Small GTPases of the Ras and Rho Families Switch on/off Signaling Pathways in Neurodegenerative Diseases

 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
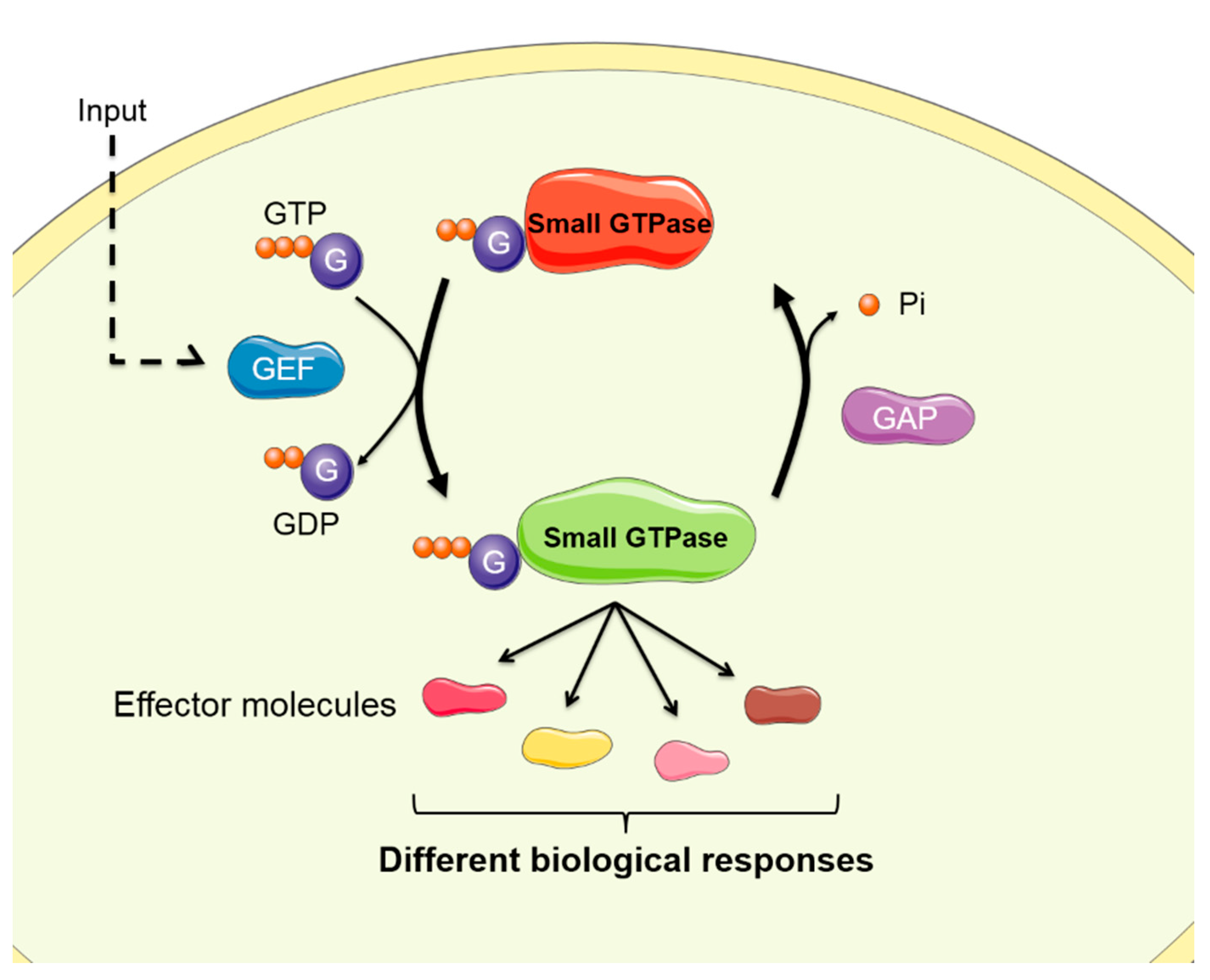
Abstract
:1. Introduction
2. Small GTPases of the Ras Family
2.1. Ras GTPase
2.2. Rap GTPase
2.2.1. Rap/MAPK/ERK
2.2.2. Rap2/JNK
2.2.3. Rap1/AF-6
2.2.4. LRRK2/EPAC-1/Rap1 Pathway
2.2.5. Epac/Rap1/APP Processing
2.3. Rheb GTPase
3. Small GTPASES of the Rho Family
3.1. RhoA
3.1.1. RhoA/ROCK
3.1.2. RhoA/NOX
3.2. Rac GTPase
3.2.1. Rac1/Toxic Peptide Accumulation
3.2.2. Rac1/PAK
3.2.3. PI3K/PDK/nPKC/Tiam-1/Rac1/Neuronal Cell Death
3.2.4. Rac1/NOX
3.2.5. Rac1/JNK
3.3. Cdc42 GTPase
3.3.1. Cdc42/ PAK
3.3.2. Cdc42/N-WASP
3.3.3. Cdc42/GSK3
3.3.4. Cdc42/Endocytosis
4. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Berridge, M.J. Calcium signalling remodelling and disease. Biochem. Soc. Trans. 2012, 40, 297–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goitre, L.; Trapani, E.; Trabalzini, L.; Retta, S.F. The Ras Superfamily of Small GTPases: The Unlocked Secrets. Methods Mol. Biol. 2014, 1120, 1–18. [Google Scholar] [PubMed]
- Song, S.; Cong, W.; Zhou, S.; Shi, Y.; Dai, W.; Zhang, H.; Wang, X.; He, B.; Zhang, Q. Small GTPases: Structure, biological function and its interaction with nanoparticles. Asian J. Pharm. Sci. 2019, 14, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Toma-Fukai, S.; Shimizu, T. Structural insights into the regulation mechanism of small GTPases by GEFs. Molecules 2019, 24, 3308. [Google Scholar] [CrossRef] [Green Version]
- Peurois, F.; Peyroche, G.; Cherfils, J. Small GTPase peripheral binding to membranes: Molecular determinants and supramolecular organization. Biochem. Soc. Trans. 2018, 47, 13–22. [Google Scholar] [CrossRef]
- Zaldua, N.; Gastineau, M.; Hoshino, M.; Lezoualc’h, F.; Zugaza, J.L. Epac signaling pathway involves STEF, a guanine nucleotide exchange factor for Rac, to regulate APP processing. FEBS Lett. 2007, 581, 5814–5818. [Google Scholar] [CrossRef] [Green Version]
- Singh, V.; Davidson, A.C.; Hume, P.J.; Humphreys, D.; Koronakis, V. Arf GTPase interplay with Rho GTPases in regulation of the actin cytoskeleton. Small GTPases 2019, 10, 411–418. [Google Scholar] [CrossRef] [Green Version]
- Aspenström, P. Activated rho GTPases in cancer—The beginning of a new paradigm. Int. J. Mol. Sci. 2018, 19, 3949. [Google Scholar] [CrossRef] [Green Version]
- Strassheim, D.; Gerasimovskaya, E.; Irwin, D.; Dempsey, E.C.; Stenmark, K.; Karoor, V. RhoGTPase in Vascular Disease. Cells 2019, 8, 551. [Google Scholar] [CrossRef] [Green Version]
- Qu, L.; Pan, C.; He, S.M.; Lang, B.; Gao, G.D.; Wang, X.L.; Wang, Y. The ras superfamily of small gtpases in non-neoplastic cerebral diseases. Front. Mol. Neurosci. 2019, 12, 121. [Google Scholar] [CrossRef] [Green Version]
- Gan, L.; Cookson, M.R.; Petrucelli, L.; La Spada, A.R. Converging pathways in neurodegeneration, from genetics to mechanisms. Nat. Neurosci. 2018, 21, 1300–1309. [Google Scholar] [CrossRef] [PubMed]
- Soria Lopez, J.A.; González, H.M.; Léger, G.C. Alzheimer’s disease. Hanb. Clin. Neurol. 2019, 167, 231–255. [Google Scholar]
- Gallardo, G.; Holtzman, D.M. Amyloid-β and Tau at the Crossroads of Alzheimer’s Disease. Adv. Exp. Med. Biol. 2019, 1184, 187–203. [Google Scholar] [PubMed]
- Yang, L.; Mao, K.; Yu, H.; Chen, J. Neuroinflammatory Responses and Parkinson’ Disease: Pathogenic Mechanisms and Therapeutic Targets. J. Neuroimmune Pharmacol. 2020. [Google Scholar] [CrossRef]
- Paisán-Ruiz, C.; Lewis, P.A.; Singleton, A.B. LRRK2: Cause, risk, and mechanism. J. Parkinsons. Dis. 2013, 3, 85–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, Parkin, and mitochondrial fidelity in parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [Green Version]
- Aguilar, B.J.; Zhu, Y.; Lu, Q. Rho GTPases as therapeutic targets in Alzheimer’s disease. Alzheimer’s Res. Ther. 2017, 9, 97. [Google Scholar] [CrossRef] [Green Version]
- Hong, L.; Sklar, L.A. Targeting GTPases in Parkinson’s disease: Comparison to the historic path of kinase drug discovery and perspectives. Front. Mol. Neurosci. 2014, 7, 52. [Google Scholar] [CrossRef] [Green Version]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef] [Green Version]
- Clapéron, A.; Therrien, M. KSR and CNK: Two scaffolds regulating RAS-mediated RAF activation. Oncogene 2007, 26, 3143–3158. [Google Scholar] [CrossRef] [Green Version]
- Kirouac, L.; Rajic, A.J.; Cribbs, D.H.; Padmanabhan, J. Activation of Ras-ERK signaling and GSK-3 by amyloid precursor protein and amyloid beta facilitates neurodegeneration in Alzheimer’s disease. eNeuro 2017, 4, 149–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McShea, A.; Zelasko, D.A.; Gerst, J.L.; Smith, M.A. Signal transduction abnormalities in Alzheimer’s disease: Evidence of a pathogenic stimuli. Brain Res. 1999, 815, 237–242. [Google Scholar] [CrossRef]
- Pei, J.J.; Braak, H.; An, W.L.; Winblad, B.; Cowburn, R.F.; Iqbal, K.; Grundke-Iqbal, I. Up-regulation of mitogen-activated protein kinases ERK1/2 and MEK1/2 is associated with the progression of neurofibrillary degeneration in Alzheimer’s disease. Mol. Brain Res. 2002, 109, 45–55. [Google Scholar] [CrossRef]
- Sung, Y.M.; Lee, T.; Yoon, H.; DiBattista, A.M.; Song, J.M.; Sohn, Y.; Moffat, E.I.; Turner, R.S.; Jung, M.; Kim, J.; et al. Mercaptoacetamide-based class II HDAC inhibitor lowers Aβ levels and improves learning and memory in a mouse model of Alzheimer’s disease. Exp. Neurol. 2013, 239, 192–201. [Google Scholar] [CrossRef] [Green Version]
- Song, J.M.; Sung, Y.M.; Nam, J.H.; Yoon, H.; Chung, A.; Moffat, E.; Jung, M.; Pak, D.T.S.; Kim, J.; Hoe, H.S. A mercaptoacetamide-based Class II histone deacetylase inhibitor increases dendritic spine density via RasGRF1/ERK pathway. J. Alzheimer’s Dis. 2016, 51, 591–604. [Google Scholar] [CrossRef]
- Cifelli, J.L.; Berg, K.R.; Yang, J. Benzothiazole amphiphiles promote RasGRF1-associated dendritic spine formation in human stem cell-derived neurons. FEBS Open Bio 2020, 10, 386–395. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.J.; Ramesha, S.; Weinstock, L.D.; Gao, T.; Ping, L.; Xiao, H.; Dammer, E.B.; Duong, D.D.; Levey, A.I.; Lah, J.J.; et al. Microglial ERK activation is a critical regulator of pro-inflammatory immune responses in Alzheimer’s disease. bioRxiv 2019, 798215. [Google Scholar]
- Haskin, J.; Szargel, R.; Shani, V.; Mekies, L.N.; Rott, R.; Lim, G.G.Y.; Lim, K.-L.; Bandopadhyay, R.; Wolosker, H.; Engelender, S. AF-6 is a positive modulator of the PINK1/parkin pathway and is deficient in Parkinson’s disease. Hum Mol Genet. 2013, 22, 2083–2096. [Google Scholar] [CrossRef]
- Eshraghi, M.; Nimrod Ramírez-Jarquín, U.; Shahani, N.; Nuzzo, T.; De Rosa, A.; Swarnkar, S.; Galli, N.; Rivera, O.; Tsaprailis, G.; Scharager-Tapia, C.; et al. RasGRP1 is a causal factor in the development of l-DOPA-induced dyskinesia in Parkinson’s disease. Sci. Adv. 2020, 6, eaaz7001. [Google Scholar] [CrossRef]
- Kumar, V.; Zhang, M.X.; Swank, M.W.; Kunz, J.; Wu, G.Y. Regulation of dendritic morphogenesis by Ras-PI3K-Akt-mTOR and Ras-MAPK signaling pathways. J. Neurosci. 2005, 25, 11288–11299. [Google Scholar] [CrossRef] [Green Version]
- Vaillant, A.R.; Mazzoni, I.; Tudan, C.; Boudreau, M.; Kaplan, D.R.; Miller, F.D. Depolarization and neurotrophins converge on the phosphatidylinositol 3- kinase-Akt pathway to synergistically regulate neuronal survival. J. Cell Biol. 1999, 146, 955–966. [Google Scholar] [CrossRef] [PubMed]
- Jaworski, J.; Spangler, S.; Seeburg, D.P.; Hoogenraad, C.C.; Sheng, M. Control of dendritic arborization by the phosphoinositide-3′-kinase- Akt-mammalian target of rapamycin pathway. J. Neurosci. 2005, 25, 11300–11312. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Na, L.; Li, Y.; Chen, L. Roles of the PI3K/AKT/mTOR signalling pathways in neurodegenerative diseases and tumours. Cell Biosci. 2020, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basil, A.H.; Sim, J.P.L.; Lim, G.G.Y.; Lin, S.; Chan, H.Y.; Engelender, S.; Lim, K.-L. AF-6 Protects Against Dopaminergic Dysfunction and Mitochondrial Abnormalities in Drosophila Models of Parkinson’s Disease. Front. Cell. Neurosci. 2017, 11, 241. [Google Scholar] [CrossRef]
- Szíber, Z.; Liliom, H.; Morales, C.O.O.; Ignácz, A.; Rátkai, A.E.; Ellwanger, K.; Link, G.; Szucs, A.; Hausser, A.; Schlett, K. Ras and Rab interactor 1 controls neuronal plasticity by coordinating dendritic filopodial motility and AMPA receptor turnover. Mol. Biol. Cell 2017, 28, 285–295. [Google Scholar] [CrossRef]
- Shirazi Fard, S.; Kele, J.; Vilar, M.; Paratcha, G.; Ledda, F. Tiam1 as a signaling mediator of Nerve Growth Factor-dependent neurite outgrowth. PLoS ONE 2010, 5, 9647. [Google Scholar] [CrossRef]
- Cajanek, L.; Ganji, R.S.; Henriques-Oliveira, C.; Theofilopoulos, S.; Konik, P.; Bryja, V.; Arenas, E. Tiam1 Regulates the Wnt/Dvl/Rac1 Signaling Pathway and the Differentiation of Midbrain Dopaminergic Neurons. Mol. Cell. Biol. 2013, 33, 59–70. [Google Scholar] [CrossRef] [Green Version]
- Yamauchi, J.; Miyamoto, Y.; Tanoue, A.; Shooter, E.M.; Chan, J.R. Ras activation of a Rac1 exchange factor, Tiam1, mediates neurotrophin-3-induced Schwann cell migration. Proc. Natl. Acad. Sci. USA 2005, 102, 14889–14894. [Google Scholar] [CrossRef] [Green Version]
- Shah, B.; Püschel, A.W. Regulation of Rap GTPases in mammalian neurons. Biol. Chem. 2016, 397, 1055–1069. [Google Scholar] [CrossRef]
- Raaijmakers, J.H.; Bos, J.L. Specificity in Ras and Rap signaling. J. Biol. Chem. 2009, 284, 10995–10999. [Google Scholar] [CrossRef] [Green Version]
- Jin, A.; Kurosu, T.; Tsuji, K.; Mizuchi, D.; Arai, A.; Fujita, H.; Hattori, M.; Minato, N.; Miura, O. BCR/ABL and IL-3 activate Rap1 to stimulate the B-Raf/MEK/Erk and Akt signaling pathways and to regulate proliferation, apoptosis, and adhesion. Oncogene 2006, 25, 4332–4340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Pak, D.; Qin, Y.; Mccormack, S.G.; Kim, M.J.; Baumgart, J.P.; Velamoor, V.; Auberson, Y.P.; Osten, P.; Van Aelst, L.; et al. Rap2-JNK removes synaptic AMPA receptors during depotentiation. Neuron 2005, 46, 905–916. [Google Scholar] [CrossRef] [Green Version]
- Woolfrey, K.M.; Srivastava, D.P.; Photowala, H.; Yamashita, M.; Barbolina, M.V.; Cahill, M.E.; Xie, Z.; Jones, K.A.; Quilliam, L.A.; Prakriya, M.; et al. Epac2 induces synapse remodeling and depression and its disease-associated forms alter spines. Nat. Neurosci. 2009, 12, 1275–1284. [Google Scholar] [CrossRef] [Green Version]
- Bacchelli, E.; Blasi, F.; Biondolillo, M.; Lamb, J.A.; Bonora, E.; Barnby, G.; Parr, J.; Beyer, K.S.; Klauck, S.M.; Poustka, A.; et al. Screening of nine candidate genes for autism on chromosome 2q reveals rare nonsynonymous variants in the cAMP-GEFII gene. Mol. Psychiatry 2003, 8, 916–924. [Google Scholar] [CrossRef] [Green Version]
- Dumbacher, M.; Van Dooren, T.; Princen, K.; De Witte, K.; Farinelli, M.; Lievens, S.; Tavernier, J.; Dehaen, W.; Wera, S.; Winderickx, J.; et al. Modifying Rap1-signalling by targeting Pde6δ is neuroprotective in models of Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 50. [Google Scholar] [CrossRef] [PubMed]
- Yarza, R.; Vela, S.; Solas, M.; Ramirez, M.J. c-Jun N-terminal kinase (JNK) signaling as a therapeutic target for Alzheimer’s disease. Front. Pharmacol. 2016, 6, 321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.; Raina, A.K.; Rottkamp, C.A.; Aliev, G.; Perry, G.; Boux, H.; Smith, M.A. Activation and redistribution of c-Jun N-terminal kinase/stress activated protein kinase in degenerating neurons in Alzheimer’s disease. J. Neurochem. 2001, 76, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.-N.; Jang, H.; Kim, G.H.; Noh, J.-E. RAPGEF2 mediates oligomeric Aβ-induced synaptic loss and cognitive dysfunction in Alzheimer’s disease. Res. Sq. 2020. [Google Scholar] [CrossRef]
- Xie, Z.; Huganir, R.L.; Penzes, P. Activity-dependent dendritic spine structural plasticity is regulated by small GTPase Rap1 and its target AF-6. Neuron 2005, 48, 605–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaudoin, G.M.J.; Schofield, C.M.; Nuwal, T.; Zang, K.; Ullian, E.M.; Huang, B.; Reichardt, L.F. Afadin, A Ras/Rap effector that controls cadherin function, promotes spine and excitatory synapse density in the hippocampus. J. Neurosci. 2012, 32, 99–110. [Google Scholar] [CrossRef]
- Levy, D.R.; Udgata, A.; Tourlomousis, P.; Symmons, M.F.; Hopkins, L.J.; Bryant, C.E.; Gay, N.J. Parkinson’s disease–associated kinase LRRK2 regulates genes required for cell adhesion, polarization, and chemotaxis in activated murine macrophages. J. Biol. Chem. 2020, 7, jbc.RA119.011842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zach, S.; Felk, S.; Gillardon, F. Signal transduction protein array analysis links LRRK2 to Ste20 kinases and PKC zeta that modulate neuronal plasticity. PLoS ONE 2010, 5, e13191. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Suaga, P.; Luzón-Toro, B.; Churamani, D.; Zhang, L.; Bloor-Young, D.; Patel, S.; Woodman, P.G.; Churchill, G.C.; Hilfiker, S. Leucine-rich repeat kinase 2 regulates autophagy through a calcium-dependent pathway involving NAADP. Hum. Mol. Genet. 2012, 21, 511–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.C.; Zhang, W.; Chua, L.L.; Chai, C.; Li, R.; Lin, L.; Cao, Z.; Angeles, D.C.; Stanton, L.W.; Peng, J.H.; et al. Phosphorylation of amyloid precursor protein by mutant LRRK2 promotes AICD activity and neurotoxicity in Parkinson’s disease. Sci. Signal. 2017, 10, eaam6790. [Google Scholar] [CrossRef] [Green Version]
- Seol, W.; Nam, D.; Son, I. Rab GTPases as physiological substrates of LRRK2 kinase. Exp. Neurobiol. 2019, 28, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.W.; Pei, Z.; Jiang, H.; Dawson, V.L.; Dawson, T.M.; Ross, C.A. Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat. Neurosci. 2006, 9, 1231–1233. [Google Scholar] [CrossRef]
- Nguyen, A.P.T.; Moore, D.J. Understanding the GTPase activity of LRRK2: Regulation, function, and neurotoxicity. Adv. Neurobiol. 2017, 14, 71–88. [Google Scholar] [PubMed] [Green Version]
- Zhang, X.; Huang, X.; Fang, C.; Li, Q.; Cui, J.; Sun, J.; Li, L. miR-124 Regulates the Expression of BACE1 in the Hippocampus Under Chronic Cerebral Hypoperfusion. Mol. Neurobiol. 2017, 54, 2498–2506. [Google Scholar] [CrossRef]
- An, F.; Gong, G.; Wang, Y.; Bian, M.; Yu, L.; Wei, C. MiR-124 acts as a target for Alzheimer’s disease by regulating BACE1. Oncotarget 2017, 8, 114065–114071. [Google Scholar] [CrossRef] [Green Version]
- Saucedo, L.J.; Gao, X.; Chiarelli, D.A.; Li, L.; Pan, D.; Edgar, B.A. Rheb promotes cell growth as a component of the insulin/TOR signalling network. Nat. Cell. Biol. 2003, 5, 566–571. [Google Scholar] [CrossRef]
- Sciarretta, S.; Zhai, P.; Shao, D.; Maejima, Y.; Robbins, J.; Volpe, M.; Condorelli, G.; Sadoshima, J. Rheb is a critical regulator of autophagy during myocardial ischemia: Pathophysiological implications in obesity and metabolic syndrome. Circulation 2012, 125, 1134–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, D.; Bai, X.; Zou, H.; Lai, Y.; Jiang, Y. Rheb GTPase controls apoptosis by regulating interaction of FKBP38 with Bcl-2 and Bcl-XL. J. Biol. Chem. 2010, 285, 8621–8627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Manning, B.D. A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem. Soc. Trans. 2009, 37, 217–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrando-Miguel, R.; Rosner, M.; Freilinger, A.; Lubec, G.; Hengstschläger, M. Tuberin—A new molecular target in Alzheimer’s disease? Neurochem. Res. 2005, 30, 1413–1419. [Google Scholar] [CrossRef]
- Habib, S.L.; Michel, D.; Masliah, E.; Thomas, B.; Ko, H.S.; Dawson, T.M.; Abboud, H.; Clark, R.A.; Imam, S.Z. Role of tuberin in neuronal degeneration. Neurochem. Res. 2008, 33, 1113–1116. [Google Scholar] [CrossRef]
- Caccamo, A.; Magrì, A.; Medina, D.X.; Wisely, E.V.; López-Aranda, M.F.; Silva, A.J.; Oddo, S. mTOR regulates tau phosphorylation and degradation: Implications for Alzheimer’s disease and other tauopathies. Aging Cell 2013, 12, 370–380. [Google Scholar] [CrossRef] [Green Version]
- Moon, G.J.; Kim, S.; Jeon, M.-T.; Lee, K.J.; Jang, I.-S.; Nakamura, M.; Kim, S.R. Therapeutic Potential of AAV1-Rheb(S16H) Transduction Against Alzheimer’s Disease. J. Clin. Med. 2019, 8, 2053. [Google Scholar] [CrossRef] [Green Version]
- Nam, J.H.; Leem, E.; Jeon, M.T.; Jeong, K.H.; Park, J.W.; Jung, U.J.; Kholodilov, N.; Burke, R.E.; Jin, B.K.; Kim, S.R. Induction of GDNF and BDNF by hRheb(S16H) Transduction of SNpc Neurons: Neuroprotective Mechanisms of hRheb(S16H) in a Model of Parkinson’s Disease. Mol. Neurobiol. 2015, 51, 487–499. [Google Scholar] [CrossRef]
- Kunimura, K.; Uruno, T.; Fukui, Y. DOCK family proteins: Key players in immune surveillance mechanisms. Int. Immunol. 2020, 32, 5–15. [Google Scholar] [CrossRef]
- Moon, G.J.; Shin, M.; Kim, S.R. Upregulation of neuronal Rheb(S16H) for hippocampal protection in the adult brain. Int. J. Mol. Sci. 2020, 21, 2023. [Google Scholar] [CrossRef] [Green Version]
- Shahani, N.; Pryor, W.; Swarnkar, S.; Kholodilov, N.; Thinakaran, G.; Burke, R.E.; Subramaniam, S. Rheb GTPase regulates β-secretase levels and amyloid β generation. J. Biol. Chem. 2014, 289, 5799–5808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahani, N.; Huang, W.C.; Varnum, M.; Page, D.T.; Subramaniam, S. Forebrain depletion of Rheb GTPase elicits spatial memory deficits in mice. Neurobiol. Aging 2017, 50, 134–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, J.L.; Delft, F.; Brennan, P.E. Targeting the Small GTPase Superfamily through Their Regulatory Proteins. Angew. Chem. Int. Ed. 2020, 59, 6342–6366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, D.H.; Cristóvão, A.C.; Guhathakurta, S.; Lee, J.; Joh, T.H.; Beal, M.F.; Kim, Y.S. NADPH oxidase 1-mediated oxidative stress leads to dopamine neuron death in Parkinson’s disease. Antioxid. Redox Signal. 2012, 16, 1033–1045. [Google Scholar] [CrossRef] [Green Version]
- Huesa, G.; Baltrons, A.; Gómez-Ramos, P.; On Morán, A.; García, A.; Hidalgo, J.; Francés, S.; Santpere, G.; Ferrer, I.; Galea, E. Altered Distribution of RhoA in Alzheimer’s Disease and AβPP Overexpressing Mice. J. Alzheimer’s Dis. 2010, 19, 37–56. [Google Scholar] [CrossRef] [Green Version]
- Tönges, L.; Frank, T.; Tatenhorst, L.; Saal, K.A.; Koch, J.C.; Va, E.’; Szego, M.; BäHr, M.; Weishaupt, J.H.; Lingor, P. Inhibition of rho kinase enhances survival of dopaminergic neurons and attenuates axonal loss in a mouse model of Parkinson’s disease. Brain 2012, 135, 3355–3370. [Google Scholar]
- Niu, M.; Xu, R.; Wang, J.; Hou, B.; Xie, A. MiR-133b ameliorates axon degeneration induced by MPP+ via targeting RhoA. Neuroscience 2016, 325, 39–49. [Google Scholar] [CrossRef]
- Amano, M.; Kaneko, T.; Maeda, A.; Nakayama, M.; Ito, M.; Yamauchi, T.; Goto, H.; Fukata, Y.; Oshiro, N.; Shinohara, A.; et al. Identification of Tau and MAP2 as novel substrates of Rho-kinase and myosin phosphatase. J. Neurochem. 2003, 87, 780–790. [Google Scholar] [CrossRef]
- Henderson, B.W.; Gentry, E.G.; Rush, T.; Troncoso, J.C.; Thambisetty, M.; Montine, T.J.; Herskowitz, J.H. Rho-associated protein kinase 1 (ROCK1) is increased in Alzheimer’s disease and ROCK1 depletion reduces amyloid-β levels in brain. J. Neurochem. 2016, 138, 525–531. [Google Scholar] [CrossRef]
- Lee, S.; Salazar, S.V.; Cox, T.O.; Strittmatter, S.M. Pyk2 signaling through graf1 and rhoA GTPase is required for amyloid-β oligomer-triggered synapse loss. J. Neurosci. 2019, 39, 1910–1929. [Google Scholar] [CrossRef]
- Zhang, X.; Ye, P.; Wang, D.; Liu, Y.; Cao, L.; Wang, Y.; Xu, Y.; Zhu, C. Involvement of RhoA/ROCK Signaling in Aβ-Induced Chemotaxis, Cytotoxicity and Inflammatory Response of Microglial BV2 Cells. Cell. Mol. Neurobiol. 2019, 39, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Scheiblich, H.; Bicker, G. Regulation of Microglial Phagocytosis by RhoA/ROCK-Inhibiting Drugs. Cell. Mol. Neurobiol. 2017, 37, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Kim, J.; Insolera, R.; Peng, X.; Fink, D.J.; Mata, M. Rho GTPase regulation of α-synuclein and VMAT2: Implications for pathogenesis of Parkinson’s disease. Mol. Cell. Neurosci. 2011, 48, 29–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, M.Y.; Kim, H.J.; Li, Y.; Kim, J.G.; Jeon, Y.J.; Won, H.Y.; Kim, J.S.; Kwon, H.Y.; Choi, I.G.; Ro, E.; et al. Involvement of small GTPase RhoA in the regulation of superoxide production in BV2 cells in response to fibrillar Aβ peptides. Cell. Signal. 2013, 25, 1861–1869. [Google Scholar] [CrossRef]
- Wilkinson, B.L.; Landreth, G.E. The microglial NADPH oxidase complex as a source of oxidative stress in Alzheimer’s disease. J. Neuroinflamm. 2006, 3. [Google Scholar] [CrossRef] [Green Version]
- Villar-Cheda, B.; Dominguez-Meijide, A.; Joglar, B.; Rodriguez-Perez, A.I.; Guerra, M.J.; Labandeira-Garcia, J.L. Involvement of microglial RhoA/Rho-Kinase pathway activation in the dopaminergic neuron death. Role of angiotensin via angiotensin type 1 receptors. Neurobiol. Dis. 2012, 47, 268–279. [Google Scholar] [CrossRef]
- Wang, S.; Chu, C.H.; Guo, M.; Jiang, L.; Nie, H.; Zhang, W.; Wilson, B.; Yang, L.; Stewart, T.; Hong, J.S.; et al. Identification of a specific α-synuclein peptide (α-Syn 29-40) capable of eliciting microglial superoxide production to damage dopaminergic neurons. J. Neuroinflamm. 2016, 13. [Google Scholar] [CrossRef] [Green Version]
- Hou, L.; Bao, X.; Zang, C.; Yang, H.; Sun, F.; Che, Y.; Wu, X.; Li, S.; Zhang, D.; Wang, Q. Integrin CD11b mediates α-synuclein-induced activation of NADPH oxidase through a Rho-dependent pathway. Redox Biol. 2018, 14, 600–608. [Google Scholar] [CrossRef]
- Wang, P.-L.; Niidome, T.; Akaike, A.; Kihara, T.; Sugimoto, H. Rac1 inhibition negatively regulates transcriptional activity of the amyloid precursor protein gene. J. Neurosci. Res. 2009, 87, 2105–2114. [Google Scholar] [CrossRef]
- Borin, M.; Saraceno, C.; Catania, M.; Lorenzetto, E.; Pontelli, V.; Paterlini, A.; Fostinelli, S.; Avesani, A.; Di Fede, G.; Zanusso, G.; et al. Rac1 activation links tau hyperphosphorylation and Aß dysmetabolism in Alzheimer’s disease. Acta Neuropathol. Commun. 2018, 6, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, Y.; Shibagaki, K.; Takata, K.; Tsuchiya, D.; Taniguchi, T.; Gebicke-Haerter, P.J.; Miki, H.; Takenawa, T.; Shimohama, S. Involvement of Wiskott-Aldrich Syndrome Protein Family Verprolin-Homologous Protein (WAVE) and Rac1 in the Phagocytosis of Amyloid-(1-42) in Rat Microglia. J. Pharmacol. Sci. 2003, 92, 115–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Calatayud, C.; Guha, S.; Fernández-Carasa, I.; Berkowitz, L.; Carballo-Carbajal, I.; Ezquerra, M.; Fernández-Santiago, R.; Kapahi, P.; Raya, Á.; et al. The Small GTPase RAC1/CED-10 Is Essential in Maintaining Dopaminergic Neuron Function and Survival Against α-Synuclein-Induced Toxicity. Mol. Neurobiol. 2018, 55, 7533–7552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, J.; Xie, Q.; Jia, J.; Liu, X.; Sun, J.; Deng, Y.; Yi, L. Integrated Analysis and Identification of Novel Biomarkers in Parkinson’s Disease. Front. Aging Neurosci. 2018, 10, 178. [Google Scholar] [CrossRef] [PubMed]
- Civiero, L.; Greggio, E. PAKs in the brain: Function and dysfunction. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 444–453. [Google Scholar] [CrossRef]
- Impey, S.; Davare, M.; Lasiek, A.; Fortin, D.; Ando, H.; Varlamova, O.; Obrietan, K.; Soderling, T.R.; Goodman, R.H.; Wayman, G.A. An activity-induced microRNA controls dendritic spine formation by regulating Rac1-PAK signaling. Mol. Cell. Neurosci. 2010, 43, 146–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, D.; Citro, A.; Cordy, J.M.; Shen, G.C.; Wolozin, B. Rac1 protein rescues neurite retraction caused by G2019s leucine-rich repeat kinase 2 (LRRK2). J. Biol. Chem. 2011, 286, 16140–16149. [Google Scholar] [CrossRef] [Green Version]
- Manterola, L.; Hernando-Rodríguez, M.; Ruiz, A.; Apraiz, A.; Arrizabalaga, O.; Vellón, L.; Alberdi, E.; Cavaliere, F.; Lacerda, H.M.; Jimenez, S.; et al. 1–42 β-amyloid peptide requires PDK1/nPKC/Rac 1 pathway to induce neuronal death. Transl. Psychiatry 2013, 3, e219. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, B.; Koenigsknecht-Talboo, J.; Grommes, C.; Lee, C.Y.D.; Landreth, G. Fibrillar β-amyloid-stimulated intracellular signaling cascades require Vav for induction of respiratory burst and phagocytosis in monocytes and microglia. J. Biol. Chem. 2006, 281, 20842–20850. [Google Scholar] [CrossRef] [Green Version]
- Wyssenbach, A.; Quintela, T.; Llavero, F.; Zugaza, J.L.; Matute, C.; Alberdi, E. Amyloid β-induced astrogliosis is mediated by β1-integrin via NADPH oxidase 2 in Alzheimer’s disease. Aging Cell 2016, 15, 1140–1152. [Google Scholar] [CrossRef]
- Silva, R.M.; Kuan, C.-Y.; Rakic, P.; Burke, R.E. Mixed lineage kinase-c-jun N-terminal kinase signaling pathway: A new therapeutic target in Parkinson’s disease. Mov. Disord. 2005, 20, 653–664. [Google Scholar] [CrossRef]
- Melendez, J.; Grogg, M.; Zheng, Y. Signaling role of Cdc42 in regulating mammalian physiology. J. Biol. Chem. 2011, 286, 2375–2381. [Google Scholar] [CrossRef] [Green Version]
- Endo, M.; Druso, J.E.; Cerione, R.A. The two splice variant forms of Cdc42 exert distinct and essential functions in neurogenesis. J. Biol. Chem. 2020, 295, 4498–4512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, M.P.; Russo, A.; O’Bryan, J.P. Emerging roles for intersectin (ITSN) in regulating signaling and disease pathways. Int. J. Mol. Sci. 2013, 14, 7829–7852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saraceno, C.; Catania, M.; Paterlini, A.; Fostinelli, S.; Ciani, M.; Zanardini, R.; Binetti, G.; Di Fede, G.; Caroppo, P.; Benussi, L.; et al. Altered Expression of circulating Cdc42 in frontotemporal lobar degeneration. J. Alzheimer’s Dis. 2018, 61, 1477–1483. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.D.; Kamiyama, D.; Kolodziej, P.; Hing, H.; Chiba, A. Isolation of Rho GTPase effector pathways during axon development. Dev. Biol. 2003, 262, 282–293. [Google Scholar] [CrossRef] [Green Version]
- Ramakers, G.J.A. Rho proteins, mental retardation and the cellular basis of cognition. Trends Neurosci. 2002, 25, 191–199. [Google Scholar] [CrossRef]
- Salminen, A.; Suuronen, T.; Kaarniranta, K. ROCK, PAK, and Toll of synapses in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2008, 371, 587–590. [Google Scholar] [CrossRef]
- Musilli, M.; Ciotti, M.T.; Pieri, M.; Martino, A.; Borrelli, S.; Dinallo, V.; Diana, G. Therapeutic effects of the Rho GTPase modulator CNF1 in a model of Parkinson’s disease. Neuropharmacology 2016, 109, 357–365. [Google Scholar] [CrossRef]
- Alekhina, O.; Burstein, E.; Billadeau, D.D. Cellular functions of WASP family proteins at a glance. J. Cell Sci. 2017, 130, 2235–2241. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, Y.; Tsuchiya, D.; Takata, K.; Shibagaki, K.; Taniguchi, T.; Smith, M.A.; Perry, G.; Miki, H.; Takenawa, T.; Shimohama, S. Possible involvement of Wiskott-Aldrich syndrome protein family in aberrant neuronal sprouting in Alzheimer’s disease. Neurosci. Lett. 2003, 346, 149–152. [Google Scholar] [CrossRef]
- Irie, F.; Yamaguchi, Y. EphB receptors regulate dendritic spine development via intersectin, Cdc42 and N-WASP. Nat. Neurosci. 2002, 5, 1117–1118. [Google Scholar] [CrossRef] [PubMed]
- Lauretti, E.; Dincer, O.; Praticò, D. Glycogen synthase kinase-3 signaling in Alzheimer’s disease. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118664. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Chung, Y.H.; Joo, K.M.; Lim, H.C.; Jeon, G.S.; Kim, D.; Lee, W.B.; Kim, Y.S.; Cha, C.I. Age-related changes in glycogen synthase kinase 3β (GSK3β) immunoreactivity in the central nervous system of rats. Neurosci. Lett. 2006, 409, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Leroy, K.; Yilmaz, Z.; Brion, J.P. Increased level of active GSK-3β in Alzheimer’s disease and accumulation in argyrophilic grains and in neurones at different stages of neurofibrillary degeneration. Neuropathol. Appl. Neurobiol. 2007, 33, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Pelleieux, S.; Picard, C.; Lamarre-Théroux, L.; Dea, D.; Leduc, V.; Tsantrizos, Y.S.; Poirier, J. Isoprenoids and tau pathology in sporadic Alzheimer’s disease. Neurobiol. Aging 2018, 65, 132–139. [Google Scholar] [CrossRef]
- Wesén, E.; Jeffries, G.D.M.; Dzebo, M.M.; Esbjörner, E.K. Endocytic uptake of monomeric amyloid-β peptides is clathrin- A nd dynamin-independent and results in selective accumulation of Aβ(1-42) compared to Aβ(1-40). Sci. Rep. 2017, 7, 1–14. [Google Scholar]
- Lichtenstein, M.P.; Carriba, P.; Masgrau, R.; Pujol, A.; Galea, E. Staging anti-inflammatory therapy in Alzheimer’s disease. Front. Aging Neurosci. 2010, 2, 142. [Google Scholar] [CrossRef] [Green Version]
- Sutton, S.S.; Magagnoli, J.; Cummings, T.; Hardin, J.W. Association between thiopurine medication exposure and Alzheimer’s disease among a cohort of patients with inflammatory bowel disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2019, 5, 809–813. [Google Scholar] [CrossRef]
- Musilli, M.; Nicolia, V.; Borrelli, S.; Scarpa, S.; Diana, G. Behavioral effects of Rho GTPase modulation in a model of Alzheimer’s disease. Behav. Brain Res. 2013, 237, 223–229. [Google Scholar] [CrossRef]
- Paintlia, A.S.; Paintlia, M.K.; Singh, A.K.; Singh, I. Inhibition of Rho family functions by lovastatin promotes myelin repair in ameliorating experimental autoimmune encephalomyelitis. Mol. Pharmacol. 2008, 73, 1381–1393. [Google Scholar] [CrossRef] [Green Version]
- Hampel, H.; Vergallo, A.; Caraci, F.; Claudio Cuello, A.; Lemercier, P.; Vellas, B.; Giudici, K.V.; Baldacci, F.; Hänisch, B.; Haberkamp, M.; et al. Alzheimer Precision Medicine Initiative (APMI). Future avenues for Alzheimer’s disease detection and therapy: Liquid biopsy, intracellular signaling modulation, systems pharmacology drug discovery. Neurophamacology 2020, 108081. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; O’Bryant, S.E.; Molinuevo, J.L.; Zetterberg, H.; Masters, C.L.; Lista, S.; Kiddle, S.J.; Batrla, R.; Blennow, K. Blood-based biomarkers for Alzheimer disease: Mapping the road to the clinic. Nat. Rev. Neurol. 2018, 14, 639–652. [Google Scholar] [CrossRef] [PubMed]
- Chahine, L.M.; Stern, M.B.; Chen-Plotkin, A. Blood-based biomarkers for Parkinson’s disease. Parkinsonism Relat. Disord. 2014, 20, 99–103. [Google Scholar] [CrossRef] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arrazola Sastre, A.; Luque Montoro, M.; Gálvez-Martín, P.; Lacerda, H.M.; Lucia, A.; Llavero, F.; Zugaza, J.L. Small GTPases of the Ras and Rho Families Switch on/off Signaling Pathways in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 6312. https://doi.org/10.3390/ijms21176312
Arrazola Sastre A, Luque Montoro M, Gálvez-Martín P, Lacerda HM, Lucia A, Llavero F, Zugaza JL. Small GTPases of the Ras and Rho Families Switch on/off Signaling Pathways in Neurodegenerative Diseases. International Journal of Molecular Sciences. 2020; 21(17):6312. https://doi.org/10.3390/ijms21176312
Chicago/Turabian StyleArrazola Sastre, Alazne, Miriam Luque Montoro, Patricia Gálvez-Martín, Hadriano M Lacerda, Alejandro Lucia, Francisco Llavero, and José Luis Zugaza. 2020. "Small GTPases of the Ras and Rho Families Switch on/off Signaling Pathways in Neurodegenerative Diseases" International Journal of Molecular Sciences 21, no. 17: 6312. https://doi.org/10.3390/ijms21176312
APA StyleArrazola Sastre, A., Luque Montoro, M., Gálvez-Martín, P., Lacerda, H. M., Lucia, A., Llavero, F., & Zugaza, J. L. (2020). Small GTPases of the Ras and Rho Families Switch on/off Signaling Pathways in Neurodegenerative Diseases. International Journal of Molecular Sciences, 21(17), 6312. https://doi.org/10.3390/ijms21176312