Application of Various Molecular Modelling Methods in the Study of Estrogens and Xenoestrogens





Abstract
:1. Introduction
1.1. Estrogens and Xenoestrogens: Types, Main Representatives, and Their Toxicity
1.2. Estrogen-Related Biomolecules as the Molecular Modelling Study Objects
2. Application of Molecular Modelling Methods in the Study of Estrogens and Xenoestrogens
2.1. Application to Estrogens
2.1.1. Ligand Docking Using Force Field Methods
- Principles of docking and re-docking
- Enzymes and receptors used as targets in estrogen-related docking studies
- Docking studies of plant-derived potential xenoestrogens
2.1.2. Quantitative Structure–Activity Relationship (QSAR)
2.1.3. Advanced Docking Using Combined Quantum Mechanics/Molecular Mechanics (QM/MM) or Molecular Dynamics (MD) Methods
2.1.4. Other MD-Based Studies of Estrogens
- Membranes
- Nanotubes
2.1.5. Density Functional Theory (DFT) Calculations in the Study of Estrogens
- Crystal structure prediction
- NMR and vibrational properties calculations
- Removal of estrogenic pollutants
2.2. Application of Various Molecular Modelling Methods in the Study of Xenoestrogens
2.2.1. Various Molecular Modelling Methods Applied in Xenoestrogen Studies
2.2.2. Bisphenol A
- Bisphenol A (BPA)–ER complex studies
- Risk assessment and removal attempts
2.2.3. Phthalates
2.2.4. Technical Aspects of Calculations Performed on (xeno)Estrogens
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 17β-HSD | 17β-hydroxysteroid dehydrogenase |
| ADMET | Adsorption distribution metabolism elimination toxicity |
| BPA | Bisphenol A |
| CoMFA | Comparative molecular field analysis |
| DBD | DNA binding domain |
| DFT | Density functional theory |
| E1 | Estrone |
| E2 | Estradiol |
| E3 | Estriol |
| E4 | Estretrol |
| ER | Estrogen receptor |
| EDCs | Endocrine-disrupting chemicals |
| FEP | Free energy perturbation |
| H-K | Hohenberg–Kohn theorems |
| K-S | Kohn–Sham theorems |
| LBD | Ligand-binding domain |
| LIE | Linear interaction energy |
| MD | Molecular dynamics |
| MM | Molecular mechanics |
| PDB | Protein Data Bank |
| QM | Quantum mechanics |
| QSAR | Quantitative structure–activity relationship |
| RBA | Relative binding affinity |
| SERMs | Selective estrogen receptor modulators |
| SHBG | Sex hormone-binding globulin |
| STS | Sulfatase |
| SULT | Sulfotransferase |
References
- Bennink, H.C.; Verhoeven, C.; Zimmerman, Y.; Visser, M.; Foidart, J.-M.; Gemzell-Danielsson, K. Clinical effects of the fetal estrogen estetrol in a multiple-rising-dose study in postmenopausal women. Matur. Eur. Menopause J. 2016, 91, 93–100. [Google Scholar] [CrossRef] [Green Version]
- Singleton, D.W.; David, W.S. Xenoestrogen exposure and mechanisms of endocrine disruption. Front. Biosci. 2003, 8, 110–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennink, F.C.; Holinka, C.F.; Visser, M.; Bennink, H.J.T.C. Maternal and fetal estetrol levels during pregnancy. Climacteric 2008, 11, 69–72. [Google Scholar] [CrossRef]
- Schreiner, W.E. The Ovary in Labhart, A. Clinical Endocrinology: Theory and Practice; Springer: Berlin/Heidelberg, Germany, 2012; p. 548. [Google Scholar]
- Kuhl, H. Pharmacology of estrogens and progestogens: Influence of different routes of administration. Climacteric 2005, 8 (Suppl. 1), 3–63. [Google Scholar] [CrossRef]
- Blackburn, S. Maternal, Fetal, & Neonatal Physiology; Elsevier: Amsterdam, The Netherlands, 2014. [Google Scholar]
- Fait, T. Menopause hormone therapy: Latest developments and clinical practice. Drugs Context 2019, 8, 1–9. [Google Scholar] [CrossRef]
- Tofovic, S.P.; Jackson, E.K. Estradiol Metabolism: Crossroads in Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2019, 21, 116. [Google Scholar] [CrossRef] [Green Version]
- EC. Corrigendum to Commission Regulation (EU) 2018/605 of 19 April 2018 Amending Annex II to Regulation (EC) No 1107/2009 by Setting Out Scientific Criteria for the Determination of Endocrine Disrupting Properties. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32018R0605&rid=210 (accessed on 25 August 2020).
- Ciubotaru, R.M.; Oyedele, J.; Zancanaro, G. Annual assessment of Echinococcus multilocularis surveillance reports submitted in 2018 in the context of Commission Regulation (EU) No 1152/2011. EFSA J. 2018, 16, 33–36. [Google Scholar] [CrossRef] [Green Version]
- OECD. Guidance Document on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption, No. 150, Update v3, Series on Testing and Assessment, ENV/JM/MONO(2012)22; OECD: Paris, France, 2017; 988p. [Google Scholar]
- OECD. Conceptual Framework for Testing and Assessment of Endocrine Disrupters. Available online: https://www.oecd.org/env/ehs/testing/oecdworkrelatedtoendocrinedisrupters.htm (accessed on 25 August 2020).
- Safe, S.; Khan, S.; Wu, F.; Li, X. Chemical-induced estrogenicity in Xenoestrogens and phytoestrogens as SERMs and implications for risk assessment (Chapter 65). In Veterinary Toxicology Basic and Clinical Principles; Elsevier: Amsterdam, The Netherlands, 2007; pp. 811–822. [Google Scholar]
- Cotterill, J.; Palazzolo, L.; Ridgway, C.; Price, N.; Rorije, E.; Moretto, A.; Peijnenburg, A.; Eberini, I. Predicting estrogen receptor binding of chemicals using a suite of in silico methods—Complementary approaches of (Q)SAR, molecular docking and molecular dynamics. Toxicol. Appl. Pharmacol. 2019, 378, 114630. [Google Scholar] [CrossRef]
- Kerdivel, G.; Habauzit, D.; Pakdel, F. Assessment and Molecular Actions of Endocrine-Disrupting Chemicals That Interfere with Estrogen Receptor Pathways. Int. J. Endocrinol. 2013, 2013, 501851. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Czapla, L.; Amaro, R.E. Molecular Simulations of Aromatase Reveal New Insights into the Mechanism of Ligand Binding. J. Chem. Inf. Model. 2013, 53, 2047–2056. [Google Scholar] [CrossRef] [Green Version]
- Lokhande, K.; Mehere, A.S.; Yadav, A.K.; Swamy, K.V. Molecular Modeling and Docking Studies of Aromatase Inhibitors with Aromatase for ERP Breast Cancer. In Proceedings of the 86th Conference of Society of Biological Chemists: Emerging Discoveries in Health and Agricultural Sciences, New Delhi, India, 16–19 November 2017. [Google Scholar] [CrossRef]
- Kang, H.; Xiao, X.; Huang, C.; Yuan, Y.; Tang, D.; Dai, X.; Zeng, X. Potent aromatase inhibitors and molecular mechanism of inhibitory action. Eur. J. Med. Chem. 2018, 143, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, M.; Singh, S.; Pandey, V.P.; Dwivedi, U.N. Molecular docking and 3D-QSAR-based virtual screening of flavonoids as potential aromatase inhibitors against estrogen-dependent breast cancer. J. Biomol. Struct. Dyn. 2014, 33, 804–819. [Google Scholar] [CrossRef] [PubMed]
- Suvannang, N.; Nantasenamat, C.; Isarankura-Na-Ayudhya, C.; Prachayasittikul, V. Molecular Docking of Aromatase Inhibitors. Molecules 2011, 16, 3597–3617. [Google Scholar] [CrossRef] [Green Version]
- Rampogu, S.; Son, M.; Park, C.; Kim, H.-H.; Suh, J.-K.; Lee, K. Sulfonanilide Derivatives in Identifying Novel Aromatase Inhibitors by Applying Docking, Virtual Screening, and MD Simulations Studies. BioMed Res. Int. 2017, 2017, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narayana, B.L.; Kishore, D.P.; Balakumar, C.; Rao, K.V.; Kaur, R.; Rao, A.R.; Murthy, J.N.; Ravikumar, M. Molecular Modeling Evaluation of Non-Steroidal Aromatase Inhibitors†. Chem. Boil. Drug Des. 2012, 79, 674–682. [Google Scholar] [CrossRef] [PubMed]
- Fischer, J.; Ganellin, C.R. Analogue-Based Drug Discovery; John Wiley & Sons: Hoboken, NJ, USA, 2006; p. 516. [Google Scholar]
- Maltais, R.; Ayan, D.; Trottier, A.; Barbeau, X.; Lagüe, P.; Bouchard, J.-E.; Poirier, D. Discovery of a Non-Estrogenic Irreversible Inhibitor of 17β-Hydroxysteroid Dehydrogenase Type 1 from 3-Substituted-16β-(m-carbamoylbenzyl)-estradiol Derivatives. J. Med. Chem. 2013, 57, 204–222. [Google Scholar] [CrossRef]
- Lespérance, M.; Barbeau, X.; Roy, J.; Maltais, R.; Lagüe, P.; Poirier, D. Chemical synthesis of C3-oxiranyl/oxiranylmethyl-estrane derivatives targeted by molecular modeling and tested as potential inhibitors of 17β-hydroxysteroid dehydrogenase type 1. Steroids 2018, 140, 104–113. [Google Scholar] [CrossRef]
- Maltais, R.; Djiemeny, A.N.; Roy, J.; Barbeau, X.; Lambert, J.-P.; Poirier, D. Design and synthesis of dansyl-labeled inhibitors of steroid sulfatase for optical imaging. Bioorg. Med. Chem. 2020, 28, 115368. [Google Scholar] [CrossRef]
- Rakers, C.; Schumacher, F.; Meinl, W.; Glatt, H.; Kleuser, B.; Wolber, G. In SilicoPrediction of Human Sulfotransferase 1E1 Activity Guided by Pharmacophores from Molecular Dynamics Simulations. J. Boil. Chem. 2015, 291, 58–71. [Google Scholar] [CrossRef] [Green Version]
- Kisselev, P.; Schunck, W.-H.; Roots, I.; Schwarz, D. Association of CYP1A1 Polymorphisms with Differential Metabolic Activation of 17 -Estradiol and Estrone. Cancer Res. 2005, 65, 2972–2978. [Google Scholar] [CrossRef] [Green Version]
- Hong, C.-C.; Tang, B.-K.; Hammond, G.L.; Tritchler, D.; Yaffe, M.; Boyd, N.F. Cytochrome P450 1A2 (CYP1A2) activity and risk factors for breast cancer: A cross-sectional study. Breast Cancer Res. 2004, 6, R352–R365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida, C.R.; Everett, S.; De Montellano, P.R.O. Specificity Determinants of CYP1B1 Estradiol Hydroxylation. Mol. Pharmacol. 2013, 84, 451–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selby, C. Sex Hormone Binding Globulin: Origin, Function and Clinical Significance. Ann. Clin. Biochem. Int. J. Lab. Med. 1990, 27, 532–541. [Google Scholar] [CrossRef]
- Dechering, K.; Boersma, C.; Mosselman, S. Estrogen receptors alpha and beta: Two receptors of a kind? Curr. Med. Chem. 2000, 7, 561–576. [Google Scholar] [CrossRef] [PubMed]
- Paterni, I.; Granchi, C.; Katzenellenbogen, J.A.; Minutolo, F. Estrogen receptors alpha (ERα) and beta (ERβ): Subtype-selective ligands and clinical potential. Steroids 2014, 90, 13–29. [Google Scholar] [CrossRef] [Green Version]
- Ruff, M.; Gangloff, M.; Wurtz, J.; Moras, D. Estrogen receptor transcription and transactivation Structure-function relationship in DNA- and ligand-binding domains of estrogen receptors. Breast Cancer Res. 2000, 2, 353–359. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Zakharov, M.N.; Khan, S.H.; Miki, R.; Jang, H.; Toraldo, G.; Singh, R.; Bhasin, S.; Jasuja, R. The Dynamic Structure of the Estrogen Receptor. J. Amino Acids 2011, 2011, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Billon-Galés, A.; Krust, A.; Fontaine, C.; Abot, A.; Flouriot, G.; Toutain, C.; Berges, H.; Gadeau, A.-P.; Lenfant, F.; Gourdy, P.; et al. Activation function 2 (AF2) of estrogen receptor-α is required for the atheroprotective action of estradiol but not to accelerate endothelial healing. Proc. Natl. Acad. Sci. USA 2011, 108, 13311–13316. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Tu, Y.; Ågren, H.; Eriksson, L.A. Characterization of Agonist Binding to His524 in the Estrogen Receptor α Ligand Binding Domain. J. Phys. Chem. B 2012, 116, 4823–4830. [Google Scholar] [CrossRef]
- Hu, G.; Wang, J. Ligand selectivity of estrogen receptors by a molecular dynamics study. Eur. J. Med. Chem. 2014, 74, 726–735. [Google Scholar] [CrossRef]
- Gu, X. Helix 12 in the human estrogen receptor (hER) is essential for the hER function by overcoming nucleosome repression in yeast. J. Cell. Biochem. 2002, 86, 224–238. [Google Scholar] [CrossRef] [PubMed]
- Shiau, A.K.; Barstad, D.; Loria, P.M.; Cheng, L.; Kushner, P.J.; Agard, D.A.; Greene, G.L. The Structural Basis of Estrogen Receptor/Coactivator Recognition and the Antagonism of This Interaction by Tamoxifen. Cell 1998, 95, 927–937. [Google Scholar] [CrossRef] [Green Version]
- Fuentes, N.; Silveyra, P. Estrogen receptor signaling mechanisms. Adv. Protein Chem. Struct. Biol. 2019, 116, 135–170. [Google Scholar] [CrossRef] [PubMed]
- Stefkovich, M.L.; Arao, Y.; Hamilton, K.J.; Korach, K.S. Experimental models for evaluating non-genomic estrogen signaling. Steroids 2018, 133, 34–37. [Google Scholar] [CrossRef] [PubMed]
- Vrtacnik, P.; Ostanek, B.; Mencej-Bedrac, S.; Marc, J. The many faces of estrogen signaling. Biochem. Medica 2014, 24, 329–342. [Google Scholar] [CrossRef] [Green Version]
- Rosano, C.; Ponassi, M.; Santolla, M.F.; Pisano, A.; Felli, L.; Vivacqua, A.; Maggiolini, M.; Lappano, R. Macromolecular Modelling and Docking Simulations for the Discovery of Selective GPER Ligands. AAPS J. 2015, 18, 41–46. [Google Scholar] [CrossRef] [Green Version]
- Bruno, A.; Aiello, F.; Costantino, G.; Radi, M. Homology Modeling, Validation and Dynamics of the G Protein-coupled Estrogen Receptor 1 (GPER-1). Mol. Inform. 2016, 35, 333–339. [Google Scholar] [CrossRef]
- Méndez-Luna, D.; Martínez-Archundia, M.; Maroun, R.C.; Ceballos, G.; Fragoso-Vázquez, M.; González-Juárez, D.; Correa-Basurto, J. Deciphering the GPER/GPR30-agonist and antagonists interactions using molecular modeling studies, molecular dynamics, and docking simulations. J. Biomol. Struct. Dyn. 2015, 33, 1–12. [Google Scholar] [CrossRef]
- Taylor, R.; Jewsbury, P.; Essex, J.W. A review of protein-small molecule docking methods. J. Comput. Mol. Des. 2002, 16, 151–166. [Google Scholar] [CrossRef]
- RCSB PDB. Available online: https://www.rcsb.org/ (accessed on 25 August 2020).
- Landeros-Martinez, L.-L.; Glossman-Mitnik, D.; Orrantia-Borunda, E.; Flores-Holguin, N. A Combined Molecular Docking and Electronic Structure Study for a Breast Cancer Drug Design. In Molecular Docking; IntechOpen: London, UK, 2018. [Google Scholar] [CrossRef] [Green Version]
- Muchtaridi, M.; Megantara, S.; Dermawan, D.; Yusuf, M. Antagonistic mechanism of α-mangostin derivatives against human estrogen receptor α of breast cancer using molecular dynamics simulation. Rasayan J. Chem. 2019, 12, 1927–1934. [Google Scholar] [CrossRef]
- Cavasotto, C.; Aucar, M.G. High-Throughput Docking Using Quantum Mechanical Scoring. Front. Chem. 2020, 8, 246. [Google Scholar] [CrossRef] [PubMed]
- Spiriti, J.; Subramanian, S.R.; Palli, R.; Wu, M.; Zuckerman, D.M. Middle-way flexible docking: Pose prediction using mixed-resolution Monte Carlo in estrogen receptor α. PLoS ONE 2019, 14, e0215694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolohan, P.; Reichert, D.E. CoMSIA and docking study of rhenium based estrogen receptor ligand analogs. Steroids 2007, 72, 247–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wierbowski, S.D.; Wingert, B.M.; Zheng, J.; Camacho, C.J. Cross-docking benchmark for automated pose and ranking prediction of ligand binding. Protein Sci. 2019, 29, 298–305. [Google Scholar] [CrossRef] [Green Version]
- Shtaiwi, A.; Adnan, R.; Khairuddean, M.; Khan, S.U. Computational investigations of the binding mechanism of novel benzophenone imine inhibitors for the treatment of breast cancer. RSC Adv. 2019, 9, 35401–35416. [Google Scholar] [CrossRef] [Green Version]
- Ling, J.; Oh, D.L.; Chia, A.Y.Y. Molecular Docking Studies of Glycyrrhizic Acid (GA), Glycyrrhetic Acid (GE) and Glabridin (GLA) with Estrogen Receptors (ERs). Biosci. Biotechnol. Res. Asia 2017, 14, 1–11. [Google Scholar] [CrossRef]
- Pang, X.; Fu, W.; Wang, J.; Kang, D.; Xu, L.; Zhao, Y.; Liu, A.; Du, G.-H. Identification of Estrogen Receptor α Antagonists from Natural Products via In Vitro and In Silico Approaches. Oxidative Med. Cell. Longev. 2018, 2018, 6040149. [Google Scholar] [CrossRef] [Green Version]
- Muchtaridi, M.; Dermawan, D.; Yusuf, M. Molecular Docking, 3D Structure-Based Pharmacophore Modeling, and ADME Prediction of Alpha Mangostin and Its Derivatives against Estrogen Receptor Alpha. J. Young Pharm. 2018, 10, 252–259. [Google Scholar] [CrossRef] [Green Version]
- Yu, E.; Xu, Y.; Shi, Y.; Yu, Q.; Liu, J.; Xu, L. Discovery of novel natural compound inhibitors targeting estrogen receptor α by an integrated virtual screening strategy. J. Mol. Model. 2019, 25, 278. [Google Scholar] [CrossRef]
- Abdelsamie, A.S.; Salah, M.; Siebenbürger, L.; Hamed, M.M.; Börger, C.; Van Koppen, C.J.; Frotscher, M.; Hartmann, R.W. Development of potential preclinical candidates with promising in vitro ADME profile for the inhibition of type 1 and type 2 17β-Hydroxysteroid dehydrogenases: Design, synthesis, and biological evaluation. Eur. J. Med. Chem. 2019, 178, 93–107. [Google Scholar] [CrossRef]
- Hasan, T.N.; B, L.G.; A Masoodi, T.; Shafi, G.; Alshatwi, A.A.; Sivashanmugham, P. Affinity of estrogens for human progesterone receptor A and B monomers and risk of breast cancer: A comparative molecular modeling study. Adv. Appl. Bioinform. Chem. 2011, 4, 29–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, X.; Wang, P.; Zhu, B.T. Characterization of the Estradiol-Binding Site Structure of Human Protein Disulfide Isomerase (PDI). PLoS ONE 2011, 6, e27185. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, A.J.; Dos Santos, E.S. Aqueous solution interactions with sex hormone-binding globulin and estradiol: A theoretical investigation. J. Boil. Phys. 2018, 44, 539–556. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.P.; Potter, B.V. The structural biology of oestrogen metabolism. J. Steroid Biochem. Mol. Boil. 2013, 137, 27–49. [Google Scholar] [CrossRef]
- Maltais, R.; Fournier, D.; Poirier, D.; Maltais, R. Quantitative Structure-Activity Relationship (QSAR) Study with a Series of 17α-Derivatives of Estradiol: Model for the Development of Reversible Steroid Sulfatase Inhibitors. QSAR Comb. Sci. 2009, 28, 1284–1299. [Google Scholar] [CrossRef]
- Granados, S.T.; Castillo, K.; Bravo-Moraga, F.; Sepúlveda, R.V.; Carrasquel-Ursulaez, W.; Rojas, M.; Carmona, E.; Lorenzo-Ceballos, Y.; González-Nilo, F.D.; González, C.; et al. The molecular nature of the 17β-Estradiol binding site in the voltage- and Ca2+-activated K+ (BK) channel β1 subunit. Sci. Rep. 2019, 9, 9965. [Google Scholar] [CrossRef]
- McCullough, C.; Neumann, T.S.; Gone, J.R.; He, Z.; Herrild, C.; (Nee Lukesh) Wondergem, J.; Pandey, R.K.; Donaldson, W.A.; Sem, D.S. Probing the human estrogen receptor-α binding requirements for phenolic mono- and di-hydroxyl compounds: A combined synthesis, binding and docking study. Bioorg. Med. Chem. 2013, 22, 303–310. [Google Scholar] [CrossRef] [Green Version]
- Kerdivel, G.; Le Guével, R.; Habauzit, D.; Brion, F.; Ait-Aissa, S.; Pakdel, F. Estrogenic Potency of Benzophenone UV Filters in Breast Cancer Cells: Proliferative and Transcriptional Activity Substantiated by Docking Analysis. PLoS ONE 2013, 8, e60567. [Google Scholar] [CrossRef] [Green Version]
- Sauvée, C.; Schäfer, A.; Sundén, H.; Ma, J.-N.; Gustavsson, A.-L.; Burstein, E.S.; Olsson, R. The A-CD analogue of 16β,17α-estriol is a potent and highly selective estrogen receptor β agonist. MedChemComm 2013, 4, 1439. [Google Scholar] [CrossRef]
- Kucinska, M.; Giron, M.D.; Piotrowska, H.; Lisiak, N.; Granig, W.H.; Lopez-Jaramillo, F.-J.; Salto, R.; Murias, M.; Erker, T. Novel Promising Estrogenic Receptor Modulators: Cytotoxic and Estrogenic Activity of Benzanilides and Dithiobenzanilides. PLoS ONE 2016, 11, e0145615. [Google Scholar] [CrossRef]
- Gonzalez, T.L.; Rae, J.M.; Colacino, J.A.; Richardson, R.J. Homology models of mouse and rat estrogen receptor-α ligand-binding domain created by in silico mutagenesis of a human template: Molecular docking with 17β-estradiol, diethylstilbestrol, and paraben analogs. Comput. Toxicol. 2019, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Yarger, J.G.; E Babine, R.; Bittner, M.; Shanle, E.; Xu, W.; Hershberger, P.; Nye, S.H. Structurally similar estradiol analogs uniquely alter the regulation of intracellular signaling pathways. J. Mol. Endocrinol. 2012, 50, 43–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maltais, R.; Trottier, A.; Barbeau, X.; Lagüe, P.; Perreault, M.; Thériault, J.-F.; Lin, S.X.; Poirier, D. Impact of structural modifications at positions 13, 16 and 17 of 16β-(m-carbamoylbenzyl)-estradiol on 17β-hydroxysteroid dehydrogenase type 1 inhibition and estrogenic activity. J. Steroid Biochem. Mol. Boil. 2016, 161, 24–35. [Google Scholar] [CrossRef] [PubMed]
- E Starkey, N.J.; Li, Y.; Drenkhahn-Weinaug, S.K.; Liu, J.; Lubahn, D.B. 27-Hydroxycholesterol Is an Estrogen Receptor β-Selective Negative Allosteric Modifier of 17β-Estradiol Binding. Endocrinology 2018, 159, 1972–1981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, K.; Munuganti, R.S.N.; Leblanc, E.; Lin, Y.L.; Leung, E.; Lallous, N.; Butler, M.S.; Cherkasov, A.; Rennie, P.S. In silico discovery and validation of potent small-molecule inhibitors targeting the activation function 2 site of human oestrogen receptor α. Breast Cancer Res. 2015, 17, 27. [Google Scholar] [CrossRef] [Green Version]
- Durrant, J.D.; Carlson, K.E.; Martin, T.A.; Offutt, T.L.; Mayne, C.G.; Katzenellenbogen, J.A.; Amaro, R.E. Neural-Network Scoring Functions Identify Structurally Novel Estrogen-Receptor Ligands. J. Chem. Inf. Model. 2015, 55, 1953–1961. [Google Scholar] [CrossRef] [Green Version]
- Ng, H.W.; Zhang, W.; Shu, M.; Luo, H.; Ge, W.; Perkins, R.; Tong, W.; Hong, H. Competitive molecular docking approach for predicting estrogen receptor subtype α agonists and antagonists. BMC Bioinform. 2014, 15, S4. [Google Scholar] [CrossRef] [Green Version]
- Amr, A.E.-G.E.; Elsayed, E.; Al-Omar, M.; Eldin, H.O.B.; Nossier, E.S.; Abdalla, M.M. Design, Synthesis, Anticancer Evaluation and Molecular Modeling of Novel Estrogen Derivatives. Molecules 2019, 24, 416. [Google Scholar] [CrossRef] [Green Version]
- Stjernschantz, E.; Reinen, J.; Meinl, W.; George, B.J.; Glatt, H.; Vermeulen, N.P.; Oostenbrink, C. Comparison of murine and human estrogen sulfotransferase inhibition in vitro and in silico—Implications for differences in activity, subunit dimerization and substrate inhibition. Mol. Cell. Endocrinol. 2010, 317, 127–140. [Google Scholar] [CrossRef] [Green Version]
- Hanson, R.N.; McCaskill, E.; Tongcharoensirikul, P.; Dilis, R.; Labaree, D.; Hochberg, R.B. Synthesis and evaluation of 17α-(dimethylphenyl)vinyl estradiols as probes of the estrogen receptor-α ligand binding domain. Steroids 2012, 77, 471–476. [Google Scholar] [CrossRef] [Green Version]
- Grande, F.; Rizzuti, B.; Occhiuzzi, M.A.; Ioele, G.; Casacchia, T.; Gelmini, F.; Guzzi, R.; Garofalo, A.; Statti, G. Identification by Molecular Docking ofHomoisoflavones from Leopoldia comosa as Ligands of Estrogen Receptors. Molecules 2018, 23, 894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powers, C.N.; Setzer, W.N. A molecular docking study of phytochemical estrogen mimics from dietary herbal supplements. Silico Pharmacol. 2015, 3, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, P.; Liang, K.; Ma, B.; Zheng, N.; Nussinov, R.; Huang, J. Multiple-Targeting and Conformational Selection in the Estrogen Receptor: Computation and Experiment. Chem. Boil. Drug Des. 2011, 78, 137–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Wang, J.; Hong, D.-Y.; Chen, L.; Zhang, Y.-Y.; Xu, Y.-N.; Pan, D.; Fu, L.-Y.; Tao, L.; Luo, H.; et al. Baicalein has protective effects on the 17β-estradiol-induced transformation of breast epithelial cells. Oncotarget 2017, 8, 10470–10484. [Google Scholar] [CrossRef] [PubMed]
- Fokialakis, N.; Alexi, X.; Aligiannis, N.; Boulaka, A.; Meligova, A.K.; Lambrinidis, G.; Kalpoutzakis, E.; Pratsinis, H.; Cheilari, A.; Mitsiou, D.J.; et al. Biological evaluation of isoflavonoids from Genista halacsyi using estrogen-target cells: Activities of glucosides compared to aglycones. PLoS ONE 2019, 14, e0210247. [Google Scholar] [CrossRef]
- Ayoub, N.M.; Siddique, A.B.; Ebrahim, H.Y.; Mohyeldin, M.M.; El Sayed, K.A. The olive oil phenolic (-)-oleocanthal modulates estrogen receptor expression in luminal breast cancer in vitro and in vivo and synergizes with tamoxifen treatment. Eur. J. Pharmacol. 2017, 810, 100–111. [Google Scholar] [CrossRef]
- Yuseran, H.; Hartoyo, E.; Nurseta, T.; Kalim, H. Molecular docking of genistein on estrogen receptors, promoter region of BCLX, caspase-3, Ki-67, cyclin D1, and telomere activity. J. Taibah Univ. Med. Sci. 2018, 14, 79–87. [Google Scholar] [CrossRef]
- Nanashima, N.; Horie, K.; Maeda, H. Phytoestrogenic Activity of Blackcurrant Anthocyanins Is Partially Mediated through Estrogen Receptor Beta. Molecules 2017, 23, 74. [Google Scholar] [CrossRef] [Green Version]
- Alam, S.; Khan, F. Virtual screening, Docking, ADMET and System Pharmacology studies on Garcinia caged Xanthone derivatives for Anticancer activity. Sci. Rep. 2018, 8, 5524. [Google Scholar] [CrossRef]
- Puranik, N.V.; Srivastava, P.; Bhatt, G.; Mary, D.J.S.J.; Limaye, A.M.; Sivaraman, J. Determination and analysis of agonist and antagonist potential of naturally occurring flavonoids for estrogen receptor (ERα) by various parameters and molecular modelling approach. Sci. Rep. 2019, 9, 7450. [Google Scholar] [CrossRef]
- Wang, T.; Wang, Y.; Zhuang, X.; Luan, F.; Zhao, C.; Cordeiro, M.N.D.S. Interaction of Coumarin Phytoestrogens with ERα and ERβ: A Molecular Dynamics Simulation Study. Molecules 2020, 25, 1165. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wang, G.-C.; Rong, J.; Wang, S.W.; Ng, T.B.; Zhang, Y.B.; Lee, K.F.; Zheng, L.; Wong, H.-K.; Yung, K.K.-L.; et al. Identification of Steroidogenic Components Derived From Gardenia jasminoides Ellis Potentially Useful for Treating Postmenopausal Syndrome. Front. Pharmacol. 2018, 9, 9. [Google Scholar] [CrossRef]
- Jeong, J.; Kim, H.; Choi, J. In Silico Molecular Docking and In Vivo Validation with Caenorhabditis elegans to Discover Molecular Initiating Events in Adverse Outcome Pathway Framework: Case Study on Endocrine-Disrupting Chemicals with Estrogen and Androgen Receptors. Int. J. Mol. Sci. 2019, 20, 1209. [Google Scholar] [CrossRef] [Green Version]
- Neves, B.J.; Braga, R.C.; Melo-Filho, C.C.; Moreira-Filho, J.T.; Muratov, E.N.; Andrade, C.H. QSAR-Based Virtual Screening: Advances and Applications in Drug Discovery. Front. Pharmacol. 2018, 9, 1275. [Google Scholar] [CrossRef] [Green Version]
- Dror, O.; Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. Novel Approach for Efficient Pharmacophore-Based Virtual Screening: Method and Applications. J. Chem. Inf. Model. 2009, 49, 2333–2343. [Google Scholar] [CrossRef] [Green Version]
- Fiser, A. Template-based protein structure modeling. Breast Cancer 2010, 673, 73–94. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Chen, M.; Huang, B.; Ji, H.; Yuan, M. Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA) Studies on α1A-Adrenergic Receptor Antagonists Based on Pharmacophore Molecular Alignment. Int. J. Mol. Sci. 2011, 12, 7022–7037. [Google Scholar] [CrossRef]
- Sharma, R.; Dhingra, N.; Patil, S. CoMFA, CoMSIA, HQSAR and Molecular Docking Analysis of Ionone-based Chalcone Derivatives as Antiprostate Cancer Activity. Indian J. Pharm. Sci. 2016, 78, 54–64. [Google Scholar] [CrossRef] [Green Version]
- Vuorinen, A.; Engeli, R.T.; Leugger, S.; Kreutz, C.R.; Schuster, D.; Odermatt, A.; Matuszczak, B. Phenylbenzenesulfonates and -sulfonamides as 17β-hydroxysteroid dehydrogenase type 2 inhibitors: Synthesis and SAR-analysis. Bioorg. Med. Chem. Lett. 2017, 27, 2982–2985. [Google Scholar] [CrossRef]
- Vuorinen, A.; Engeli, R.; Meyer, A.; Bachmann, F.; Griesser, U.J.; Schuster, D.; Odermatt, A. Ligand-Based Pharmacophore Modeling and Virtual Screening for the Discovery of Novel 17β-Hydroxysteroid Dehydrogenase 2 Inhibitors. J. Med. Chem. 2014, 57, 5995–6007. [Google Scholar] [CrossRef]
- Chang, Y.-H.; Chen, J.-Y.; Hor, C.-Y.; Chuang, Y.-C.; Yang, C.-B.; Yang, C.-N. Computational Study of Estrogen Receptor-Alpha Antagonist with Three-Dimensional Quantitative Structure-Activity Relationship, Support Vector Regression, and Linear Regression Methods. Int. J. Med. Chem. 2013, 2013, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Sodero, A.C.R.; Romeiro, N.C.; Da Cunha, E.F.F.; Magalhães, U.D.O.; De Alencastro, R.B.; Rodrigues, C.R.; Cabral, L.M.; Castro, H.C.; Albuquerque, M.G. Application of 4D-QSAR Studies to a Series of Raloxifene Analogs and Design of Potential Selective Estrogen Receptor Modulators. Molecules 2012, 17, 7415–7439. [Google Scholar] [CrossRef]
- Wang, P.; McInnes, C.; Zhu, B.T. Structural Characterization of the Binding Interactions of Various Endogenous Estrogen Metabolites with Human Estrogen Receptor α and β Subtypes: A Molecular Modeling Study. PLoS ONE 2013, 8, e74615. [Google Scholar] [CrossRef] [Green Version]
- Bhhatarai, B.; Wilson, D.M.; Price, P.S.; Marty, S.; Parks, A.K.; Carney, E. Evaluation of OASIS QSAR Models Using ToxCast™ in Vitro Estrogen and Androgen Receptor Binding Data and Application in an Integrated Endocrine Screening Approach. Environ. Health Perspect. 2016, 124, 1453–1461. [Google Scholar] [CrossRef] [Green Version]
- Bohari, M.; Srivastava, H.K.; Sastry, G.N. Analogue-based approaches in anti-cancer compound modelling: The relevance of QSAR models. Org. Med. Chem. Lett. 2011, 1, 3. [Google Scholar] [CrossRef] [Green Version]
- Dems, M.A.E.; Laib, S.; Latelli, N.; Ouddai, N. A DFT-based Quantitative structure activity relationship Study of organometallic estradiol derivatives. JCPS 2017, 10, 483–487. [Google Scholar]
- Zhang, T.; Wei, D.-Q.; Chou, K.-C. A pharmacophore model specific to active site of CYP1A2 with a novel molecular modeling explorer and CoMFA. Med. Chem. 2012, 8, 198–207. [Google Scholar]
- Poirier, D.; Roy, J.; Cortes-Benitez, F.; Dutour, R. Targeting cytochrome P450 (CYP) 1B1 with steroid derivatives. Bioorg. Med. Chem. Lett. 2016, 26, 5272–5276. [Google Scholar] [CrossRef]
- Kar, S.; Roy, K.; Leszczynski, J. Impact of Pharmaceuticals on the Environment: Risk Assessment Using QSAR Modeling Approach. Methods Mol. Biol. 2018, 1800, 395–443. [Google Scholar] [CrossRef]
- Colosi, L.M.; Huang, Q.; Weber, W.J. QSAR-assisted design of an environmental catalyst for enhanced estrogen remediation. Chemosphere 2010, 81, 897–903. [Google Scholar] [CrossRef] [Green Version]
- Rokhina, E.V.; Suri, R. Application of density functional theory (DFT) to study the properties and degradation of natural estrogen hormones with chemical oxidizers. Sci. Total Environ. 2012, 417, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Warshel, A.; Levitt, M. Theoretical studies of enzymic reactions: Dielectric, electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme. J. Mol. Boil. 1976, 103, 227–249. [Google Scholar] [CrossRef]
- Jorgensen, W. The Many Roles of Computation in Drug Discovery. Science 2004, 303, 1813–1818. [Google Scholar] [CrossRef] [PubMed]
- Simonson, T.; Archontis, G.; Karplus, M. Free Energy Simulations Come of Age: Protein−Ligand Recognition. Acc. Chem. Res. 2002, 35, 430–437. [Google Scholar] [CrossRef] [Green Version]
- Fratev, F.; Steinbrecher, T.; Jónsdóttir, S.O. Prediction of Accurate Binding Modes Using Combination of Classical and Accelerated Molecular Dynamics and Free-Energy Perturbation Calculations: An Application to Toxicity Studies. ACS Omega 2018, 3, 4357–4371. [Google Scholar] [CrossRef] [Green Version]
- Schindler, C.; Rippmann, F.; Kuhn, D. Relative binding affinity prediction of farnesoid X receptor in the D3R Grand Challenge 2 using FEP+. J. Comput. Mol. Des. 2017, 32, 265–272. [Google Scholar] [CrossRef]
- Olsson, M.A.; García-Sosa, A.T.; Ryde, U. Binding affinities of the farnesoid X receptor in the D3R Grand Challenge 2 estimated by free-energy perturbation and docking. J. Comput. Mol. Des. 2017, 32, 211–224. [Google Scholar] [CrossRef]
- Åqvist, J.; Luzhkov, V.B.; Brandsdal, B.O. Ligand Binding Affinities from MD Simulations. Acc. Chem. Res. 2002, 35, 358–365. [Google Scholar] [CrossRef]
- Van Lipzig, M.M.H.; Ter Laak, A.M.; Jongejan, A.; Vermeulen, N.P.; Wamelink, M.; Geerke, D.P.; Meerman, J.H.N. Prediction of Ligand Binding Affinity and Orientation of Xenoestrogens to the Estrogen Receptor by Molecular Dynamics Simulations and the Linear Interaction Energy Method. J. Med. Chem. 2004, 47, 1018–1030. [Google Scholar] [CrossRef]
- Rifai, E.A.; Van Dijk, M.; Vermeulen, N.P.; Geerke, D.P. Binding free energy predictions of FXR agonists using LIE with reliability estimation: Application to the D3R Grand Challenge 2. J. Comput. Mol. Des. 2017, 32, 239–249. [Google Scholar] [CrossRef] [Green Version]
- Simonson, T. Protein: Ligand recognition: Simple models for electrostatic effects. Curr. Pharm. Des. 2013, 19, 4241–4256. [Google Scholar] [CrossRef] [PubMed]
- Gelpí, J.L.; Hospital, A.; Goñi, J.R.; Orozco, M. Molecular dynamics simulations: Advances and applications. Adv. Appl. Bioinform. Chem. 2015, 8, 37–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freindorf, M.; Furlani, T.R.; Kong, J.; Cody, V.; Davis, F.B.; Davis, P.J. Combined QM/MM Study of Thyroid and Steroid Hormone Analogue Interactions with αvβ3 Integrin. J. Biomed. Biotechnol. 2012, 2012, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kalaiarasi, C.; Manjula, S.; Kumaradhas, P. Combined quantum mechanics/molecular mechanics (QM/MM) methods to understand the charge density distribution of estrogens in the active site of estrogen receptors. RSC Adv. 2019, 9, 40758–40771. [Google Scholar] [CrossRef] [Green Version]
- Costa, A.H.L.; Clemente, W.S.; Bezerra, K.S.; Neto, J.X.L.; De Albuquerque, É.L.; Fulco, U.; Clemente, W.S., Jr. Computational biochemical investigation of the binding energy interactions between an estrogen receptor and its agonists. New J. Chem. 2018, 42, 19801–19810. [Google Scholar] [CrossRef]
- Hilder, T.A.; Hodgkiss, J.M. Molecular Mechanism of Binding between 17β-Estradiol and DNA. Comput. Struct. Biotechnol. J. 2016, 15, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Eisold, A.; LaBudde, D. Detailed Analysis of 17β-Estradiol-Aptamer Interactions: A Molecular Dynamics Simulation Study. Molecules 2018, 23, 1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakhin, A.V.; Tarantul, V.Z.; Gening, L.V. Aptamers: Problems, Solutions and Prospects. Acta Naturae 2013, 5, 34–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutkiewicz, Z.; Mikstacka, R. Structure-Based Drug Design for Cytochrome P450 Family 1 Inhibitors. Bioinorg. Chem. Appl. 2018, 2018, 3924608. [Google Scholar] [CrossRef] [PubMed]
- Moldogazieva, N.T.; Ostroverkhova, D.S.; Kuzmich, N.N.; Kadochnikov, V.V.; Terentiev, A.A.; Porozov, Y. Elucidating Binding Sites and Affinities of ERα Agonists and Antagonists to Human Alpha-Fetoprotein by In Silico Modeling and Point Mutagenesis. Int. J. Mol. Sci. 2020, 21, 893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, K.; Saha, P.K.; Chan, L.; Moore, D.D. Farnesoid X receptor is essential for normal glucose homeostasis. J. Clin. Investig. 2006, 116, 1102–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Huang, X.; Yi, T.; Yen, Y.; Moore, D.D.; Huang, W. Spontaneous Development of Liver Tumors in the Absence of the Bile Acid Receptor Farnesoid X Receptor. Cancer Res. 2007, 67, 863–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levin, E.R. Cellular Functions of the Plasma Membrane Estrogen Receptor. Trends Endocrinol. Metab. 1999, 10, 374–377. [Google Scholar] [CrossRef]
- Soltysik, K.; Czekaj, P. Membrane estrogen receptors - is it an alternative way of estrogen action? J. Physiol. Pharmacol. Off. J. Pol. Physiol. Soc. 2013, 64, 129–142. [Google Scholar]
- Guo, J.J.; Yang, D.-P.; Tian, X.; Vemuri, V.K.; Yin, D.; Li, C.; Duclos, R.I.; Shen, L.; Ma, X.; Janero, D.R.; et al. 17β-estradiol (E2) in membranes: Orientation and dynamic properties. Biochim. Biophys. Acta (BBA) Biomembr. 2016, 1858, 344–353. [Google Scholar] [CrossRef]
- Vogel, A.; Scheidt, H.A.; Feller, S.E.; Metso, J.; Badeau, R.M.; Tikkanen, M.J.; Wähälä, K.; Jauhiainen, M.; Huster, D. The Orientation and Dynamics of Estradiol and Estradiol Oleate in Lipid Membranes and HDL Disc Models. Biophys. J. 2014, 107, 114–125. [Google Scholar] [CrossRef] [Green Version]
- Goh, J.Y.; Goh, K.S.; Yip, Y.M.; Ng, C.K. High salinity enhances adsorption of 17α-ethinyl estradiol by polyethersulfone membrane: Isotherm modelling and molecular simulation. engrXiv Preprints 2019. [Google Scholar] [CrossRef]
- Lakshmanan, S.; Kanwal, A.; Liu, S.; Patlolla, A.; Iqbal, Z.; Mitra, S.; Thomas, G.A.; Fagan, J.A.; Farrow, R.C. Improved Electrophoretic Deposition of Vertical Single Wall Carbon Nanotubes with Nanoscopic Electrostatic Lenses. Micromachines 2020, 11, 324. [Google Scholar] [CrossRef] [Green Version]
- Ulissi, Z.W.; Zhang, J.; Sresht, V.; Blankschtein, D.; Strano, M.S. 2D Equation-of-State Model for Corona Phase Molecular Recognition on Single-Walled Carbon Nanotube and Graphene Surfaces. Langmuir 2014, 31, 628–636. [Google Scholar] [CrossRef]
- Sun, W.; Li, M.; Zhang, W.; Wei, J.; Chen, B.; Wang, C. Sediments inhibit adsorption of 17β-estradiol and 17α-ethinylestradiol to carbon nanotubes and graphene oxide. Environ. Sci. Nano 2017, 4, 1900–1910. [Google Scholar] [CrossRef]
- Jiang, L.; Liu, Y.; Liu, S.; Zeng, G.; Hu, X.-J.; Hu, X.; Guo, Z.; Tan, X.; Wang, L.; Wu, Z. Adsorption of Estrogen Contaminants by Graphene Nanomaterials under Natural Organic Matter Preloading: Comparison to Carbon Nanotube, Biochar, and Activated Carbon. Environ. Sci. Technol. 2017, 51, 6352–6359. [Google Scholar] [CrossRef] [PubMed]
- Boateng, L.K.; Heo, J.; Flora, J.R.V.; Park, Y.-G.; Yoon, Y. Molecular level simulation of the adsorption of bisphenol A and 17α-ethinyl estradiol onto carbon nanomaterials. Sep. Purif. Technol. 2013, 116, 471–478. [Google Scholar] [CrossRef]
- Zaib, Q.; Khan, I.A.; Saleh, N.B.; Flora, J.R.V.; Park, Y.-G.; Yoon, Y. Removal of Bisphenol A and 17β-Estradiol by Single-Walled Carbon Nanotubes in Aqueous Solution: Adsorption and Molecular Modeling. Water Air Soil Pollut. 2012, 223, 3281–3293. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864. [Google Scholar] [CrossRef] [Green Version]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S. Density functional theory with London dispersion corrections. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 211–228. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Krist. Cryst. Mater. 2005, 220, 567–570. [Google Scholar] [CrossRef] [Green Version]
- Amusia, M.Y.; Msezane, A.Z.; Shaginyan, V.R. Density Functional Theory versus the Hartree–Fock Method: Comparative Assessment. Phys. Scr. 2003, 68, C133–C140. [Google Scholar] [CrossRef] [Green Version]
- Mota, K.; Neto, J.X.L.; Costa, A.L.; Oliveira, J.; Bezerra, K.; De Albuquerque, É.L.; Caetano, E.W.S.; Freire, V.; Fulco, U. A quantum biochemistry model of the interaction between the estrogen receptor and the two antagonists used in breast cancer treatment. Comput. Theor. Chem. 2016, 1089, 21–27. [Google Scholar] [CrossRef]
- Mazurek, A.H.; Szeleszczuk, Ł.; Pisklak, D.M. Periodic DFT Calculations—Review of Applications in the Pharmaceutical Sciences. Pharmaceutics 2020, 12, 415. [Google Scholar] [CrossRef]
- Stevenson, E.L.; Lancaster, R.W.; Buanz, A.B.M.; Price, L.S.; Tocher, D.A.; Price, S.L. The solid state forms of the sex hormone 17-β-estradiol. CrystEngComm 2019. [Google Scholar] [CrossRef] [Green Version]
- Du, R.; Xu, J.; Zhang, L.; Ning, L.; Li, S. Ethinyl estradiol cocrystals assembled by chain structures: Improvement in stability and solubility. New J. Chem. 2019, 43, 16889–16897. [Google Scholar] [CrossRef]
- Wang, J.-R.; Yang, Y.; Chen, X.; Mei, X. Solid-state characterization of 17β-estradiol co-crystals presenting improved dissolution and bioavailability. CrystEngComm 2016, 18, 3498–3505. [Google Scholar] [CrossRef]
- Watanabe, H.; Okiyama, Y.; Nakano, T.; Tanaka, S. Incorporation of solvation effects into the fragment molecular orbital calculations with the Poisson–Boltzmann equation. Chem. Phys. Lett. 2010, 500, 116–119. [Google Scholar] [CrossRef]
- Szeleszczuk, Ł.; Pisklak, D.M.; Zielińska-Pisklak, M.; Wawer, I. Effects of structural differences on the NMR chemical shifts in cinnamic acid derivatives: Comparison of GIAO and GIPAW calculations. Chem. Phys. Lett. 2016, 653, 35–41. [Google Scholar] [CrossRef]
- Charpentier, T. The PAW/GIPAW approach for computing NMR parameters: A new dimension added to NMR study of solids. Solid State Nucl. Magn. Reson. 2011, 40, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Elyashberg, M.E. Identification and structure elucidation by NMR spectroscopy. TrAC Trends Anal. Chem. 2015, 69, 88–97. [Google Scholar] [CrossRef]
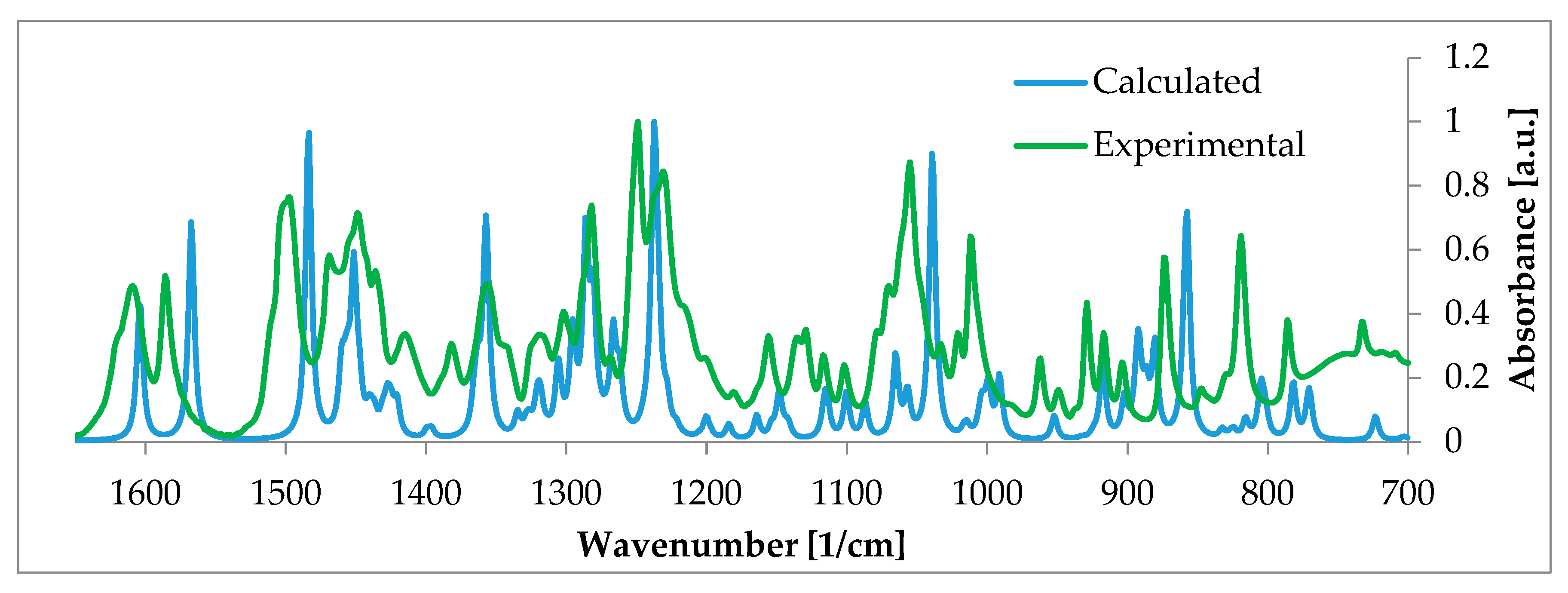
- Szeleszczuk, Ł.; Pisklak, D.M.; Zielińska-Pisklak, M.; Jurczak, E. A new polymorph of 17-β-estradiol and the application of different analytical techniques (ssNMR, PXRD, DSC, and FTIR) for its study. J. Mol. Struct. 2019, 1183, 274–280. [Google Scholar] [CrossRef]
- Singh, H.; Singh, S.; Srivastava, A.; Tandon, P.; Bharti, P.; Kumar, S.; Maurya, R. Conformational analysis and vibrational study of daidzein by using FT-IR and FT-Raman spectroscopies and DFT calculations. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 120, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Machado, N.; De Carvalho, L.A.E.B.; Otero, J.C.; Marques, M.P.M. A conformational study of hydroxyflavones by vibrational spectroscopy coupled to DFT calculations. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2013, 109, 116–124. [Google Scholar] [CrossRef] [Green Version]
- A Minaeva, V.; Minaev, B.F.; Hovorun, D.M. Vibrational spectra of the steroid hormones, estradiol and estriol, calculated by density functional theory. The role of low-frequency vibrations. Ukr. Biokhimichnyi Zhurnal 2009, 80, 82–95. [Google Scholar]
- Scheidt, H.A.; Badeau, R.M.; Huster, D. Investigating the membrane orientation and transversal distribution of 17β-estradiol in lipid membranes by solid-state NMR. Chem. Phys. Lipids 2010, 163, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Vedad, J.; Mojica, E.-R.E.; Desamero, R.Z. Raman spectroscopic discrimination of estrogens. Vib. Spectrosc. 2018, 96, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Oren, I.; Fleishman, S.J.; Kessel, A.; Tal, N.B. Free Diffusion of Steroid Hormones Across Biomembranes: A Simplex Search with Implicit Solvent Model Calculations. Biophys. J. 2004, 87, 768–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellena, J.; De Paula, K.; De Melo, C.C.; Da Silva, C.C.P.; Bezerra, B.P.; Venâncio, T.; Ayala, A.P. Temperature-Driven Isosymmetric Reversible Phase Transition of the Hormone Estradiol 17β Valerate. Cryst. Growth Des. 2014, 14, 5700–5709. [Google Scholar] [CrossRef]
- Morishima, F.; Inokuchi, Y.; Ebata, T. Laser Spectroscopic Study of β-Estradiol and Its Monohydrated Clusters in a Supersonic Jet. J. Phys. Chem. A 2012, 116, 8201–8208. [Google Scholar] [CrossRef] [Green Version]
- Borah, M.M.; Devi, T.G. The vibrational spectroscopic studies and molecular property analysis of Estradiol, Tamoxifen and their interaction by density functional theory. J. Mol. Struct. 2018, 1163, 205–220. [Google Scholar] [CrossRef]
- Cherkasova, O.; Nazarov, M.; Mankova, A.; Fedulova, E.; Volodin, V.; Minaeva, V.A.; Minaev, B.F.; Baryshnikov, G.V. Terahertz time-domain spectroscopy of testosterone, estradiol and estriol. In Proceedings of the 2010 International Kharkov Symposium on Physics and Engineering of Microwaves, Millimeter and Submillimeter Waves, Kharkiv, Ukraine, 21–26 June 2010; pp. 1–3. [Google Scholar]
- Hafizi, R.; Taheri, R.A.; Moghimi, H. Liquid phase extraction of nanosized biologically active estrogenic pollutants by using an efficient adsorbent. J. Mol. Liq. 2018, 266, 535–539. [Google Scholar] [CrossRef]
- Donini, C.A.; Silva, M.K.L.; Simões, R.; Cesarino, I. Reduced graphene oxide modified with silver nanoparticles for the electrochemical detection of estriol. J. Electroanal. Chem. 2018, 809, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Klamt, A.; Schüürmann, G. COSMO: A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin Trans. 2 1993, 2, 799–805. [Google Scholar] [CrossRef]
- Mennucci, B.; Tomasi, J.; Cammi, R.; Cheeseman, J.R.; Frisch, M.J.; Devlin, F.J.; Gabriel, S.; Stephens, P.J. Polarizable Continuum Model (PCM) Calculations of Solvent Effects on Optical Rotations of Chiral Molecules. J. Phys. Chem. A 2002, 106, 6102–6113. [Google Scholar] [CrossRef]
- Chinnasamy, K.; Kumaradhas, P. Intermolecular interactions and charge density distribution of endocrine-disrupting molecules (xenoestrogens) with ERα: QM/MM perspective. Struct. Chem. 2020, 31, 1013–1028. [Google Scholar] [CrossRef]
- Ruiz, P.; Ingale, K.; Wheeler, J.S.; Mumtaz, M. 3D QSAR studies of hydroxylated polychlorinated biphenyls as potential xenoestrogens. Chemosphere 2016, 144, 2238–2246. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Li, L.Y.; Grace, J.R. Predictability of physicochemical properties of polychlorinated dibenzo-p-dioxins (PCDDs) based on single-molecular descriptor models. Environ. Pollut. 2016, 213, 99–111. [Google Scholar] [CrossRef]
- Eguchi, A.; Hanazato, M.; Suzuki, N.; Matsuno, Y.; Todaka, E.; Mori, C. Maternal–fetal transfer rates of PCBs, OCPs, PBDEs, and dioxin-like compounds predicted through quantitative structure–activity relationship modeling. Environ. Sci. Pollut. Res. 2015, 25, 7212–7222. [Google Scholar] [CrossRef]
- Delfosse, V.; Grimaldi, M.; Cavaillès, V.; Balaguer, P.; Bourguet, W. Structural and Functional Profiling of Environmental Ligands for Estrogen Receptors. Environ. Health Perspect. 2014, 122, 1306–1313. [Google Scholar] [CrossRef] [Green Version]
- Nwachukwu, J.C.; Srinivasan, S.; Bruno, N.E.; Nowak, J.; Wright, N.J.; Minutolo, F.; Rangarajan, E.S.; Izard, T.; Yao, X.-Q.; Grant, B.J.; et al. Systems Structural Biology Analysis of Ligand Effects on ERα Predicts Cellular Response to Environmental Estrogens and Anti-hormone Therapies. Cell Chem. Boil. 2017, 24, 35–45. [Google Scholar] [CrossRef]
- Cozzini, P.; Dellafiora, L. In silico approach to evaluate molecular interaction between mycotoxins and the estrogen receptors ligand binding domain: A case study on zearalenone and its metabolites. Toxicol. Lett. 2012, 214, 81–85. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, W.; Song, Y.; Hou, L.; Li, T.; Guan, T.; Zhang, T.; Wang, Y. Homogeneous assay for zearalenone analogues and their docking studies with apo-/holo-estrogen receptors. Anal. Methods 2019, 11, 192–199. [Google Scholar] [CrossRef]
- Dellafiora, L.; Oswald, I.P.; Dorne, J.-L.; Galaverna, G.; Battilani, P.; Dall’Asta, C. An in silico structural approach to characterize human and rainbow trout estrogenicity of mycotoxins: Proof of concept study using zearalenone and alternariol. Food Chem. 2020, 312, 126088. [Google Scholar] [CrossRef]
- Yang, J.; Hu, C.T.; Zhu, X.; Zhu, Q.; Ward, M.D.; Kahr, B. DDT Polymorphism and the Lethality of Crystal Forms. Angew. Chem. 2017, 129, 10299–10303. [Google Scholar] [CrossRef]
- Zhang, H.; He, W.; Luo, X.; Lin, X.; Lu, X. Adsorption of 2,3,7,8-tetrochlorodibenzo-p-dioxins on intrinsic, defected, and Ti (N, Ag) doped graphene: A DFT study. J. Mol. Model. 2014, 20, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Li, H.; Johnston, C.T.; Boyd, S.A.; Teppen, B.J. Relating Clay Structural Factors to Dioxin Adsorption by Smectites: Molecular Dynamics Simulations. Soil Sci. Soc. Am. J. 2012, 76, 110–120. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, W.; Wang, Y.; Xing, X.; Zhong, S.; Guan, T.; Zhang, T.; Hou, L.; Li, T. Estrogen receptor-based fluorescence polarization assay for bisphenol analogues and molecular modeling study of their complexation mechanism. Anal. Chim. Acta 2018, 1032, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Obiorah, I.; Maximov, P.; Curpan, R.; Jordan, V.C. Molecular mechanism of action of bisphenol and bisphenol A mediated by oestrogen receptor alpha in growth and apoptosis of breast cancer cells. Br. J. Pharmacol. 2013, 169, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Qu, K.; Hai, Y.; Zhao, C. Bisphenol A (BPA) binding on full-length architectures of estrogen receptor. J. Cell. Biochem. 2018, 119, 6784–6794. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, T.; Wang, T.; Yuan, C.; Zhong, S.; Guan, T.; Li, Z.; Wang, Y.; Yu, H.; Luo, Q.; et al. Estrogenicity of halogenated bisphenol A: In vitro and in silico investigations. Arch. Toxicol. 2017, 92, 1215–1223. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Wang, F.; Liang, Y.; Wang, H.; Zhang, A.; Song, M. Experimental and computational insights on the recognition mechanism between the estrogen receptor α with bisphenol compounds. Arch. Toxicol. 2017, 91, 3897–3912. [Google Scholar] [CrossRef]
- Zhuang, S.; Zhang, C.; Liu, W. Atomic Insights into Distinct Hormonal Activities of Bisphenol A Analogues toward PPARγ and ERα Receptors. Chem. Res. Toxicol. 2014, 27, 1769–1779. [Google Scholar] [CrossRef]
- Li, L.; Wang, Q.; Zhang, Y.; Niu, Y.; Yao, X.; Liu, H. The Molecular Mechanism of Bisphenol A (BPA) as an Endocrine Disruptor by Interacting with Nuclear Receptors: Insights from Molecular Dynamics (MD) Simulations. PLoS ONE 2015, 10, e0120330. [Google Scholar] [CrossRef]
- Delfosse, V.; Grimaldi, M.; Pons, J.-L.; Boulahtouf, A.; Le Maire, A.; Cavaillès, V.; Labesse, G.; Bourguet, W.; Balaguer, P. Structural and mechanistic insights into bisphenols action provide guidelines for risk assessment and discovery of bisphenol A substitutes. Proc. Natl. Acad. Sci. USA 2012, 109, 14930–14935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, D.; Li, J.; Chen, Z.; Liang, L.; Ma, J.; Wei, M.; Ai, Y.; Wang, X. Understanding bisphenol-A adsorption in magnetic modified covalent organic frameworks: Experiments coupled with DFT calculations. J. Mol. Liq. 2020, 301, 112431. [Google Scholar] [CrossRef]
- Bao, S.; Wu, S.; Huang, L.; Xu, X.; Xu, R.; Li, Y.; Liang, Y.; Yang, M.; Yoon, D.K.; Lee, M.; et al. Supramolecular Nanopumps with Chiral Recognition for Moving Organic Pollutants from Water. ACS Appl. Mater. Interfaces 2019, 11, 31220–31226. [Google Scholar] [CrossRef] [PubMed]
- Díaz, I.; Díez, E.; Camacho, J.; León, S.; Ovejero, G.; Cabanillas, S.L. Comparison between three predictive methods for the calculation of polymer solubility parameters. Fluid Phase Equilibria 2013, 337, 6–10. [Google Scholar] [CrossRef]
- Wang, C.-Y.; Zhang, Y.-J.; Wang, W.-K.; Pei, D.-N.; Huang, G.-X.; Chen, J.-J.; Zhang, X.; Yu, H.-Q. Enhanced photocatalytic degradation of bisphenol A by Co-doped BiOCl nanosheets under visible light irradiation. Appl. Catal. B Environ. 2018, 221, 320–328. [Google Scholar] [CrossRef]
- Motta, A.; La Mantia, F.P.; Ascione, L.; Mistretta, M. Theoretical study on the decomposition mechanism of bisphenol A polycarbonate induced by the combined effect of humidity and UV irradiation. J. Mol. Graph. Model. 2020, 99, 107622. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, Y.; Liu, Z.; Du, F.; Qin, G.; Li, G.; Hu, X.; Xu, Z.; Cai, Z. Supramolecularly imprinted polymeric solid phase microextraction coatings for synergetic recognition nitrophenols and bisphenol A. J. Hazard. Mater. 2019, 368, 358–364. [Google Scholar] [CrossRef]
- Dvorakova, M.; Kejlová, K.; Rucki, M.; Jírová, D. Selected bisphenols and phthalates screened for estrogen and androgen disruption by in silico and in vitro methods. Neuro Endocrinol. Lett. 2018, 39, 409–416. [Google Scholar]
- Zhu, Q.; Liu, L.; Zhou, X.; Ma, M. In silico study of molecular mechanisms of action: Estrogenic disruptors among phthalate esters. Environ. Pollut. 2019, 255, 113193. [Google Scholar] [CrossRef]
- Josh, M.S.; Pradeep, S.; Adarsh, V.; Amma, K.V.; Devi, R.S.; Balachandran, S.; Sreejith, M.; Jaleel, U.A.; Benjamin, S. In silicoevidences for the binding of phthalates onto human estrogen receptor α, β subtypes and human estrogen-related receptor γ. Mol. Simul. 2013, 40, 408–417. [Google Scholar] [CrossRef]
- Sheikh, I.A.; Turki, R.F.; Abuzenadah, A.M.; Damanhouri, G.A.; A Beg, M. Endocrine Disruption: Computational Perspectives on Human Sex Hormone-Binding Globulin and Phthalate Plasticizers. PLoS ONE 2016, 11, e0151444. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, S.; Chen, M.; Xu, D.; Tang, L.; Wang, S. Elucidating Adsorption Mechanisms of Phthalate Esters upon Carbon Nanotubes/Graphene and Natural Organic Acid Competitive Effects in Water by DFT and MD Calculations. Bull. Korean Chem. Soc. 2015, 36, 1631–1636. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, R.; Wang, X.; Sun, P.; Chen, W.; Shen, J.; Xue, G. Hydrogenation induced deviation of temperature and concentration dependences of polymer-solvent interactions in poly(vinyl chloride) and a new eco-friendly plasticizer. Eur. Phys. J. Plus 2015, 130, 11. [Google Scholar] [CrossRef]
- Jacob, R.B.; Andersen, T.; McDougal, O.M. Accessible High-Throughput Virtual Screening Molecular Docking Software for Students and Educators. PLoS Comput. Boil. 2012, 8, e1002499. [Google Scholar] [CrossRef] [Green Version]
- Chaput, L.; Mouawad, L. Efficient conformational sampling and weak scoring in docking programs? Strategy of the wisdom of crowds. J. Chem. 2017, 9, 37. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Software Website. GOLD—Protein Ligand Docking Software. Available online: www.ccdc.cam.ac.uk/solutions/csd-discovery/components/gold/ (accessed on 25 August 2020).
- Clark, M.; Cramer, R.D.; Van Opdenbosch, N. Validation of the general purpose tripos 5.2 force field. J. Comput. Chem. 1989, 10, 982–1012. [Google Scholar] [CrossRef]
- Software Website. Available online: https://www.charmm.org/ (accessed on 25 August 2020).
- Brooks, B.R.; Brooks, C.L.; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- Software Website. Available online: http://www.swissdock.ch/ (accessed on 25 August 2020).
- Grosdidier, A.; Zoete, V.; Michielin, O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011, 39, W270–W277. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; Beer, T.A.P.D.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Dominguez, C.; Boelens, R.; Bonvin, A.M.J.J. HADDOCK: A Protein−Protein Docking Approach Based on Biochemical or Biophysical Information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jian, Y.; He, Y.; Yang, J.; Han, W.; Zhai, X.; Zhao, Y.; Li, Y. Molecular Modeling Study for the Design of Novel Peroxisome Proliferator-Activated Receptor Gamma Agonists Using 3D-QSAR and Molecular Docking. Int. J. Mol. Sci. 2018, 19, 630. [Google Scholar] [CrossRef] [Green Version]
- Software Website. Available online: https://www.schrodinger.com/maestro (accessed on 25 August 2020).
- McAliley, J.H.; Bruce, D.A. Development of Force Field Parameters for Molecular Simulation of Polylactide. J. Chem. Theory Comput. 2011, 7, 3756–3767. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Compt. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Software Website. Available online: https://gaussian.com/ (accessed on 25 August 2020).
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational protein–ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Software Website. Available online: http://autodock.scripps.edu/ (accessed on 25 August 2020).
- Lipparini, F.; Mennucci, B. Perspective: Polarizable continuum models for quantum-mechanical descriptions. J. Chem. Phys. 2016, 144, 160901. [Google Scholar] [CrossRef] [Green Version]
- Li, D.-D.; Meng, X.-F.; Wang, Q.; Yu, P.; Zhao, L.-G.; Zhang, Z.-P.; Wang, Z.; Xiao, W. Consensus scoring model for the molecular docking study of mTOR kinase inhibitor. J. Mol. Graph. Model. 2018, 79, 81–87. [Google Scholar] [CrossRef]
- Software Website. Available online: https://ambermd.org/ (accessed on 25 August 2020).
- Case, D.A.; Iii, T.E.C.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C.L. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins Struct. Funct. Bioinform. 2006, 65, 712–725. [Google Scholar] [CrossRef] [Green Version]
- Available online: http://www.cosmo-model.org/ (accessed on 25 August 2020).
- Grubmüller, H.; Heller, H.; Windemuth, A.; Schulten, K. Generalized Verlet Algorithm for Efficient Molecular Dynamics Simulations with Long-range Interactions. Mol. Simul. 1991, 6, 121–142. [Google Scholar] [CrossRef]
- Softwae Website. Available online: www.gromacs.org (accessed on 25 August 2020).
- Softwae Website. Available online: https://www.vasp.at/ (accessed on 25 August 2020).
- Softwae Website. Available online: https://www.ks.uiuc.edu/Research/namd/ (accessed on 25 August 2020).
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karamertzanis, P.G.; Pantelides, C.C. Ab initio crystal structure prediction. I. Rigid molecules. J. Comput. Chem. 2005, 26, 304–324. [Google Scholar] [CrossRef] [PubMed]
- Karamertzanis, P.G.; Pantelides, C.C. Ab initio crystal structure prediction. II. Flexible molecules. Mol. Phys. 2007, 105, 273–291. [Google Scholar] [CrossRef]
- Vasileiadis, M.; Pantelides, C.C.; Adjiman, C.S. Prediction of the crystal structures of axitinib, a polymorphic pharmaceutical molecule. Chem. Eng. Sci. 2015, 121, 60–76. [Google Scholar] [CrossRef] [Green Version]
- Delley, B. DFT studies: From molecules and molecular environments to surfaces and solids. Comput. Mater. Sci. 2000, 17, 122–126. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Softwae Website. Available online: http://www.castep.org/ (accessed on 25 August 2020).
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Mark, P.; Nilsson, L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Substrate | Enzyme | Product | Ref. |
|---|---|---|---|
| Testosterone | Aromatase (CYP219A1) | Estradiol | [16] |
| Estradiol | 17β-OH-dehydrogenase (17β-HSD) | Estrone | [24,25] |
| Estradiol | CYP1B1 | 4-OH-hydroxylated estradiol | [30] |
| Estradiol | CYP1A1, CYP1A2 | 2-OH-hydroxylated estradiol | [28,29] |
| Estradiol | Sulfotransferase (SULT) | Inactivated estradiol (sulfated) | [27] |
| Inactivated (sulfated) estradiol | Sulfatase (STS) | Activated estradiol | [26] |
| Protein | RCSB PDB Reference Code | Resolution (Å) | Incorporated Ligands |
|---|---|---|---|
| βER | 5TOA | 2.5 | Estradiol |
| βER-LBD | 1QKM | 1.8 | Genistein |
| Phosphorylated βER-LBD | 3OLL | 1.5 | Estradiol, N-peptide linking |
| αER-LBD | 3UUC | 2.1 | Bisphenol C |
| αER-LBD mutant | 4Q50 | 3.07 | 4-hydroxytamoxifen |
| αER-LBD mutant | 2QXS | 1.7 | Raloxifene |
| αER-LBD | 2R6Y | 2.0 | SERM |
| 17β-HSD | 1IOL | 2.30 | 17β-estradiol |
| 17β-HSD | 6MNC | 2.40 | Estrone |
| 17β-HSD | 6MNE | 1.86 | Estrone, NADP+ |
| 17β-HSD | 3DHE | 2.30 | Dehydroepiandrosterone (DHEA) |
| 17β-HSD | 4FJ0 | 2.2 | 3,7-dihydroxy flavone |
| 17β-HSD | 4FJ1 | 2.3 | Genistein |
| SULT1E1 | 1AQU | 1.6 | Estradiol, PAP cofactor |
| SULT1E1 | 4JVM | 1.994 | Flame retardant, PAP cofactor |
| Placental E1/DHEA STS | 1P49 | 2.6 | l-octylglucoside, N-acetylo-d-glucosamine, Ca2+, PO43− |
| CYP1B1 | 6OyV | 3.101 | Estradiol |
| N° | Code/Software Used | Force Field or DFT Functional and Basis Set | Type of Calculation | Ref. Method | Ref. in Article |
|---|---|---|---|---|---|
| 1 | GOLD | Molecular docking | [208] | [57,69] | |
| 2 | -Ghemical 2.95 -Swiss Dock | -Tripos 5.2 -CHARMM | -Geometry optimization -Molecular docking | [209,210,211,212] | [70] |
| 3 | -Swiss model -Hex 8.0, HADDOCK | -OPLS | -Homology of receptors -Molecular docking | [212,213,214,215] | [87] |
| 4 | -Swiss model -SybylX | -Tripos 5.2 | -Homology of receptors -Molecular docking | [209,214,216] | [84] |
| 5 | Maestro Schrödinger | OPLS 2005, Glide SP, XP | Molecular docking | [217,218] | [83,85,86] |
| 6 | Maestro Schrödinger | MMFF94 | Geometry optimization, molecular docking | [217,218,219] | |
| 7 | -Gaussian09W -AutoDockTools | -B3LYP/6-31G(d) -AutoDockZN | -Geometry optimization -Molecular docking | [220,221,222,223] | [169,184] |
| 8 | Gaussian03 | B3LYP/6-311++g**, PCM | Hydration enthalpy | [220,224] | [183] |
| 9 | Maestro Schrödinger | ZINC database OPLS 2005, Glide SP eHiTS docking module consensus score | Energy minimization HTVS rank | [205,217,218,225] | [75] |
| 10 | Maestro Schrödinger | OPLS 2005, Glide SP, XP | HTVS | [205,217,218] | [76] |
| 11 | Maestro Schrödinger | OPLS 2005, Glide SP, XP | Segregation: agonists/antagonists | [217,218] | [77] |
| 12 | Maestro Schrödinger | -OPLS 2005, Grid (Glide) -Desmond OPLS 2005 | -Molecular docking, MD -ADMET parameters | [217,218] | [90] |
| 13 | -Maestro Schrödinger -AMBER14 -AMBER14 | -OPLS 2005, Glide -FF03 (protein) GAFF (ligand) -MMPBSA, MMGBSA | -Docking -MD -Binding free energy, decomposition energy | [207,217,218,226,227,228] | [91] |
| 14 | -MOPAC2016 -Gaussian09 -Gabedit package | -PM6 in HF, COSMO model -B3LYP, PCM -Verlet algorithm | -Pre-optimization, solvent model -Optimization (DFT), solvent model -MD | [156,220,224,229,230] | [169] |
| 15 | -GOLD -GROMACS -Swiss Param Tool | -CHARMM27 -CHARMM27 | -Molecular docking -MD -Ligand parametrization | [209,210,211,231] | [180] |
| 16 | -SybylX -AMBER11 -AutoDock 4.0 | -Tripos 5.2 -AMBER -AutoDockZN | -Geometry optimization -MD -Molecular docking | [216,221,222,223,226,227,228] | [186] |
| 17 | -Gaussian09 -LeDock -AMBER12 -AmberTools14 | -B3LYP/-cc-pVTZ -CHARMM -AMBER -MM/GBSA | -Geometry optimization -Molecular docking -MD -Binding free energy | [207,220,221,222,223,226,227,228] | [188] |
| 18 | -Gaussian09 -Molegro Virtual Docker -AMBER Tools | -B3LYP/6-311++G(d,p) -AMBER -AMBER03 | -Molecular electrostatic potential -Molecular docking -MD | [220,226,227,228] | [187] |
| 19 | -Maestro Schrödinger -Gaussian09 -AMBER10 | -OPLS 2005 -HF, 6–31G* -GAFF (ligand), ff03 (protein) | -Molecular docking -Geometry optimization -MD | [217,218,220,226,227,228] | [189] |
| 20 | -VASP -GROMACS | -PBE GGA (DFT-D3) -GROMOS96 | -Geometry optimization -MD | [230,232] | [192] |
| 21 | -GROMACS -AutoDock Tools -AutoDock Vina, Hex8.0.0 GROMACS | -AutoDockZN -AutoDock Vina, GROMOS96 | -Energy minimization -Molecular docking -MD | [221,222,223,231] | [199] |
| 22 | -NAMD -Spartan04 | -Charm CMAP FF -HF 3-21G | -MD -QM | [233,234] | [125] |
| 23 | -Gaussian 03 -AutoDock -AMBBER | -B3LYP/6311**G -AutoDockZN -PM3/Amberff14SB FF | -Geometry optimization -Molecular docking -QM/MM | [220,221,222,223,226,227,228] | [114] |
| 24 | -GROMACS -Gaussian 09 | -CHARMM (MM) -GGA-D2 (QM) | -Geometry optimization -DFT calculations | [210,211,220,221,222,223,229] | [115] |
| 25 | -Maestro Schrödinger -AMBER Tools | -OPLS 2005 Glide -B3LYP/Amberff14SB | -Protein, ligand preparation (geometry optimization), molecular docking -QM/MM | [217,218,226,227,228] | [124] |
| 26 | -Crystal Predictor -Crystal Optimizer (Gaussian) -DMACRYS | -PBE0/631G(d,p) | -Conformations -Geometry optimization CSP -Intermolecular lattice energies | [220,235,236,237] | [150] |
| 27 | -GULP, DFTB+ -VASP | -optB88 level | -Geometry pre-optimization CSP -Geometry re-optimization | [232,235,236,237] | [181] |
| 28 | DMol3 | DNP basis set, PBE GGA | Geometry, energy optimization | [238,239] | [182] |
| 29 | CASTEP | GGA PBE | DFT, NMR | [239,240] | [157] |
| 30 | CASTEP | GGA PBE | DFT, structure parameters calculation | [239,240] | [195] |
| 31 | Gaussian09W | B3LYP/631G(d) | DFT, IR | [238,241] | [164] |
| 32 | Gaussian09 | M05-2X/6-311++G** | DFT, IR | [238,241] | [165] |
| 33 | Gaussian09W | B3LYP/6-31G (d,p) | DFT, Raman | [238,241] | [166] |
| 34 | Gaussian | B3LYP/6-31G(d,p) | DFT, IR | [238,241] | [167] |
| Calculation Method | Pros and Capabilities | Cons and Limitations |
|---|---|---|
| Molecular docking | -Explanation of a molecular basis for protein–ligand binding -Relatively short calculation time -Enables virtual screening for active compounds | -Lower accuracy when compared to QM methods -Significant increase in time and complexity of calculations when combined with QM (QM/MM) |
| QSAR | -Evaluation of estrogenicity -No protein preparation needed | -No receptor–ligand binding data -Large set of high-quality experimental data needed to obtain accurate results |
| QM (DFT-D) | -High accuracy of calculations -Simulation of IR, Raman, NMR spectra -Thermodynamic calculations | -Long calculation time -A lot of computational power needed -Usually limited to small molecules and systems such as estrogen complexes, salts, co-crystals, etc. |
| QM/MM | -High accuracy of calculations in the binding area (QM) -Consideration of a whole complex (protein–ligand) with emphasis on the binding pocket | -Calculation time elongated due to QM -Limitation of the QM-calculated area |
| MD | -Simulation of dynamical processes -Possibility to perform DFT-MD | -Significantly longer time required |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazurek, A.H.; Szeleszczuk, Ł.; Simonson, T.; Pisklak, D.M. Application of Various Molecular Modelling Methods in the Study of Estrogens and Xenoestrogens. Int. J. Mol. Sci. 2020, 21, 6411. https://doi.org/10.3390/ijms21176411
Mazurek AH, Szeleszczuk Ł, Simonson T, Pisklak DM. Application of Various Molecular Modelling Methods in the Study of Estrogens and Xenoestrogens. International Journal of Molecular Sciences. 2020; 21(17):6411. https://doi.org/10.3390/ijms21176411
Chicago/Turabian StyleMazurek, Anna Helena, Łukasz Szeleszczuk, Thomas Simonson, and Dariusz Maciej Pisklak. 2020. "Application of Various Molecular Modelling Methods in the Study of Estrogens and Xenoestrogens" International Journal of Molecular Sciences 21, no. 17: 6411. https://doi.org/10.3390/ijms21176411
APA StyleMazurek, A. H., Szeleszczuk, Ł., Simonson, T., & Pisklak, D. M. (2020). Application of Various Molecular Modelling Methods in the Study of Estrogens and Xenoestrogens. International Journal of Molecular Sciences, 21(17), 6411. https://doi.org/10.3390/ijms21176411