir. Dis. 2016, 10, 275–288. [Google Scholar] [CrossRef] [Green Version]
- Vogelzang, N.J.; Rusthoven, J.J.; Symanowski, J.; Denham, C.; Kaukel, E.; Ruffie, P.; Gatzemeier, U.; Boyer, M.; Emri, S.; Manegold, C.; et al. Phase III study of pemetrexed in combination with cisplatin versus cisplatin alone in patients with malignant pleural mesothelioma. J. Clin. Oncol. 2003, 21, 2636–2644. [Google Scholar] [CrossRef]
- Matt Mauney FDA Approves First New Treatment for Mesothelioma in 15 Years. Available online: https://www.asbestos.com/news/2019/05/24/fda-approves-new-mesothelioma-treatment/ (accessed on 7 July 2020).
- U.S. Food & Drug Administration. NovoTTFTM-100L System-H180002. Available online: https://www.fda.gov/medical-devices/recently-approved-devices/novottftm-100l-system-h180002 (accessed on 7 July 2020).
- Lemen, R.A. Mesothelioma from asbestos exposures: Epidemiologic patterns and impact in the United States. J. Toxicol. Environ. Health Part B 2016, 19, 250–265. [Google Scholar] [CrossRef] [PubMed]
- Rath, E.M.; Cheng, Y.Y.; Pinese, M.; Sarun, K.H.; Hudson, A.L.; Weir, C.; Wang, Y.D.; Håkansson, A.P.; Howell, V.M.; Liu, G.J.; et al. BAMLET kills chemotherapy-resistant mesothelioma cells, holding oleic acid in an activated cytotoxic state. PLoS ONE 2018, 13, e0203003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudd, R.M. Malignant mesothelioma. Br. Med. Bull. 2010, 93, 105–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parvathaneni, V.; Kulkarni, N.S.; Muth, A.; Gupta, V. Drug repurposing: A promising tool to accelerate the drug discovery process. Drug Discov. Today 2019. [Google Scholar] [CrossRef] [PubMed]
- Sleire, L.; Førde, H.E.; Netland, I.A.; Leiss, L.; Skeie, B.S.; Enger, P.Ø. Drug repurposing in cancer. Pharmacol. Res. 2017, 124, 74–91. [Google Scholar] [CrossRef]
- Cha, Y.; Erez, T.; Reynolds, I.J.; Kumar, D.; Ross, J.; Koytiger, G.; Kusko, R.; Zeskind, B.; Risso, S.; Kagan, E.; et al. Drug repurposing from the perspective of pharmaceutical companies. Br. J. Pharmacol. 2018, 175, 168–180. [Google Scholar] [CrossRef] [Green Version]
- Solomon, V.R.; Pundir, S.; Le, H.-T.; Lee, H. Design and synthesis of novel quinacrine-(1,3)-thiazinan-4-one hybrids for their anti-breast cancer activity. Eur. J. Med. Chem. 2018, 143, 1028–1038. [Google Scholar] [CrossRef]
- Kalogera, E.; Roy, D.; Khurana, A.; Mondal, S.; Weaver, A.L.; He, X.; Dowdy, S.C.; Shridhar, V. Quinacrine in endometrial cancer: Repurposing an old antimalarial drug. Gynecol. Oncol. 2017, 146, 187–195. [Google Scholar] [CrossRef]
- Das, S.; Tripathi, N.; Preet, R.; Siddharth, S.; Nayak, A.; Bharatam, P.V.; Kundu, C.N. Quinacrine induces apoptosis in cancer cells by forming a functional bridge between TRAIL-DR5 complex and modulating the mitochondrial intrinsic cascade. Oncotarget 2017, 8, 248–267. [Google Scholar] [CrossRef] [Green Version]
- Khurana, A.; Roy, D.; Kalogera, E.; Mondal, S.; Wen, X.; He, X.; Dowdy, S.; Shridhar, V. Quinacrine promotes autophagic cell death and chemosensitivity in ovarian cancer and attenuates tumor growth. Oncotarget 2015, 6, 36354–36369. [Google Scholar] [CrossRef] [Green Version]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Ye, L.; Ma, S.; Robinson, B.W.; Creaney, J. Immunotherapy strategies for mesothelioma—The role of tumor specific neoantigens in a new era of precision medicine. Expert Rev. Respir. Med. 2019, 13, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Bibby, A.C.; Tsim, S.; Kanellakis, N.; Ball, H.; Talbot, D.C.; Blyth, K.G.; Maskell, N.A.; Psallidas, I. Malignant pleural mesothelioma: An update on investigation, diagnosis and treatment. Eur. Respir. Rev. 2016, 25, 472–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.G.; Kim, C.W.; Lee, D.H.; Lee, J.-S.; Oh, E.-T.; Park, H.J. Quinacrine-Mediated Inhibition of Nrf2 Reverses Hypoxia-Induced 5-Fluorouracil Resistance in Colorectal Cancer. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaidya, B.; Kulkarni, N.S.; Shukla, S.K.; Parvathaneni, V.; Chauhan, G.; Damon, J.K.; Sarode, A.; Garcia, J.V.; Kunda, N.; Mitragotri, S.; et al. Development of inhalable quinacrine loaded bovine serum albumin modified cationic nanoparticles: Repurposing quinacrine for lung cancer therapeutics. Int. J. Pharm. 2020, 577, 118995. [Google Scholar] [CrossRef]
- Tsao, A.; Nakano, T.; Nowak, A.K.; Popat, S.; Scagliotti, G.V.; Heymach, J. Targeting angiogenesis for patients with unresectable malignant pleural mesothelioma. Semin. Oncol. 2019, 46, 145–154. [Google Scholar] [CrossRef]
- Follo, C.; Cheng, Y.; Richards, W.G.; Bueno, R.; Broaddus, V.C. Inhibition of autophagy initiation potentiates chemosensitivity in mesothelioma. Mol. Carcinog. 2018, 57, 319–332. [Google Scholar] [CrossRef]
- Galani, V.; Varouktsi, A.; Papadatos, S.S.; Mitselou, A.; Sainis, I.; Constantopoulos, S.; Dalavanga, Y. The role of apoptosis defects in malignant mesothelioma pathogenesis with an impact on prognosis and treatment. Cancer Chemother. Pharmacol. 2019, 84, 241–253. [Google Scholar] [CrossRef]
- Li, Q.; Verschraegen, C.F.; Mendoza, J.; Hassan, R. Cytotoxic activity of the recombinant anti-mesothelin immunotoxin, SS1(dsFv)PE38, towards tumor cell lines established from ascites of patients with peritoneal mesotheliomas. Anticancer Res. 2004, 24, 1327–1335. [Google Scholar]
- Batirel, H.F.; Metintas, M.; Caglar, H.B.; Ak, G.; Yumuk, P.F.; Ahiskali, R.; Bozkurtlar, E.; Bekiroglu, N.; Lacin, T.; Yildizeli, B.; et al. Macroscopic complete resection is not associated with improved survival in patients with malignant pleural mesothelioma. J. Thorac. Cardiovasc. Surg. 2018, 155, 2724–2733. [Google Scholar] [CrossRef]
- Halezeroğlu, S.; Migliore, M. Management of recurrence after initial surgery for malignant pleural mesothelioma: A mini-review. Future Oncol. 2015, 11, 23–27. [Google Scholar] [CrossRef]
- Kostron, A.; Friess, M.; Crameri, O.; Inci, I.; Schneiter, D.; Hillinger, S.; Stahel, R.; Weder, W.; Opitz, I. Relapse pattern and second-line treatment following multimodality treatment for malignant pleural mesothelioma. Eur. J. Cardiothorac. Surg. 2016, 49, 1516–1523. [Google Scholar] [CrossRef] [PubMed]
- De Perrot, M.; Dong, Z.; Bradbury, P.; Patsios, D.; Keshavjee, S.; Leighl, N.B.; Hope, A.; Feld, R.; Cho, J. Impact of tumour thickness on survival after radical radiation and surgery in malignant pleural mesothelioma. Eur. Respir. J. 2017, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertoglio, P.; Fanucchi, O.; Ricciardi, S.; Chella, A.; Lucchi, M.; Mussi, A. Chest wall resection for mesothelioma recurrence after surgery. Asian Cardiovasc. Thorac. Ann. 2016, 24, 893–895. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, B.; Parvathaneni, V.; Kulkarni, N.S.; Shukla, S.K.; Damon, J.K.; Sarode, A.; Kanabar, D.; Garcia, J.V.; Mitragotri, S.; Muth, A.; et al. Cyclodextrin modified erlotinib loaded PLGA nanoparticles for improved therapeutic efficacy against non-small cell lung cancer. Int. J. Biol. Macromol. 2019, 122, 338–347. [Google Scholar] [CrossRef]
- Sohn, E.J.; Won, G.; Lee, J.; Yoon, S.W.; Lee, I.; Kim, H.J.; Kim, S.-H. Blockage of epithelial to mesenchymal transition and upregulation of let 7b are critically involved in ursolic acid induced apoptosis in malignant mesothelioma cell. Int. J. Biol. Sci. 2016, 12, 1279–1288. [Google Scholar] [CrossRef] [Green Version]
- Nomura, Y.; Tashiro, H.; Hisamatsu, K. In Vitro clonogenic growth and metastatic potential of human operable breast cancer. Cancer Res. 1989, 49, 5288–5293. [Google Scholar]
- Liang, C.-C.; Park, A.Y.; Guan, J.-L. In Vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2007, 2, 329–333. [Google Scholar] [CrossRef] [Green Version]
- Zidovetzki, R.; Sherman, I.W.; Atiya, A.; De Boeck, H. A nuclear magnetic resonance study of the interactions of the antimalarials chloroquine, quinacrine, quinine and mefloquine with dipalmitoylphosphatidylcholine bilayers. Mol. Biochem. Parasitol. 1989, 35, 199–207. [Google Scholar] [CrossRef]
- Parks, A.; Charest-Morin, X.; Boivin-Welch, M.; Bouthillier, J.; Marceau, F. Autophagic flux inhibition and lysosomogenesis ensuing cellular capture and retention of the cationic drug quinacrine in murine models. PeerJ 2015, 3, e1314. [Google Scholar] [CrossRef]
- Marceau, F.; Bawolak, M.-T.; Lodge, R.; Bouthillier, J.; Gagné-Henley, A.; C.-Gaudreault, R.; Morissette, G. Cation trapping by cellular acidic compartments: Beyond the concept of lysosomotropic drugs. Toxicol. Appl. Pharmacol. 2012, 259, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Zaguilan, R.; Lynch, R.M.; Martinez, G.M.; Gillies, R.J. Vacuolar-type H(+)-ATPases are functionally expressed in plasma membranes of human tumor cells. Am. J. Physiol. Cell Physiol. 1993, 265, C1015–C1029. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, N.S.; Parvathaneni, V.; Shukla, S.K.; Barasa, L.; Perron, J.C.; Yoganathan, S.; Muth, A.; Gupta, V. Tyrosine kinase inhibitor conjugated quantum dots for non-small cell lung cancer (NSCLC) treatment. Eur. J. Pharm. Sci. 2019, 133, 145–159. [Google Scholar] [CrossRef] [PubMed]
- McCombe, A.W.; Binnington, J.; McCombe, T.S. Hearing protection for motorcyclists. Clin. Otolaryngol. Allied Sci. 1993, 18, 465–469. [Google Scholar] [CrossRef]
- Kondo, J.; Ekawa, T.; Endo, H.; Yamazaki, K.; Tanaka, N.; Kukita, Y.; Okuyama, H.; Okami, J.; Imamura, F.; Ohue, M.; et al. High-throughput screening in colorectal cancer tissue-originated spheroids. Cancer Sci. 2019, 110, 345–355. [Google Scholar] [CrossRef]
- Brüningk, S.C.; Rivens, I.; Box, C.; Oelfke, U.; ter Haar, G. 3D tumour spheroids for the prediction of the effects of radiation and hyperthermia treatments. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Elbatanony, R.S.; Parvathaneni, V.; Kulkarni, N.S.; Shukla, S.K.; Chauhan, G.; Kunda, N.K.; Gupta, V. Afatinib-loaded inhalable PLGA nanoparticles for localized therapy of non-small cell lung cancer (NSCLC)—Development and in-vitro efficacy. Drug Deliv. Transl. Res. 2020. [Google Scholar] [CrossRef]
- Vandermeers, F.; Hubert, P.; Delvenne, P.; Mascaux, C.; Grigoriu, B.; Burny, A.; Scherpereel, A.; Willems, L. Valproate, in Combination with Pemetrexed and Cisplatin, Provides Additional Efficacy to the Treatment of Malignant Mesothelioma. Clin. Cancer Res. 2009, 15, 2818–2828. [Google Scholar] [CrossRef] [Green Version]
- Hoarau-Véchot, J.; Rafii, A.; Touboul, C.; Pasquier, J. Halfway between 2D and Animal Models: Are 3D Cultures the Ideal Tool to Study Cancer-Microenvironment Interactions? Int. J. Mol. Sci. 2018, 19, 181. [Google Scholar] [CrossRef] [Green Version]
- Ehsanian, R.; Van Waes, C.; Feller, S.M. Beyond DNA binding—A review of the potential mechanisms mediating quinacrine’s therapeutic activities in parasitic infections, inflammation, and cancers. Cell Commun. Signal. 2011, 9, 13. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.; Bian, A.; Gao, X.; Li, H.; Chen, Z.; Liu, X. Novel applications for an established antimalarial drug: Tumoricidal activity of quinacrine. Future Oncol. 2018, 14, 1511–1520. [Google Scholar] [CrossRef]
- Cao, Y.; Arbiser, J.; D’Amato, R.J.; D’Amore, P.A.; Ingber, D.E.; Kerbel, R.; Klagsbrun, M.; Lim, S.; Moses, M.A.; Zetter, B.; et al. Forty-Year Journey of Angiogenesis Translational Research. Sci. Transl. Med. 2011, 3, 114rv3. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Li, K.; Wang, Q.; Zhang, L.; Shi, J.; Wang, Z.; Cheng, Y.; He, J.; Shi, Y.; Zhao, Y.; et al. Effect of Anlotinib as a Third-Line or Further Treatment on Overall Survival of Patients With Advanced Non–Small Cell Lung Cancer: The ALTER 0303 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2018, 4, 1569–1575. [Google Scholar] [CrossRef] [PubMed]
- Gregorc, V.; Gaafar, R.M.; Favaretto, A.; Grossi, F.; Jassem, J.; Polychronis, A.; Bidoli, P.; Tiseo, M.; Shah, R.; Taylor, P.; et al. NGR-hTNF in combination with best investigator choice in previously treated malignant pleural mesothelioma (NGR015): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet Oncol. 2018, 19, 799–811. [Google Scholar] [CrossRef]
- Tsao, A.S.; Moon, J.; Wistuba, I.I.; Vogelzang, N.J.; Kalemkerian, G.P.; Redman, M.W.; Gandara, D.R.; Kelly, K. Phase I Trial of Cediranib in Combination with Cisplatin and Pemetrexed in Chemonaive Patients with Unresectable Malignant Pleural Mesothelioma (SWOG S0905). J. Thorac. Oncol. 2017, 12, 1299–1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pass, H.I.; Brewer, G.J.; Dick, R.; Carbone, M.; Merajver, S. A Phase II Trial of Tetrathiomolybdate after Surgery for Malignant Mesothelioma: Final Results. Ann. Thorac. Surg. 2008, 86, 383–390. [Google Scholar] [CrossRef]
- Wheatley-Price, P.; Chu, Q.; Bonomi, M.; Seely, J.; Gupta, A.; Goss, G.; Hilton, J.; Feld, R.; Lee, C.W.; Goffin, J.R.; et al. A Phase II Study of PF-03446962 in Patients with Advanced Malignant Pleural Mesothelioma. CCTG Trial IND.207. J. Thorac. Oncol. 2016, 11, 2018–2021. [Google Scholar] [CrossRef] [Green Version]
- Satapathy, S.R.; Siddharth, S.; Das, D.; Nayak, A.; Kundu, C.N. Enhancement of Cytotoxicity and Inhibition of Angiogenesis in Oral Cancer Stem Cells by a Hybrid Nanoparticle of Bioactive Quinacrine and Silver: Implication of Base Excision Repair Cascade. Mol. Pharm. 2015, 12, 4011–4025. [Google Scholar] [CrossRef]
- Kubota, Y.; Kleinman, H.K.; Martin, G.R.; Lawley, T.J. Role of laminin and basement membrane in the morphological differentiation of human endothelial cells into capillary-like structures. J. Cell Biol. 1988, 107, 1589–1598. [Google Scholar] [CrossRef]
- DeCicco-Skinner, K.L.; Henry, G.H.; Cataisson, C.; Tabib, T.; Gwilliam, J.C.; Watson, N.J.; Bullwinkle, E.M.; Falkenburg, L.; O’Neill, R.C.; Morin, A.; et al. Endothelial cell tube formation assay for the in vitro study of angiogenesis. J. Vis. Exp. 2014, e51312. [Google Scholar] [CrossRef]
- Yap, T.A.; Aerts, J.G.; Popat, S.; Fennell, D.A. Novel insights into mesothelioma biology and implications for therapy. Nat. Rev. Cancer 2017, 17, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Zalcman, G.; Mazieres, J.; Margery, J.; Greillier, L.; Audigier-Valette, C.; Moro-Sibilot, D.; Molinier, O.; Corre, R.; Monnet, I.; Gounant, V.; et al. Bevacizumab for newly diagnosed pleural mesothelioma in the Mesothelioma Avastin Cisplatin Pemetrexed Study (MAPS): A randomised, controlled, open-label, phase 3 trial. Lancet 2016, 387, 1405–1414. [Google Scholar] [CrossRef]
- Onorati, A.V.; Dyczynski, M.; Ojha, R.; Amaravadi, R.K. Targeting autophagy in cancer. Cancer 2018, 124, 3307–3318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grootaert, M.O.J.; Roth, L.; Schrijvers, D.M.; De Meyer, G.R.Y.; Martinet, W. Defective Autophagy in Atherosclerosis: To Die or to Senesce? Oxid. Med. Cell Longev. 2018, 2018, 7687083. [Google Scholar] [CrossRef] [PubMed]
- Zhan, L.; Li, J.; Wei, B. Autophagy in endometriosis: Friend or foe? Biochem. Biophys. Res. Commun. 2018, 495, 60–63. [Google Scholar] [CrossRef] [PubMed]
- Nagar, R. Autophagy: A brief overview in perspective of dermatology. Indian J. Dermatol. Venereol. Leprol. 2017, 83, 290–297. [Google Scholar] [CrossRef]
- Scrivo, A.; Bourdenx, M.; Pampliega, O.; Cuervo, A.M. Selective autophagy as a potential therapeutic target for neurodegenerative disorders. Lancet Neurol. 2018, 17, 802–815. [Google Scholar] [CrossRef]
- Hwang, K.-E.; Kim, Y.-S.; Jung, J.-W.; Kwon, S.-J.; Park, D.-S.; Cha, B.-K.; Oh, S.-H.; Yoon, K.-H.; Jeong, E.-T.; Kim, H.-R. Inhibition of autophagy potentiates pemetrexed and simvastatin-induced apoptotic cell death in malignant mesothelioma and non-small cell lung cancer cells. Oncotarget 2015, 6, 29482–29496. [Google Scholar] [CrossRef]
- Bresciani, A.; Spiezia, M.C.; Boggio, R.; Cariulo, C.; Nordheim, A.; Altobelli, R.; Kuhlbrodt, K.; Dominguez, C.; Munoz-Sanjuan, I.; Wityak, J.; et al. Quantifying autophagy using novel LC3B and p62 TR-FRET assays. PLoS ONE 2018, 13, e0194423. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-R.; Cho, K.-H.; Hwang, K.-E.; Jeong, E.-T. Inhibition of autophagy potentiates pemetrexed and simvastatin-induced apoptotic cell death in malignant mesothelioma and non-small cell lung cancer cells. In Proceedings of the 11.1 Lung Cancer; European Respiratory Society: Plymouth, UK, 2015; p. PA4263. [Google Scholar]
- Echeverry, N.; Ziltener, G.; Barbone, D.; Weder, W.; Stahel, R.A.; Broaddus, V.C.; Felley-Bosco, E. Inhibition of autophagy sensitizes malignant pleural mesothelioma cells to dual PI3K/mTOR inhibitors. Cell Death Dis. 2015, 6, e1757. [Google Scholar] [CrossRef] [Green Version]
- Barbone, D.; Follo, C.; Echeverry, N.; Gerbaudo, V.H.; Klabatsa, A.; Bueno, R.; Felley-Bosco, E.; Broaddus, V.C. Autophagy Correlates with the Therapeutic Responsiveness of Malignant Pleural Mesothelioma in 3D Models. PLoS ONE 2015, 10, e0134825. [Google Scholar] [CrossRef] [Green Version]
- Wlodkowic, D.; Telford, W.; Skommer, J.; Darzynkiewicz, Z. Apoptosis and Beyond: Cytometry in Studies of Programmed Cell Death. In Methods in Cell Biology; Elsevier: Amsterdam, The Netherlands, 2011; Volume 103, pp. 55–98. ISBN 978-0-12-385493-3. [Google Scholar]
- Dermawan, J.K.T.; Gurova, K.; Pink, J.; Dowlati, A.; De, S.; Narla, G.; Sharma, N.; Stark, G.R. Quinacrine overcomes resistance to erlotinib by inhibiting FACT, NF-κB, and cell-cycle progression in non-small cell lung cancer. Mol. Cancer Ther. 2014, 13, 2203–2214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dent, P. Not so WEE: Targeting G₂/M to kill mesothelioma cells. Cancer Biol. 2014, 15, 351–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, M.; Madokoro, H.; Yamada, K.; Nishida, H.; Morimoto, C.; Sakamoto, M.; Yamada, T. A humanized anti-CD26 monoclonal antibody inhibits cell growth of malignant mesothelioma via retarded G2/M cell cycle transition. Cancer Cell Int. 2016, 16, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalilzadeh, B.; Shadjou, N.; Kanberoglu, G.S.; Afsharan, H.; de la Guardia, M.; Charoudeh, H.N.; Ostadrahimi, A.; Rashidi, M.-R. Advances in nanomaterial based optical biosensing and bioimaging of apoptosis via caspase-3 activity: A review. Microchim. Acta 2018, 185. [Google Scholar] [CrossRef] [PubMed]
- Mazor, R.; Onda, M.; Park, D.; Addissie, S.; Xiang, L.; Zhang, J.; Hassan, R.; Pastan, I. Dual B- and T-cell de-immunization of recombinant immunotoxin targeting mesothelin with high cytotoxic activity. Oncotarget 2016, 7, 29916–29926. [Google Scholar] [CrossRef] [Green Version]
- Mazor, R.; Zhang, J.; Xiang, L.; Addissie, S.; Awuah, P.; Beers, R.; Hassan, R.; Pastan, I. Recombinant Immunotoxin with T-cell Epitope Mutations That Greatly Reduce Immunogenicity for Treatment of Mesothelin-Expressing Tumors. Mol. Cancer Ther. 2015, 14, 2789–2796. [Google Scholar] [CrossRef] [Green Version]
- Geissmann, Q. OpenCFU, a new free and open-source software to count cell colonies and other circular objects. PLoS ONE 2013, 8, e54072. [Google Scholar] [CrossRef] [Green Version]
- Hebert, L.; Bellanger, D.; Guillas, C.; Campagne, A.; Dingli, F.; Loew, D.; Fievet, A.; Jacquemin, V.; Popova, T.; Jean, D.; et al. Modulating BAP1 expression affects ROS homeostasis, cell motility and mitochondrial function. Oncotarget 2017, 8, 72513–72527. [Google Scholar] [CrossRef] [Green Version]
- Vaidya, B.; Shukla, S.K.; Kolluru, S.; Huen, M.; Mulla, N.; Mehra, N.; Kanabar, D.; Palakurthi, S.; Ayehunie, S.; Muth, A.; et al. Nintedanib-cyclodextrin complex to improve bio-activity and intestinal permeability. Carbohydr. Polym. 2019, 204, 68–77. [Google Scholar] [CrossRef]
- Parvathaneni, V.; Kulkarni, N.S.; Shukla, S.K.; Farrales, P.T.; Kunda, N.K.; Muth, A.; Gupta, V. Systematic Development and Optimization of Inhalable Pirfenidone Liposomes for Non-Small Cell Lung Cancer Treatment. Pharmaceutics 2020, 12, 206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, S.K.; Kulkarni, N.S.; Farrales, P.; Kanabar, D.D.; Parvathaneni, V.; Kunda, N.K.; Muth, A.; Gupta, V. Sorafenib Loaded Inhalable Polymeric Nanocarriers against Non-Small Cell Lung Cancer. Pharm. Res. 2020, 37. [Google Scholar] [CrossRef] [PubMed]
- Viallard, C.; Larrivée, B. Tumor angiogenesis and vascular normalization: Alternative therapeutic targets. Angiogenesis 2017, 20, 409–426. [Google Scholar] [CrossRef] [PubMed]
- Parvathaneni, V.; Kulkarni, N.S.; Chauhan, G.; Shukla, S.K.; Elbatanony, R.; Patel, B.; Kunda, N.K.; Muth, A.; Gupta, V. Development of pharmaceutically scalable inhaled anti-cancer nanotherapy—Repurposing amodiaquine for non-small cell lung cancer (NSCLC). Mater. Sci. Eng. C 2020, 115, 111139. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.K.; Kulkarni, N.S.; Chan, A.; Parvathaneni, V.; Farrales, P.; Muth, A.; Gupta, V. Metformin-Encapsulated Liposome Delivery System: An Effective Treatment Approach against Breast Cancer. Pharmaceutics 2019, 11, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, S.; Chen, Z.; Wang, L.; Peng, D.; Belkhiri, A.; Lockhart, A.C.; El-Rifai, W. A Combination of SAHA and Quinacrine Is Effective in Inducing Cancer Cell Death in Upper Gastrointestinal Cancers. Clin. Cancer Res. 2018, 24, 1905–1916. [Google Scholar] [CrossRef] [Green Version]
- Bryant, J.; Batis, N.; Franke, A.C.; Clancey, G.; Hartley, M.; Ryan, G.; Brooks, J.; Southam, A.D.; Barnes, N.; Parish, J.; et al. Repurposed quinacrine synergizes with cisplatin, reducing the effective dose required for treatment of head and neck squamous cell carcinoma. Oncotarget 2019, 10, 5229–5244. [Google Scholar] [CrossRef] [Green Version]
- Shannon, J.A.; Earle, D.P.; Brodie, B.B.; Taggart, J.V.; Berliner, R.W. The Pharmacological Basis for the Rational Use of Atabrine in the Treatment of Malaria. J. Pharm. Exp. 1944, 81, 307–330. [Google Scholar]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kulkarni, N.S.; Vaidya, B.; Parvathaneni, V.; Bhanja, D.; Gupta, V. Repurposing Quinacrine for Treatment of Malignant Mesothelioma: In-Vitro Therapeutic and Mechanistic Evaluation. Int. J. Mol. Sci. 2020, 21, 6306. https://doi.org/10.3390/ijms21176306
Kulkarni NS, Vaidya B, Parvathaneni V, Bhanja D, Gupta V. Repurposing Quinacrine for Treatment of Malignant Mesothelioma: In-Vitro Therapeutic and Mechanistic Evaluation. International Journal of Molecular Sciences. 2020; 21(17):6306. https://doi.org/10.3390/ijms21176306
Chicago/Turabian StyleKulkarni, Nishant S., Bhuvaneshwar Vaidya, Vineela Parvathaneni, Debarati Bhanja, and Vivek Gupta. 2020. "Repurposing Quinacrine for Treatment of Malignant Mesothelioma: In-Vitro Therapeutic and Mechanistic Evaluation" International Journal of Molecular Sciences 21, no. 17: 6306. https://doi.org/10.3390/ijms21176306
APA StyleKulkarni, N. S., Vaidya, B., Parvathaneni, V., Bhanja, D., & Gupta, V. (2020). Repurposing Quinacrine for Treatment of Malignant Mesothelioma: In-Vitro Therapeutic and Mechanistic Evaluation. International Journal of Molecular Sciences, 21(17), 6306. https://doi.org/10.3390/ijms21176306