Updated Understanding of Cancer as a Metabolic and Telomere-Driven Disease, and Proposal for Complex Personalized Treatment, a Hypothesis

 ,
, 

Abstract
: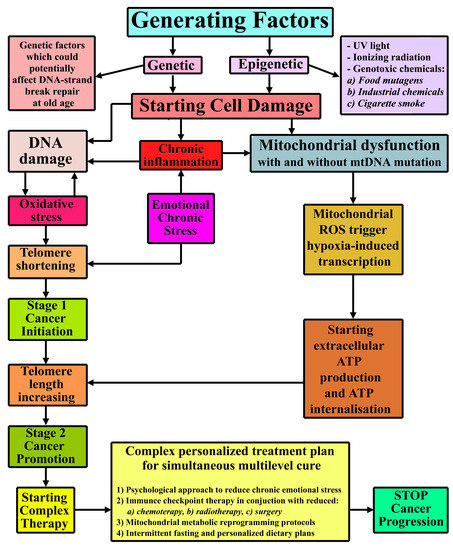
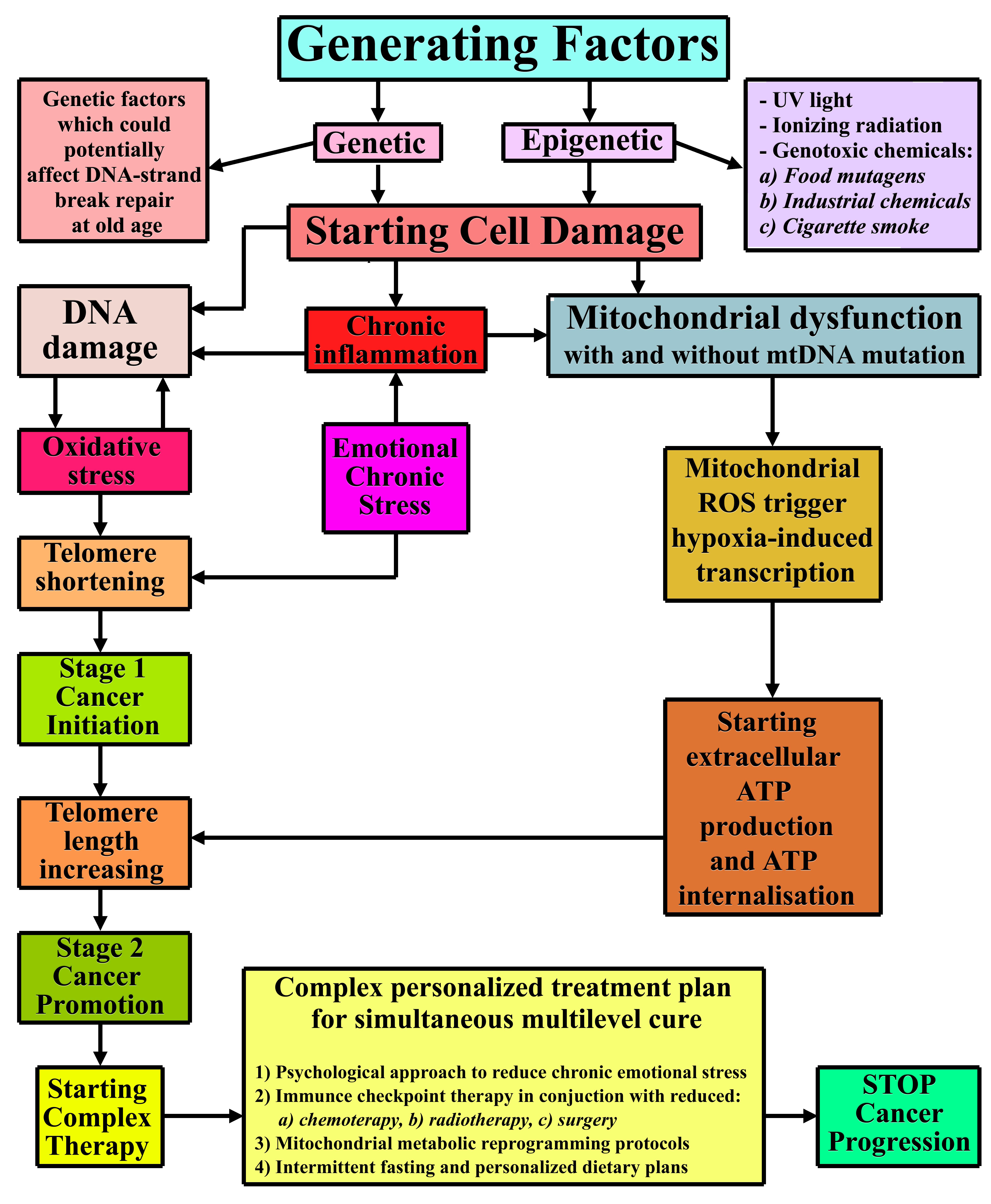{kind=link}
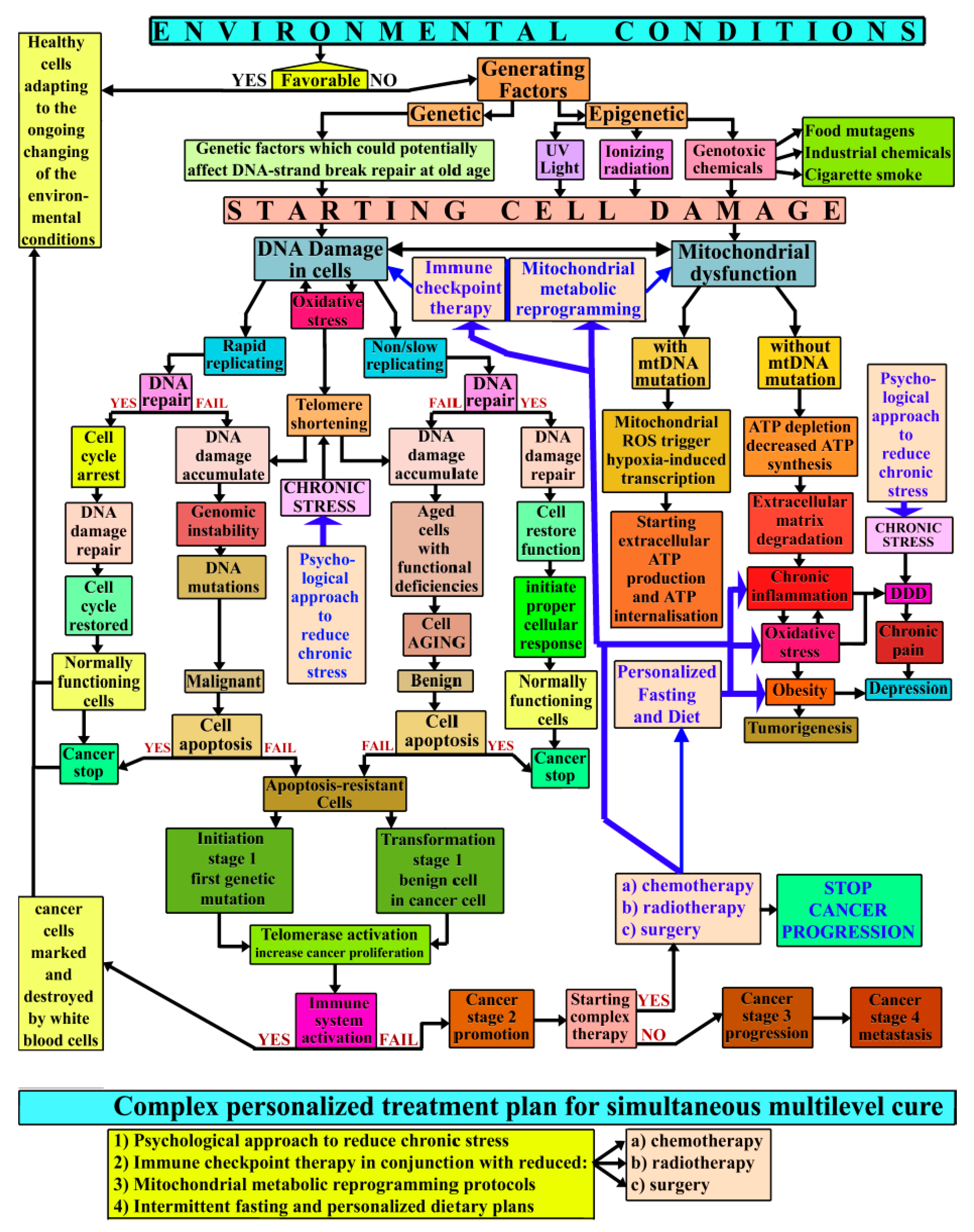{kind=link}
1. Introduction
2. Cancer Mechanism
2.1. DNA Cell Mutations and Telomere Length
2.2. Cancer Complex Mechanism Schematics
2.3. Cancer and the Mitochondria Metabolic Dysfunction
3. Cancer Cell Behavior
4. Autophagy as a Potential Treatment
4.1. Main Autophagic Mechanisms
4.2. Autophagy and Cancer Treatment
5. Novel Cancer Immune Checkpoint Therapy
6. Mitochondrial Metabolic Reprogramming in Cancer Therapies
7. Cancer and Growth Factors Therapies
8. Nutrition as an Adjuvant in Cancer Therapy
9. The Role of Chemotherapy and Radiotherapy in Cancer Treatment and Their Outcome
10. Discussions and Conclusions for a Complex Multilevel Personalized Treatment
- -
- Obtain an accurate history of the patient’s dramatic and stressful events during his or her lifetime and advanced psychological counseling to evaluate ways to make the patient aware of the consequences of chronic stress and means of coping with and, ideally, discontinuing current stress mechanisms.
- -
- Utilize immune checkpoint therapy with chemotherapy, radiotherapy, and surgery performed with minimal aggressiveness toward the cancer itself in order to avoid cancer resistance and to reduce the danger of tumor(s) threatening vital organs. Radiotherapies and chemotherapies should be used in such a way to minimize the impact on the immune system.
- -
- Use intermittent fasting, before and after medical interventions, but without interfering with the other proposed therapies.
- -
- Employ the mitochondrial metabolic reprogramming protocol, according to the patient’s medical conditions and identifying the oncogenic metabolites.
- -
- If the patient’s cancer have been caused or correlated with a viral/bacterial chronic infection, [190,191] then additional therapies may be needed. Also, there are other bacteria which could be used to treat cancer [192], therefore, the complex personalized cancer therapy should take into consideration all possible choices.
- -
- Provide personalized dietary restrictions to maintain the best metabolic equilibrium for avoiding cancer reoccurrence or relapse.
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| DNA | Deoxyribonucleic acid |
| ATP | Adenosine triphosphate |
| ROS | Reactive oxygen species |
| OS | Oxidative stress |
| DDD | Degenerative disc disease |
| Akt1 | RAC-alpha serine/threonine-protein kinase |
| CTLA-4 | Cytotoxic T-lymphocyte-associated protein |
| PD-1 | Programmed cell death protein 1 |
| PD-L1 | Programmed cell death protein ligand 1 |
| Beclin1 | Protein encoded by the BECN1 gene |
| HDAC | Histone deacetylase |
| TGF-β | Transforming growth factor beta |
| MR1 | Major histocompatibility complex class I-related gene protein |
| TCR | T-cell receptor |
| HIF-1 | Hypoxia-inducible factor 1 |
| miRNAs | microRNAs |
| mtDNA | Mitochondrial DNA (Deoxyribonucleic acid) |
| OS | Oxidative stress |
| DDD | Degenerative disc disease |
| Akt1 | RAC-alpha serine/threonine-protein kinase |
| CTLA-4 | Cytotoxic T-lymphocyte-associated protein |
References
- National Institutes of Health (US); Biological Sciences Curriculum Study. NIH Curriculum Supplement Series [Internet]; Understanding Cancer; National Institutes of Health (US): Bethesda, MD, USA, 2007. Available online: https://www.ncbi.nlm.nih.gov/books/NBK20362 (accessed on 16 August 2020).
- Cooper, G.M. The Cell: A Molecular Approach, 2nd ed.; The Development and Causes of Cancer; Sinauer Associates: Sunderland, MA, USA, 2000. Available online: https://www.ncbi.nlm.nih.gov/books/NBK9963 (accessed on 16 August 2020).
- Clark, W.H. Tumour progression and the nature of cancer. Br. J. Cancer 1991, 64, 631–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arvelo, F.; Sojo, F.; Cotte, C. Tumour progression and metastasis. Ecancermedicalscience 2016, 10, 617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, J.C. Mechanisms of multistep carcinogenesis and carcinogen risk assessment. Environ. Health Perspect. 1993, 100, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.C.; Wiseman, R.W. Cellular and molecular mechanisms of multistep carcinogenesis: Relevance to carcinogen risk assessment. Environ. Health Perspect. 1987, 76, 65–70. [Google Scholar] [CrossRef]
- St. Jude Children’s Research Hospital. Acute Lymphoblastic Leukemia (ALL). 2020. Available online: https://www.stjude.org/disease/acute-lymphoblastic-leukemia-all.html (accessed on 16 August 2020).
- American Cancer Society. Cancer Facts & Figures. Atlanta: American Cancer Society. 2019. Available online: https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2019.html (accessed on 16 August 2020).
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [Green Version]
- Lord, C.J.; Ashworth, A. The DNA damage response and cancer therapy. Nature 2012, 481, 287–294. [Google Scholar] [CrossRef]
- Chen, J.; Miller, B.F.; Furano, A.V. Repair of naturally occurring mismatches can induce mutations in flanking DNA. Elife 2014, 3, e02001. [Google Scholar] [CrossRef] [Green Version]
- Bui, D.T.; Friedrich, A.; Al-Sweel, N.; Liti, G.; Schacherer, J.; Aquadro, C.F.; Alani, E. Mismatch Repair Incompatibilities in Diverse Yeast Populations. Genetics 2017, 205, 1459–1471. [Google Scholar] [CrossRef] [Green Version]
- Rodgers, K.; McVey, M. Error-Prone Repair of DNA Double-Strand Breaks. J. Cell. Physiol. 2016, 231, 15–24. [Google Scholar] [CrossRef] [Green Version]
- Goodman, M.F. Error-prone repair DNA polymerases in prokaryotes and eukaryotes. Annu. Rev. Biochem. 2002, 71, 17–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoeijmakers, J.H.J. DNA damage, aging, and cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef]
- Ames, B.N. Endogenous oxidative DNA damage, aging, and cancer. Free Radic. Res. Commun. 1989, 7, 121–128. [Google Scholar] [CrossRef]
- Freitas, A.A.; de Magalhães, J.P. A review and appraisal of the DNA damage theory of ageing. Mutat. Res. 2011, 728, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Setlow, R.B. DNA repair, aging, and cancer. Natl. Cancer Inst. Monogr. 1982, 60, 249–255. [Google Scholar] [PubMed]
- Jafri, M.A.; Ansari, S.A.; Alqahtani, M.H.; Shay, J.W. Roles of telomeres and telomerase in cancer, and advances in telomerase-targeted therapies. Genome Med. 2016, 8, 69. [Google Scholar] [CrossRef] [Green Version]
- Shay, J.W.; Wright, W.E. Role of telomeres and telomerase in cancer. Semin. Cancer Biol. 2011, 21, 349–353. [Google Scholar] [CrossRef] [Green Version]
- Shay, J.W. Role of Telomeres and Telomerase in Aging and Cancer. Cancer Discov. 2016, 6, 584–593. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Rane, G.; Dai, X.; Shanmugam, M.K.; Arfuso, F.; Samy, R.P.; Lai, M.K.; Kappei, D.; Kumar, A.P.; Sethi, G. Ageing and the telomere connection: An intimate relationship with inflammation. Ageing Res. Rev. 2016, 25, 55–69. [Google Scholar] [CrossRef]
- Blackburn, E.H.; Epel, E.S. Telomeres and adversity: Too toxic to ignore. Nature 2012, 490, 169–171. [Google Scholar] [CrossRef]
- Lyon, D.E.; Starkweather, A.R.; Montpetit, A.; Menzies, V.; Jallo, N. A biobehavioral perspective on telomere length and the exposome. Biol. Res. Nurs. 2014, 16, 448–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchino, B.N.; Cawthon, R.M.; Smith, T.W.; Kent, R.G.; Bowen, K.; Light, K.C. A cross-sectional analysis of the association between perceived network social control and telomere length. Health Psychol. 2015, 34, 531–538. [Google Scholar] [CrossRef]
- Tyrka, A.R.; Parade, S.H.; Price, L.H.; Kao, H.T.; Porton, B.; Philip, N.S.; Welch, E.S.; Carpenter, L.L. Alterations of Mitochondrial DNA Copy Number and Telomere Length with Early Adversity and Psychopathology. Biol. Psychiatry 2016, 79, 78–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Révész, D.; Verhoeven, J.E.; Milaneschi, Y.; de Geus, E.J.; Wolkowitz, O.M.; Penninx, B.W. Dysregulated physiological stress systems and accelerated cellular aging. Neurobiol. Aging 2014, 35, 1422–1430. [Google Scholar] [CrossRef] [PubMed]
- Schaakxs, R.; Wielaard, I.; Verhoeven, J.E.; Beekman, A.T.; Penninx, B.W.; Comijs, H.C. Early and recent psychosocial stress and telomere length in older adults. Int. Psychogeriatr. 2016, 28, 405–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathur, M.B.; Epel, E.; Kind, S.; Desai, M.; Parks, C.G.; Sandler, D.P.; Khazeni, N. Perceived stress and telomere length: A systematic review, meta-analysis, and methodologic considerations for advancing the field. Brain Behav. Immun. 2016, 54, 158–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, E.; Girgis, A.; Lambert, S.; Sylvie, L.; Levesque, J.; Pickett, H. Telomeres and Stress: Promising Avenues for Research in Psycho-Oncology. Asia-Pac. J. Oncol. Nurs. 2016, 3, 137–147. [Google Scholar] [CrossRef]
- Shin, D.; Shin, J.; Lee, K.W. Effects of Inflammation and Depression on Telomere Length in Young Adults in the United States. J. Clin. Med. 2019, 8, 711. [Google Scholar] [CrossRef] [Green Version]
- Boccardi, M.; Boccardi, V. Psychological Wellbeing and Healthy Aging: Focus on Telomeres. Geriatrics 2019, 4, 25. [Google Scholar] [CrossRef] [Green Version]
- Kiecolt-Glaser, J.K.; Glaser, R. Psychological stress, telomeres, and telomerase. Brain Behav. Immun. 2010, 24, 529–530. [Google Scholar] [CrossRef] [Green Version]
- Price, L.H.; Kao, H.T.; Burgers, D.E.; Carpenter, L.L.; Tyrka, A.R. Telomeres and early-life stress: An overview. Biol. Psychiatry 2013, 73, 15–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiecolt-Glaser, J.K.; Jaremka, L.M.; Derry, H.M.; Glaser, R. Telomere length: A marker of disease susceptibility? Brain Behav. Immun. 2013, 34, 29–30. [Google Scholar] [CrossRef] [Green Version]
- Deng, T.; Lyon, C.J.; Bergin, S.; Caligiuri, M.A.; Hsueh, W.A. Obesity, Inflammation, and Cancer. Annu. Rev. Pathol. 2016, 11, 421–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyengar, N.M.; Gucalp, A.; Dannenberg, A.J.; Hudis, C.A. Obesity and Cancer Mechanisms: Tumor Microenvironment and Inflammation. J. Clin. Oncol. 2016, 34, 4270–4276. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Nino, M.E. The role of chronic inflammation in obesity-associated cancers. ISRN Oncol. 2013, 697521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Cancer Institute. Obesity and Cancer. 2017. Available online: https://www.cancer.gov/about-cancer/causes-prevention/risk/obesity/obesity-fact-sheet#q4 (accessed on 16 August 2020).
- Gallagher, E.J.; LeRoith, D. Obesity and diabetes: The increased risk of cancer and cancer-related mortality. Physiol. Rev. 2015, 95, 727–748. [Google Scholar] [CrossRef] [Green Version]
- Sheflin, A.M.; Whitney, A.K.; Weir, T.L. Cancer-promoting effects of microbial dysbiosis. Curr. Oncol. Rep. 2014, 16, 406. [Google Scholar] [CrossRef] [Green Version]
- Kolb, R.; Sutterwala, F.S.; Zhang, W. Obesity and cancer: Inflammation bridges the two. Curr. Opin. Pharmacol. 2016, 29, 77–89. [Google Scholar] [CrossRef] [Green Version]
- Iyengar, N.M.; Hudis, C.A.; Dannenberg, A.J. Obesity and inflammation: New insights into breast cancer development and progression. Am. Soc. Clin. Oncol. Educ. Book 2013, 46–51. [Google Scholar] [CrossRef]
- Salaün, H.; Thariat, J.; Vignot, M.; Merrouche, Y.; Vignot, S. Obesity and cancer. Bull. Cancer 2017, 104, 30–41. [Google Scholar] [CrossRef]
- Fujita, K.; Hayashi, T.; Matsushita, M.; Uemura, M.; Nonomura, N. Obesity, Inflammation, and Prostate Cancer. J. Clin. Med. 2019, 8, 201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, T.W.; McPherson, M.; Gail, D.L. Obesity and Cancer: Existing and New Hypotheses for a Causal Connection. EBioMedicine 2018, 30, 14–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, N.A. Obesity and cancer pathogenesis. Ann. N. Y. Acad. Sci. 2014, 1311, 57–76. [Google Scholar] [CrossRef] [PubMed]
- Nowsheen, S.; Yang, E.S. The intersection between DNA damage response and cell death pathways. Exp. Oncol. 2012, 34, 243–254. [Google Scholar]
- Borges, H.L.; Linden, R.; Wang, J.Y. DNA damage-induced cell death: Lessons from the central nervous system. Cell Res. 2008, 18, 17–26. [Google Scholar] [CrossRef]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013, 332, 237–248. [Google Scholar] [CrossRef]
- Bernstein, C.; Bernstein, H.; Payne, C.M.; Garewal, H. DNA repair/pro-apoptotic dual-role proteins in five major DNA repair pathways: Fail-safe protection against carcinogenesis. Mutat. Res. 2002, 511, 145–178. [Google Scholar] [CrossRef]
- De Zio, D.; Cianfanelli, V.; Cecconi, F. New insights into the link between DNA damage and apoptosis. Antioxid. Redox Signal. 2013, 19, 559–571. [Google Scholar] [CrossRef] [Green Version]
- de Mello, A.H.; Costa, A.B.; Engel, J.D.G.; Rezin, G.T. Mitochondrial dysfunction in obesity. Life Sci. 2018, 192, 26–32. [Google Scholar] [CrossRef]
- Bournat, J.C.; Brown, C.W. Mitochondrial dysfunction in obesity. Curr. Opin. Endocrinol. Diabetes Obes. 2010, 17, 446–452. [Google Scholar] [CrossRef] [Green Version]
- Guerra, F.; Arbini, A.A.; Moro, L. Mitochondria and cancer chemoresistance. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 686–699. [Google Scholar] [CrossRef]
- Guaragnella, N.; Giannattasio, S.; Moro, L. Mitochondrial dysfunction in cancer chemoresistance. Biochem. Pharmacol. 2014, 92, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.C.; Tseng, L.M.; Lee, H.C. Role of mitochondrial dysfunction in cancer progression. Exp. Biol. Med. 2016, 241, 1281–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Księżakowska-Łakoma, K.; Żyła, M.; Wilczyński, J.R. Mitochondrial dysfunction in cancer. Prz. Menopauzalny 2014, 13, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Chiu, H.Y.; Tay, E.X.Y.; Ong, D.S.T.; Taneja, R. Mitochondrial Dysfunction at the Center of Cancer Therapy. Antioxid. Redox Signal. 2020, 32, 309–330. [Google Scholar] [CrossRef]
- Seyfried, T.N. Cancer as a mitochondrial metabolic disease. Front. Cell Dev. Biol. 2015, 3, 43. [Google Scholar] [CrossRef] [Green Version]
- Seyfried, T.N.; Flores, R.E.; Poff, A.M.; D’Agostino, D.P. Cancer as a metabolic disease: Implications for novel therapeutics. Carcinogenesis 2014, 35, 515–527. [Google Scholar] [CrossRef]
- Neagu, M.; Constantin, C.; Popescu, I.D.; Zipeto, D.; Tzanakakis, G.; Nikitovic, D.; Fenga, C.; Stratakis, C.A.; Spandidos, D.A.; Tsatsakis, A.M. Inflammation and Metabolism in Cancer Cell-Mitochondria Key Player. Front. Oncol. 2019, 9, 348. [Google Scholar] [CrossRef] [Green Version]
- Poljsak, B.; Kovac, V.; Dahmane, R.; Levec, T.; Starc, A. Cancer Etiology: A Metabolic Disease Originating from Life’s Major Evolutionary Transition? Oxid. Med. Cell. Longev. 2019, 2019, 7831952. [Google Scholar] [CrossRef] [Green Version]
- Israel, B.A.; Schaeffer, W.I. Citoplasmic suppression of malignancy. In Vitro Cell. Dev. Biol. 1987, 23, 627–632. [Google Scholar] [CrossRef]
- Shay, J.W.; Liu, Y.N.; Werbin, H. Cytoplasmic suppression of tumor progression in reconstituted cells. Somat. Cell Mol. Genet. 1988, 14, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Israel, B.A.; Schaeffer, W.I. Cytoplasmic mediation of malignancy. In Vitro Cell. Dev. Biol. 1988, 24, 487–490. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N.; Kiebish, M.A.; Marsh, J.; Shelton, L.M.; Huysentruyt, L.C.; Mukherjee, P. Metabolic management of brain cancer. Biochim. Biophys. Acta 2011, 1807, 577–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seyfried, T.N.; Shelton, L.M. Cancer as a metabolic disease. Nutr. Metab. 2010, 7, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coller, H.A. Is cancer a metabolic disease? Am. J. Pathol. 2014, 184, 4–17. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y. Book Review: Cancer as a metabolic disease: On the origin, management, and prevention of cancer. J. Gynecol. Oncol. 2014, 25, 260–261. [Google Scholar] [CrossRef] [Green Version]
- Guha, M.; Avadhani, N.G. Mitochondrial retrograde signaling at the crossroads of tumor bioenergetics, genetics and epigenetics. Mitochondrion 2013, 13, 577–591. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Kim, J. Mitochondrial Retrograde Signalling and Metabolic Alterations in the Tumour Microenvironment. Cells 2019, 8, 275. [Google Scholar] [CrossRef] [Green Version]
- Kulawiec, M.; Safina, A.; Desouki, M.M.; Still, I.; Matsui, S.; Bakin, A.; Singh, K.K. Tumorigenic transformation of human breast epithelial cells induced by mitochondrial DNA depletion. Cancer Biol. Ther. 2008, 7, 1732–1743. [Google Scholar] [CrossRef] [Green Version]
- Hertweck, K.L.; Dasgupta, S. The Landscape of mtDNA Modifications in Cancer: A Tale of Two Cities. Front. Oncol. 2017, 7, 262. [Google Scholar] [CrossRef]
- Moiseeva, O.; Bourdeau, V.; Roux, A.; Deschênes-Simard, X.; Ferbeyre, G. Mitochondrial dysfunction contributes to oncogene-induced senescence. Mol. Cell. Biol. 2009, 29, 4495–4507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasileiou, P.V.S.; Evangelou, K.; Vlasis, K.; Fildisis, G.; Panayiotidis, M.I.; Chronopoulos, E.; Passias, P.G.; Kouloukoussa, M.; Gorgoulis, V.G.; Havaki, S. Mitochondrial Homeostasis and Cellular Senescence. Cells 2019, 8, 686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korolchuk, V.I.; Miwa, S.; Carroll, B.; von Zglinicki, T. Mitochondria in Cell Senescence: Is Mitophagy the Weakest Link? EBioMedicine 2017, 21, 7–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef] [Green Version]
- Aliev, G.; Priyadarshini, M.; Reddy, V.P.; Grieg, N.H.; Kaminsky, Y.; Cacabelos, R.; Ashraf, G.M.; Jabir, N.R.; Kamal, M.A.; Nikolenko, V.N.; et al. Oxidative stress mediated mitochondrial and vascular lesions as markers in the pathogenesis of Alzheimer disease. Curr. Med. Chem. 2014, 21, 2208–2217. [Google Scholar] [CrossRef]
- Gonzales-Ebsen, A.C.; Gregersen, N.; Olsen, R.K. Linking telomere loss and mitochondrial dysfunction in chronic disease. Front. Biosci. 2017, 22, 117–127. [Google Scholar] [CrossRef] [Green Version]
- Finkel, T. Telomeres and mitochondrial function. Circ. Res. 2011, 108, 903–904. [Google Scholar] [CrossRef]
- Zheng, Q.; Huang, J.; Wang, G. Mitochondria, Telomeres and Telomerase Subunits. Front. Cell Dev. Biol. 2019, 7, 274. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Trimarchi, J.R.; Smith, P.J.; Keefe, D.L. Mitochondrial dysfunction leads to telomere attrition and genomic instability. Aging Cell 2002, 1, 40–46. [Google Scholar] [CrossRef] [Green Version]
- Fossel, M. Telomerase and Cancer. 2013. Available online: http://www.michaelfossel.com/blog/?p=47 (accessed on 16 August 2020).
- Sahin, E.; Depinho, R.A. Linking functional decline of telomeres, mitochondria and stem cells during ageing. Nature 2010, 464, 520–528. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Upadhyay, M.; Samal, J.; Kandpal, M.; Singh, O.V.; Vivekanandan, P. The Warburg effect: Insights from the past decade. Pharmacol. Ther. 2013, 137, 318–330. [Google Scholar] [CrossRef]
- Fung, J. The Paradox of Cancer’s Warburg Effect. 2018. Available online: https://medium.com/@drjasonfung/the-paradox-of-cancers-warburg-effect-7fb572364b81 (accessed on 16 August 2020).
- Cluntun, A.A.; Lukey, M.J.; Cerione, R.A.; Locasale, J.W. Glutamine Metabolism in Cancer: Understanding the Heterogeneity. Trends Cancer 2017, 3, 169–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Le, A. Glutamine Metabolism in Cancer. Adv. Exp. Med. Biol. 2018, 1063, 13–32. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.K.; Park, K.G. Targeting Glutamine Metabolism for Cancer Treatment. Biomol. Ther. 2018, 26, 19–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Yu, L.; Chen, X.; Sun, X.; Wang, L.; Chen, S. The Glycolytic Switch in Tumors: How Many Players Are Involved? J. Cancer 2017, 8, 3430–3440. [Google Scholar] [CrossRef]
- Warburg, O. The metabolism of carcinoma cells. J. Cancer Res. 1925, 9, 148–163. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O.; Posener, K.; Negelein, E. Ueber den stoffwechsel der tumoren. Biochem. Z. 1924, 152, 319–344. [Google Scholar]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [Green Version]
- Kumari, S.; Badana, A.K.; Murali, M.G.; Shailender, G.; Malla, R. Reactive Oxygen Species: A Key Constituent in Cancer Survival. Biomark. Insights 2018, 13, 1177271918755391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, C.K.; Sindhu, K.K. Oxidative stress and metabolic syndrome. Life Sci. 2009, 84, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Rani, V.; Deep, G.; Singh, R.K.; Palle, K.; Yadav, U.C. Oxidative stress and metabolic disorders: Pathogenesis and therapeutic strategies. Life Sci. 2016, 148, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.S.; Sun, L.; Tan, R.; Gao, Y.; Yang, L.; Chen, H.; Teng, Y.; Lan, L. The oxidative DNA damage response: A review of research undertaken with Tsinghua and Xiangya students at the University of Pittsburgh. Sci. China Life Sci. 2017, 60, 1077–1080. [Google Scholar] [CrossRef]
- The Nobel Prize in Physiology or Medicine 2018. NobelPrize.org. Nobel Media AB 2020. 2020. Available online: https://www.nobelprize.org/prizes/medicine/2018/press-release/ (accessed on 16 August 2020).
- The Nobel Prize in Physiology or Medicine 2016. NobelPrize.org. Nobel Media AB 2020. 2020. Available online: https://www.nobelprize.org/prizes/medicine/2016/summary/ (accessed on 16 August 2020).
- Takeshige, K.; Baba, M.; Tsuboi, S.; Noda, T.; Ohsumi, Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J. Cell Biol. 1992, 119, 301–311. [Google Scholar] [CrossRef] [Green Version]
- Thumm, M.; Egner, R.; Koch, B.; Schlumpberger, M.; Straub, M.; Veenhuis, M.; Wolf, D.H. Isolation of autophagocytosis mutants of Saccharomyces cerevisiae. FEBS Lett. 1994, 349, 275–280. [Google Scholar] [CrossRef] [Green Version]
- Tsukada, M.; Ohsumi, Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993, 333, 169–174. [Google Scholar] [CrossRef] [Green Version]
- Kondo, Y.; Kondo, S. Autophagy and Cancer Therapy. Autophagy 2006, 2, 85–90. [Google Scholar] [CrossRef]
- Klionsky, D.J. Autophagy revisited: A conversation with Christian de Duve. Autophagy 2008, 4, 740–743. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [Green Version]
- Maycotte, P.; Thorburn, A. Autophagy and cancer therapy. Cancer Biol. Ther. 2011, 11, 127–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gélinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef] [Green Version]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Czaja, M.J. Regulation of lipid stores and metabolism by lipophagy. Cell Death Differ. 2013, 20, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro-Obregon, S. The Discovery of Lysosomes and Autophagy. What do cells do when they are “hungry”? Eukaryotic cells cope with starving conditions by eating their own components, a process called autophagy. Nat. Educ. 2010, 3, 49. Available online: https://www.nature.com/scitable/topicpage/the-discovery-of-lysosomes-and-autophagy-14199828. (accessed on 16 August 2020).
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef]
- Bandyopadhyay, U.; Kaushik, S.; Varticovski, L.; Cuervo, A.M. The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol. Cell. Biol. 2008, 28, 5747–5763. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef]
- Levine, B.; Klionsky, D.J. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev. Cell 2004, 6, 463–477. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Bergamini, E.; Brunk, U.T.; Dröge, W.; Ffrench, M.; Terman, A. Autophagy and aging: The importance of maintaining “clean” cells. Autophagy 2005, 1, 131–140. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.J.; Lei, Y.H.; Yao, N.; Wang, C.R.; Hu, N.; Ye, W.C.; Zhang, D.M.; Chen, Z.S. Autophagy and multidrug resistance in cancer. Chin. J. Cancer 2017, 36, 52. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, Y.; Shimizu, S. Another way to die: Autophagic programmed cell death. Cell Death Differ. 2005, 12, 1528–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez, M.G.; Master, S.S.; Singh, S.B.; Taylor, G.A.; Colombo, M.I.; Deretic, V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 2004, 119, 753–766. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Paglin, S.; Hollister, T.; Delohery, T.; Hackett, N.; McMahill, M.; Sphicas, E.; Domingo, D.; Yahalom, J. A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res. 2001, 61, 439–444. [Google Scholar]
- Elgendy, M.; Sheridan, C.; Brumatti, G.; Martin, S.J. Oncogenic Ras-induced expression of Noxa and Beclin-1 promotes autophagic cell death and limits clonogenic survival. Mol. Cell 2011, 42, 23–35. [Google Scholar] [CrossRef] [Green Version]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef] [Green Version]
- Amaravadi, R.; Kimmelman, A.C.; White, E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016, 30, 1913–1930. [Google Scholar] [CrossRef]
- Rosenfeldt, M.T.; O’Prey, J.; Morton, J.P.; Nixon, C.; MacKay, G.; Mrowinska, A.; Au, A.; Rai, T.S.; Zheng, L.; Ridgway, R.; et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature 2013, 504, 296–300. [Google Scholar] [CrossRef]
- Antunes, F.; Erustes, A.G.; Costa, A.J.; Nascimento, A.C.; Bincoletto, C.; Ureshino, R.P.; Pereira, G.; Smaili, S.S. Autophagy and intermittent fasting: The connection for cancer therapy? Clinics 2018, 73, e814s. [Google Scholar] [CrossRef]
- Wu, W.K.; Coffelt, S.B.; Cho, C.H.; Wang, X.J.; Lee, C.W.; Chan, F.K.; Yu, J.; Sung, J.J. The autophagic paradox in cancer therapy. Oncogene 2012, 31, 939–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanarek, N.; Petrova, B.; Sabatini, D.M. Dietary modifications for enhanced cancer therapy. Nature 2020, 579, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Soldati, L.; Di Renzo, L.; Jirillo, E.; Ascierto, P.A.; Marincola, F.M.; De Lorenzo, A. The influence of diet on anti-cancer immune responsiveness. J. Transl. Med. 2018, 16, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Pietrocola, F.; Kroemer, G. Nutrition, inflammation and cancer. Nat. Immunol. 2017, 18, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Berrigan, D.; Perkins, S.N.; Haines, D.C.; Hursting, S.D. Adult-onset calorie restriction and fasting delay spontaneous tumorigenesis in p53-deficient mice. Carcinogenesis 2002, 23, 817–822. [Google Scholar] [CrossRef] [Green Version]
- Brandhorst, S.; Longo, V.D. Fasting and caloric restriction in cancer prevention and treatment. Recent Results Cancer Res. 2016, 207, 241–266. [Google Scholar] [CrossRef]
- Weber, D.D.; Aminazdeh-Gohari, S.; Kohler, B. Ketogenic diet in cancer therapy. Aging 2018, 10, 164–165. [Google Scholar] [CrossRef] [Green Version]
- Lettieri-Barbato, D.; Cannata, S.M.; Casagrande, V.; Ciriolo, M.R.; Aquilano, K. Time-controlled fasting prevents aging-like mitochondrial changes induced by persistent dietary fat overload in skeletal muscle. PLoS ONE 2018, 13, e0195912. [Google Scholar] [CrossRef] [Green Version]
- de Groot, S.; Pijl, H.; van der Hoeven, J.J.M.; Kroep, J.R. Effects of short-term fasting on cancer treatment. J. Exp. Clin. Cancer Res. 2019, 38, 209. [Google Scholar] [CrossRef] [Green Version]
- Caccialanza, R.; Cereda, E.; De Lorenzo, F.; Farina, G.; Pedrazzoli, P.; AIOM-SINPE-FAVO Working Group. To fast, or not to fast before chemotherapy, that is the question. BMC Cancer 2018, 18, 337. [Google Scholar] [CrossRef] [Green Version]
- O’Flanagan, C.H.; Smith, L.A.; McDonell, S.B.; Hursting, S.D. When less may be more: Calorie restriction and response to cancer therapy. BMC Med. 2017, 15, 106. [Google Scholar] [CrossRef] [PubMed]
- Caccialanza, R.; Aprile, G.; Cereda, E.; Pedrazzoli, P. Fasting in oncology: A word of caution. Nat. Rev. Cancer 2019, 19, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epel, E.S.; Blackburn, E.H.; Lin, J.; Dhabhar, F.S.; Adler, N.E.; Morrow, J.D.; Cawthon, R.M. Accelerated telomere shortening in response to life stress. Proc. Natl. Acad. Sci. USA 2004, 101, 17312–17315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cea, M.; Cagnetta, A.; Gobbi, M.; Patrone, F.; Richardson, P.G.; Hideshima, T.; Anderson, K.C. New insights into the treatment of multiple myeloma with histone deacetylase inhibitors. Curr. Pharm. Des. 2013, 19, 734–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreu-Vieyra, C.V.; Berenson, J.R. The potential of panobinostat as a treatment option in patients with relapsed and refractory multiple myeloma. Ther. Adv. Hematol. 2014, 5, 197–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Springer Nature Limited. Nobel Prize in Physiology or Medicine. Nature Collection 2018. Available online: https://www.nature.com/collections/gqznlfngkz (accessed on 16 August 2020).
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef]
- Press Release: The Nobel Prize in Physiology or Medicine 2018. Available online: https://www.nobelprize.org/prizes/medicine/2018/press-release (accessed on 16 August 2020).
- Kruger, S.; Ilmer, M.; Kobold, S.; Cadilha, B.L.; Endres, S.; Ormanns, S.; Schuebbe, G.; Renz, B.W.; D’Haese, J.G.; Schloesser, H.; et al. Advances in cancer immunotherapy 2019—Latest trends. J. Exp. Clin. Cancer Res. 2019, 38, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.M.; Fowler, D.W.; Smith, P.; Dalgleish, A.G. Pre-treatment with chemotherapy can enhance the antigenicity and immunogenicity of tumours by promoting adaptive immune responses. Br. J. Cancer 2010, 102, 115–123. [Google Scholar] [CrossRef]
- Brix, N.; Tiefenthaller, A.; Anders, H.; Belka, C.; Lauber, K. Abscopal, immunological effects of radiotherapy: Narrowing the gap between clinical and preclinical experiences. Immunol. Rev. 2017, 280, 249–279. [Google Scholar] [CrossRef]
- Friedman, C.F.; Snyder, A. Atypical autoimmune adverse effects with checkpoint blockade therapies. Ann. Oncol. 2017, 28, 206–207. [Google Scholar] [CrossRef]
- Tauriello, D.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crowther, M.D.; Dolton, G.; Legut, M.; Caillaud, M.E.; Lloyd, A.; Attaf, M.; Galloway, S.; Rius, C.; Farrell, C.P.; Szomolay, B.; et al. Genome-wide CRISPR–Cas9 screening reveals ubiquitous T cell cancer targeting via the monomorphic MHC class I-related protein MR1. Nat. Immunol. 2020, 21, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Gherardin, N.A.; Keller, A.N.; Woolley, R.E.; Le Nours, J.; Ritchie, D.S.; Neeson, P.J.; Birkinshaw, R.W.; Eckle, S.; Waddington, J.N.; Liu, L.; et al. Diversity of T Cells Restricted by the MHC Class I-Related Molecule MR1 Facilitates Differential Antigen Recognition. Immunity 2016, 44, 32–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- University of Washington. Cancer Cells More Likely to Genetically Mutate. ScienceDaily 2007. Available online: www.sciencedaily.com/releases/2007/02/070218194439.htm (accessed on 16 August 2020).
- Knapton, S. Cancer Cells Programmed Back to Normal by US Scientists. The Telegraph 2015. Available online: https://www.telegraph.co.uk/news/science/science-news/11821334/Cancer-cells-programmed-back-to-normal-by-US-scientists.html (accessed on 16 August 2020).
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even Warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [Green Version]
- Valencia, T.; Kim, J.Y.; Abu-Baker, S.; Moscat-Pardos, J.; Ahn, C.S.; Reina-Campos, M.; Duran, A.; Castilla, E.A.; Metallo, C.M.; Diaz-Meco, M.T.; et al. Metabolic reprogramming of stromal fibroblasts through p62-mTORC1 signaling promotes inflammation and tumorigenesis. Cancer Cell 2014, 26, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Sousa, M.I.; Rodrigues, A.S.; Pereira, S.; Perestrelo, T.; Correia, M.; Ramalho-Santos, J. Mitochondrial Mechanisms of Metabolic Reprogramming in Proliferating Cells. Curr. Med. Chem. 2015, 22, 2493–2504. [Google Scholar] [CrossRef]
- Herst, P.M.; Grasso, C.; Berridge, M.V. Metabolic reprogramming of mitochondrial respiration in metastatic cancer. Cancer Metastasis Rev. 2018, 37, 643–653. [Google Scholar] [CrossRef]
- Marelli-Berg, F.M.; Fu, H.; Mauro, C. Molecular mechanisms of metabolic reprogramming in proliferating cells: Implications for T-cell-mediated immunity. Immunology 2012, 136, 363–369. [Google Scholar] [CrossRef]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar] [CrossRef] [Green Version]
- Pellegatti, P.; Raffaghello, L.; Bianchi, G.; Piccardi, F.; Pistoia, V.; Di Virgilio, F. Increased Level of Extracellular ATP at Tumor Sites: In Vivo Imaging with Plasma Membrane Luciferase. PLoS ONE 2008, 3, e2599. [Google Scholar] [CrossRef]
- Ohta, A.; Gorelik, E.; Prasad, S.J.; Ronchese, F.; Lukashev, D.; Wong, M.K.K.; Huang, X.; Caldwell, S.; Liu, K.; Smith, P.; et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13132–13137. [Google Scholar] [CrossRef] [Green Version]
- Hirai, K.; Aliev, G.; Nunomura, A.; Fujioka, H.; Russell, R.L.; Atwood, C.S.; Johnson, A.B.; Kress, Y.; Vinters, H.V.; Tabaton, M.; et al. Mitochondrial Abnormalities in Alzheimer’s Disease. J. Neurosci. 2001, 21, 3017–3023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aliev, G.; Palacios, H.H.; Gasimov, E.; Obrenovich, M.E.; Morales, L.; Leszek, J.; Bragin, V.; Solís Herrera, A.; Gokhman, D. Oxidative Stress Induced Mitochondrial Failure and Vascular Hypoperfusion as a Key Initiator for the Development of Alzheimer Disease. Pharmaceuticals 2010, 3, 158–187. [Google Scholar] [CrossRef] [Green Version]
- Aliev, G.; Palacios, H.H.; Walrafen, B.; Lipsitt, A.E.; Obrenovich, M.E.; Morales, L. Brain mitochondria as a primary target in the development of treatment strategies for Alzheimer disease. Int. J. Biochem. Cell Biol. 2009, 41, 1989–2004. [Google Scholar] [CrossRef] [PubMed]
- Sporn, M.B.; Todaro, G.J. Autocrine secretion and malignant transformation of cells. N. Engl. J. Med. 1980, 303, 878–880. [Google Scholar] [CrossRef] [PubMed]
- Asahara, T.; Murohara, T.; Sullivan, A.; Silver, M.; van der Zee, R.; Li, T.; Witzenbichler, B.; Schatteman, G.; Isner, J.M. Isolation of putative progenitor endothelial cells for angiogenesis. Science 1997, 275, 964–967. [Google Scholar] [CrossRef]
- Muresanu, C.; Somasundaram, S.; Vissarionov, S.V.; Bragin, V.; Kirkland, C.E.; Aliev, G. Hypothetical Role of Growth Factors to Prevent Degenerative Disc Diseases, through Educated Biological Transformations. Curr. Pharm. Des. 2020. [Google Scholar]
- Muresanu, C.; Somasundaram, S.G.; Kirkland, C.E. Biological Transformations Controlled by the Mind—The Science & the Practice; In Progress Preparation; Salem University: Salem, WV, USA, 2020; Volume 2. [Google Scholar]
- Wiseman, M.J. Nutrition and cancer: Prevention and survival. Br. J. Nutr. 2019, 122, 481–487. [Google Scholar] [CrossRef] [Green Version]
- Grosso, G.; Bella, F.; Godos, J.; Sciacca, S.; Del Rio, D.; Ray, S.; Galvano, F.; Giovannucci, E.L. Possible role of diet in cancer: Systematic review and multiple meta-analyses of dietary patterns, lifestyle factors, and cancer risk. Nutr. Rev. 2017, 75, 405–419. [Google Scholar] [CrossRef]
- Gerson, M. The cure of advanced cancer by diet therapy: A summary of 30 years of clinical experimentation. Physiol. Chem. Phys. 1978, 10, 449–464. [Google Scholar]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Levine, B.; Green, D.R.; Kroemer, G. Pharmacological modulation of autophagy: Therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 2017, 16, 487–511. [Google Scholar] [CrossRef] [Green Version]
- Fulda, S. Autophagy in Cancer Therapy. Front. Oncol. 2017, 7, 128. [Google Scholar] [CrossRef] [PubMed]
- Somasundaram, S.; Edmund, N.A.; Moore, D.T.; Small, G.W.; Shi, Y.Y.; Orlowski, R.Z. Dietary curcumin inhibits chemotherapy-induced apoptosis in models of human breast cancer. Cancer Res. 2002, 62, 3868–3875. [Google Scholar] [PubMed]
- Campbell, T.C. Cancer Prevention and Treatment by Wholistic Nutrition. J. Nat. Sci. 2017, 3, e448. [Google Scholar] [PubMed]
- Pal, D.; Banerjee, S.; Ghosh, A.K. Dietary-induced cancer prevention: An expanding research arena of emerging diet related to healthcare system. J. Adv. Pharm. Technol. Res. 2012, 3, 16–24. [Google Scholar] [CrossRef]
- Aghajanpour, M.; Nazer, M.R.; Obeidavi, Z.; Akbari, M.; Ezati, P.; Kor, N.M. Functional foods and their role in cancer prevention and health promotion: A comprehensive review. Am. J. Cancer Res. 2017, 7, 740–769. [Google Scholar]
- Shingler, E.; Perry, R.; Mitchell, A.; England, C.; Perks, C.; Herbert, G.; Ness, A.; Atkinson, C. Dietary restriction during the treatment of cancer: Results of a systematic scoping review. BMC Cancer 2019, 19, 811. [Google Scholar] [CrossRef] [Green Version]
- Iorio, M.V.; Croce, C.M. MicroRNAs in cancer: Small molecules with a huge impact. J. Clin. Oncol. 2009, 27, 5848–5856. [Google Scholar] [CrossRef]
- Zhou, K.; Liu, M.; Cao, Y. New Insight into microRNA Functions in Cancer: Oncogene-microRNA-Tumor Suppressor Gene Network. Front. Mol. Biosci. 2017, 4, 46. [Google Scholar] [CrossRef] [Green Version]
- Gandellini, P.; Profumo, V.; Folini, M.; Zaffaroni, N. MicroRNAs as new therapeutic targets and tools in cancer. Expert Opin. Ther. Targets 2011, 15, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, C.; Sharma, A.R.; Sharma, G.; Sarkar, B.K.; Lee, S.S. The novel strategies for next-generation cancer treatment: MiRNA combined with chemotherapeutic agents for the treatment of cancer. Oncotarget 2018, 9, 10164–10174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fruehauf, J.P.; Meyskens, F.L., Jr. Reactive oxygen species: A breath of life or death? Clin. Cancer Res. 2007, 13, 789–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamran, S.C.; Berrington de Gonzalez, A.; Ng, A.; Haas-Kogan, D.; Viswanathan, A.N. Therapeutic radiation and the potential risk of second malignancies. Cancer 2016, 122, 1809–1821. [Google Scholar] [CrossRef]
- Dracham, C.B.; Shankar, A.; Madan, R. Radiation induced secondary malignancies: A review article. Radiat. Oncol. J. 2018, 36, 85–94. [Google Scholar] [CrossRef]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Parsonnet, J. Bacterial infection as a cause of cancer. Environ. Health Perspect. 1995, 103, 263–268. [Google Scholar] [CrossRef] [Green Version]
- Sedighi, M.; Zahedi, B.A.; Hamblin, M.R.; Ohadi, E.; Asadi, A.; Halajzadeh, M.; Lohrasbi, V.; Mohammadzadeh, N.; Amiriani, T.; Krutova, M.; et al. Therapeutic bacteria to combat cancer; current advances, challenges, and opportunities. Cancer Med. 2019, 8, 3167–3181. [Google Scholar] [CrossRef]
- Muresanu, C.; Somasundaram, S.G. Biological Transformations Controlled by the Mind; AlphaGraphics Sugar Land: Houston, TX, USA, 2013; Volume 1. [Google Scholar]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muresanu, C.; Somasundaram, S.G.; Vissarionov, S.V.; Torres Solis, L.F.; Solís Herrera, A.; Kirkland, C.E.; Aliev, G. Updated Understanding of Cancer as a Metabolic and Telomere-Driven Disease, and Proposal for Complex Personalized Treatment, a Hypothesis. Int. J. Mol. Sci. 2020, 21, 6521. https://doi.org/10.3390/ijms21186521
Muresanu C, Somasundaram SG, Vissarionov SV, Torres Solis LF, Solís Herrera A, Kirkland CE, Aliev G. Updated Understanding of Cancer as a Metabolic and Telomere-Driven Disease, and Proposal for Complex Personalized Treatment, a Hypothesis. International Journal of Molecular Sciences. 2020; 21(18):6521. https://doi.org/10.3390/ijms21186521
Chicago/Turabian StyleMuresanu, Cristian, Siva G. Somasundaram, Sergey V. Vissarionov, Luis Fernando Torres Solis, Arturo Solís Herrera, Cecil E. Kirkland, and Gjumrakch Aliev. 2020. "Updated Understanding of Cancer as a Metabolic and Telomere-Driven Disease, and Proposal for Complex Personalized Treatment, a Hypothesis" International Journal of Molecular Sciences 21, no. 18: 6521. https://doi.org/10.3390/ijms21186521
APA StyleMuresanu, C., Somasundaram, S. G., Vissarionov, S. V., Torres Solis, L. F., Solís Herrera, A., Kirkland, C. E., & Aliev, G. (2020). Updated Understanding of Cancer as a Metabolic and Telomere-Driven Disease, and Proposal for Complex Personalized Treatment, a Hypothesis. International Journal of Molecular Sciences, 21(18), 6521. https://doi.org/10.3390/ijms21186521