Regulation of Functional Protein Aggregation by Multiple Factors: Implications for the Amyloidogenic Behavior of the CAP Superfamily Proteins




Abstract
:
{kind=link}
{kind=link}
{kind=link}
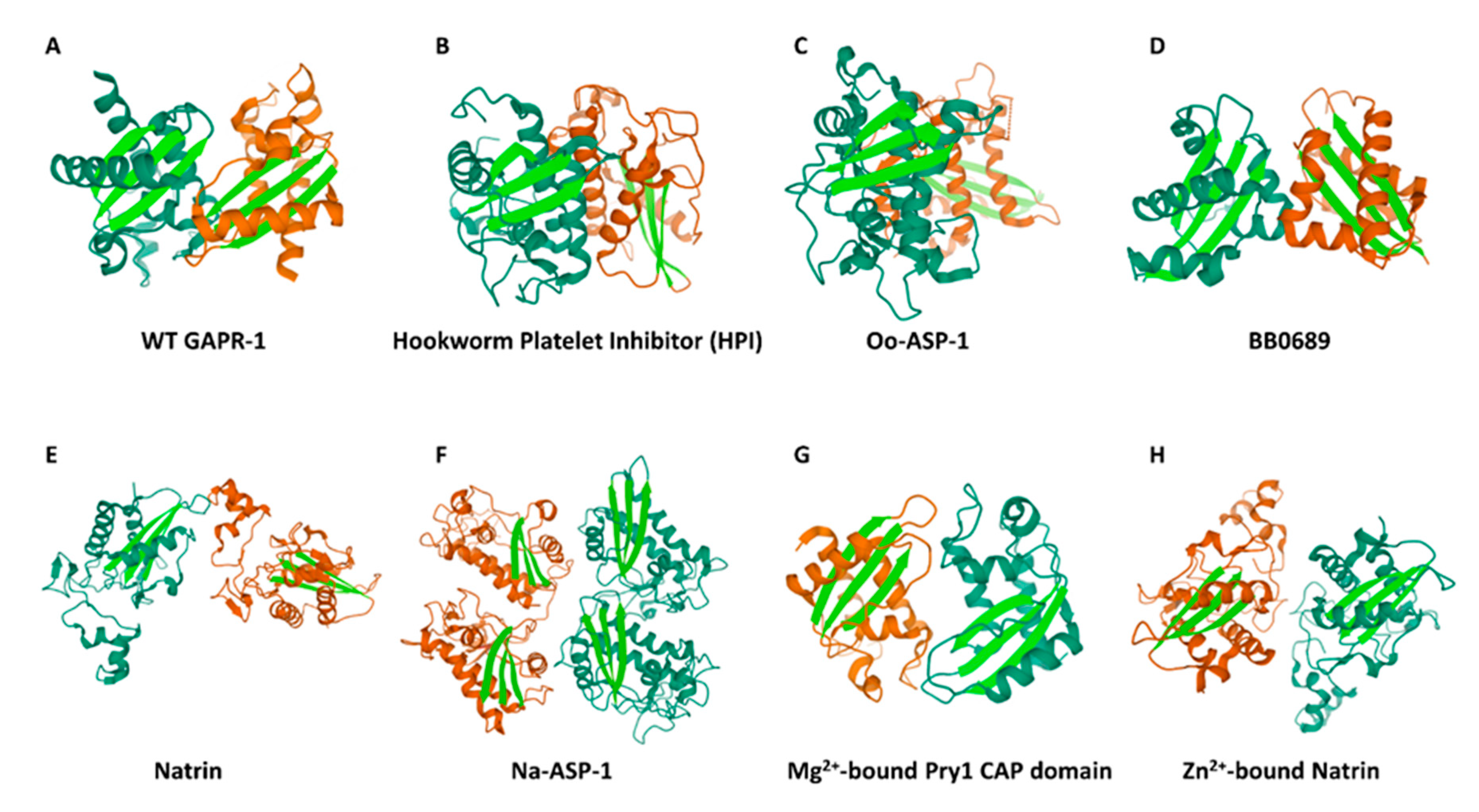{kind=link}
1. Introduction to Protein Aggregates: Two Sides of a Coin
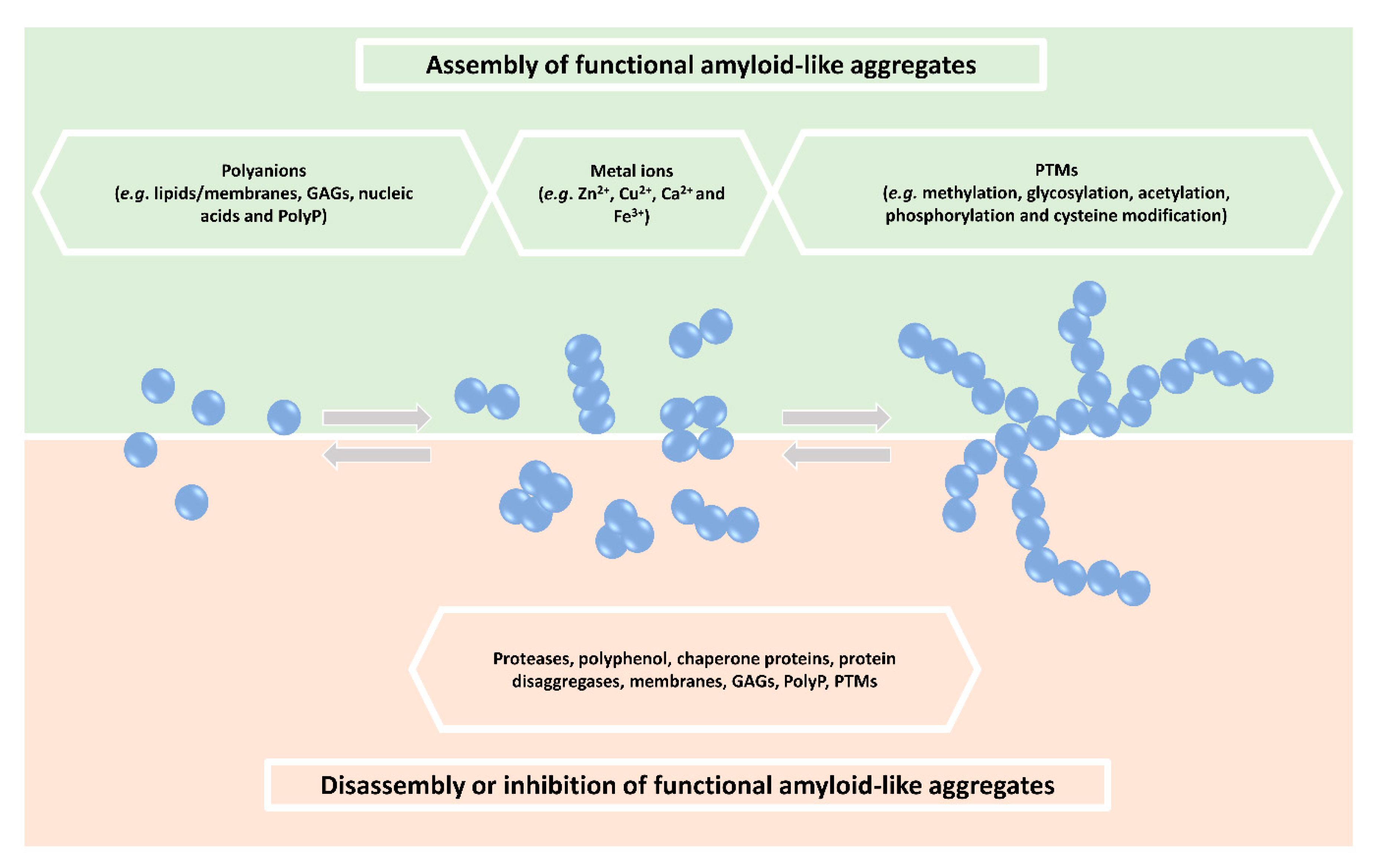
2. Regulation of Functional Amyloid-Like Aggregate Formation
2.1. Kinetics
2.2. Reversibility
3. Other Factors Regulating Functional Protein Aggregation
3.1. Polyanions
3.1.1. Lipids/Membranes
3.1.2. GAGs
3.1.3. Nucleic Acids
3.1.4. Polyphosphate (PolyP)
3.2. Metal Ions
3.3. Post-Translational Modifications
3.4. Emerging Factors Affecting Protein Aggregation
4. Crosstalk between Different Factors Affecting Amyloidogenesis of GAPR-1 and Members of the CAP Superfamily Proteins
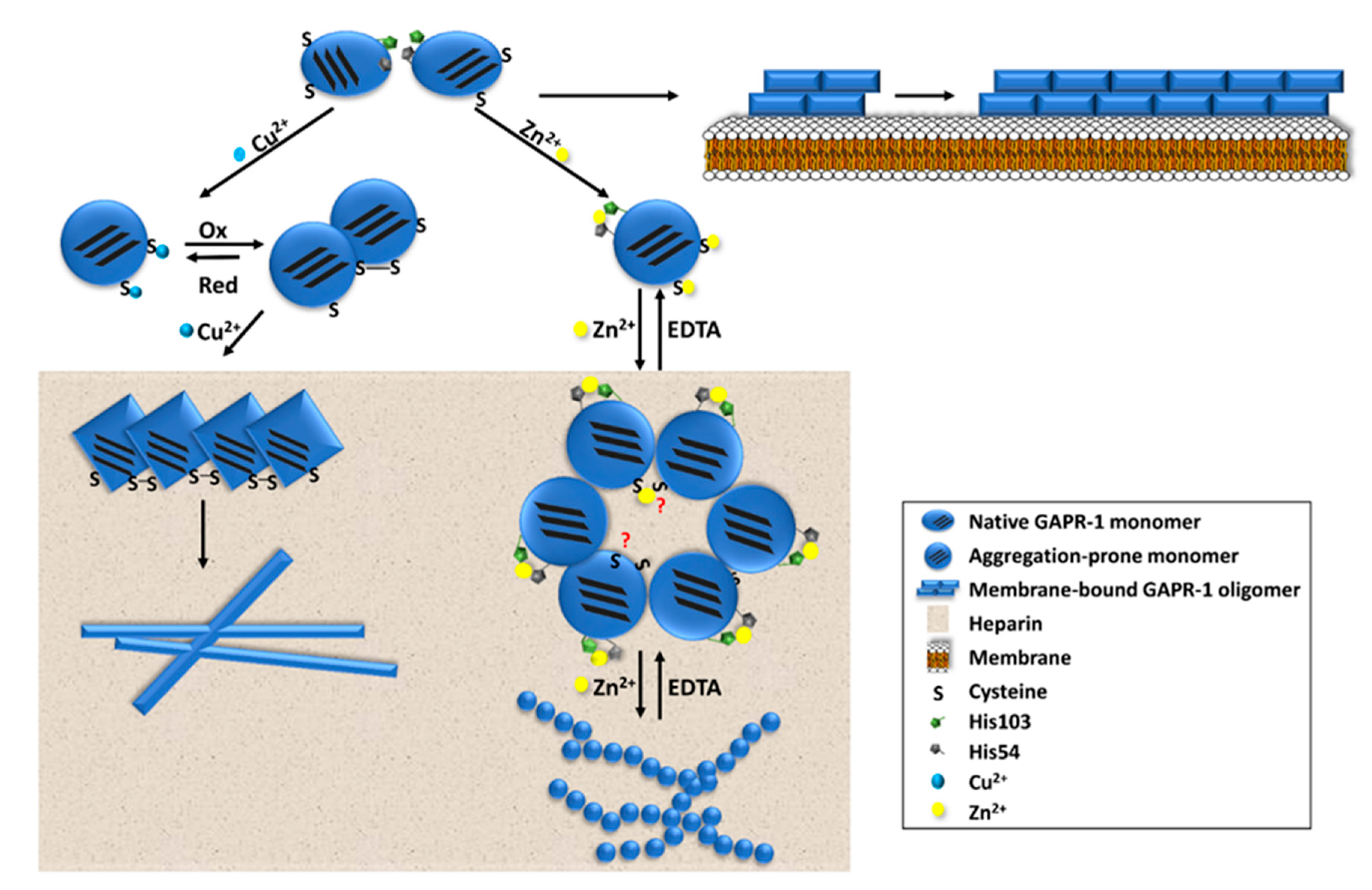
4.1. Amyloid-Like Aggregation of GAPR-1
4.2. Potential Regulation of Amyloid-Like Aggregation of CAP Family Members
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| GAGs | glycosaminoglycans |
| PTMs | post-translational modifications |
| HSPs | heat shock proteins |
| polyP | polyphosphate |
| PMEL | pre-melanosomal protein |
| PG-1 | protegrin-1 |
| CRESs | cystatin-related epididymal spermatogenic proteins |
| PLD | prion-like domain |
| A-bodies | amyloid bodies |
| P-bodies | processing bodies |
| HSGs | heat-shock granules |
| SGs | stress granules |
| GH | growth hormone |
| AM | acrosomal matrix |
| USP | ubiquitin-proteasome system |
| RPT | repeat domain |
| HS | heparan sulfate |
| IAPP | islet amyloid polypeptide |
| AD | Alzheimer’s disease |
| PD | Parkinson’s disease |
| ALS | amyotrophic lateral sclerosis |
| HypF-N | Escherichia coli HypF |
| KCTD1 | potassium channel tetramerization domain containing 1 |
| LCR | low complexity region |
| Httex1 | phosphorylation of mutant huntingtin exon1 |
| SOD1 | superoxide dismutase 1 |
| PrP | prion protein |
| KLD | Kringle-like domain |
| SST-14 | somatostatin-14 |
| RIPK1 | receptor-interacting protein kinase 1 |
| MLKL | mixed-lineage kinase domain-like protein |
| ZP | sperm-zona pellucida |
| β-CD | β-cyclodextrin |
| GAPR-1 | golgi-associated pathogenesis-related protein 1 |
| CRISP 1 | cysteine-rich secretory protein 1 |
| ECD | evolutionary conserved domain |
| CAP | cysteine-rich secretory proteins, antigen 5 and pathogenesis-related proteins group 1 |
| APRs | aggregation-prone regions |
| PR-1 | pathogenesis related protein 1 |
| GLIPR1 | glioma pathogenesis-related protein 1 |
References
- Stefani, M.; Dobson, C.M. Protein aggregation and aggregate toxicity: New insights into protein folding, misfolding diseases and biological evolution. J. Mol. Med. 2003, 81, 678–699. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; O’Coonor, T. Protein aggregation diseases: Pathogenicity and therapeutic perspectives. Nat. Rev. 2010, 9, 237–248. [Google Scholar] [CrossRef]
- Cereghetti, G.; Saad, S.; Dechant, R.; Peter, M. Reversible, functional amyloids: Towards an understanding of their regulation in yeast and humans. Cell Cycle 2018, 17, 1545–1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iadanza, M.G.; Jackson, M.P.; Hewitt, E.W.; Ranson, N.A.; Radford, S.E. A new era for understanding amyloid structures and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 755–773. [Google Scholar] [CrossRef] [PubMed]
- Sawaya, M.R.; Sambashivan, S.; Nelson, R.; Ivanova, M.I.; Sievers, S.A.; Apostol, M.I.; Thompson, M.J.; Balbirnie, M.; Wiltzius, J.J.W.; Mcfarlane, H.T.; et al. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature 2007, 447, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Protein misfolding, amyloid formation, and human disease: A summary of progress over the last decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef] [PubMed]
- Maji, S.K.; Perrin, M.H.; Sawaya, M.R.; Jessberger, S.; Vadodaria, K.; Rissman, R.A.; Singru, P.S.; Nilsson, K.P.R.; Simon, R.; Schubert, D.; et al. Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science 2009, 325, 328–332. [Google Scholar] [CrossRef] [Green Version]
- Jacob, R.S.; Das, S.; Ghosh, S.; Anoop, A.; Jha, N.N.; Khan, T.; Singru, P.; Kumar, A.; Maji, S.K. Amyloid formation of growth hormone in presence of zinc: Relevance to its storage in secretory granules. Sci. Rep. 2016, 6, 23370. [Google Scholar] [CrossRef] [Green Version]
- Roan, N.R.; Sandi-Monroy, N.; Kohgadai, N.; Usmani, S.M.; Hamil, K.G.; Neidleman, J.; Montano, M.; Ständker, L.; Röcker, A.; Cavrois, M.; et al. Semen amyloids participate in spermatozoa selection and clearance. eLife 2017, 6, e24888. [Google Scholar] [CrossRef]
- Egge, N.; Muthusubramanian, A.; Cornwall, G.A. Amyloid properties of the mouse egg zona pellucida. PLoS ONE 2015, 10, e0129907. [Google Scholar] [CrossRef] [Green Version]
- Hewetson, A.; Do, H.Q.; Myers, C.; Muthusubramanian, A.; Sutton, R.B.; Wylie, B.J.; Cornwall, G.A. Functional amyloids in reproduction. Biomolecules 2017, 7, 46. [Google Scholar] [CrossRef] [PubMed]
- Bissig, C.; Rochin, L.; Niel, G. van PMEL amyloid fibril formation: The bright steps of pigmentation. Int. J. Mol. Sci. 2016, 17, 1438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Liu, H.; Johnston, A.; Hanna-Addams, S.; Reynoso, E.; Xiang, Y.; Wang, Z. MLKL forms disulfide bond-dependent amyloid-like polymers to induce necroptosis. Proc. Natl. Acad. Sci. USA 2017, 114, E7450–E7459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, H.; Arce, F.T.; Mustata, M.; Ramachandran, S.; Capone, R.; Nussinov, R.; Lal, R. Antimicrobial protegrin-1 forms amyloid-like fibrils with rapid kinetics suggesting a functional link. Biophys. J. 2011, 100, 1775–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saad, S.; Cereghetti, G.; Feng, Y.; Picotti, P.; Peter, M.; Dechant, R. Reversible protein aggregation is a protective mechanism to ensure cell cycle restart after stress. Nat. Cell Biol. 2017, 19, 1202–1213. [Google Scholar] [CrossRef]
- Audas, T.E.; Audas, D.E.; Jacob, M.D.; Ho, J.J.D.; Khacho, M.; Wang, M.; Perera, J.K.; Gardiner, C.; Bennett, C.A.; Head, T.; et al. Adaptation to stressors by systemic protein amyloidogenesis. Dev. Cell. 2016, 39, 155–168. [Google Scholar] [CrossRef] [Green Version]
- Woodruff, J.B.; Hyman, A.A.; Boke, E. Organization and function of non-dynamic biomolecular condensates. Trends Biochem. Sci. 2018, 43, 81–94. [Google Scholar] [CrossRef]
- Boke, E.; Ruer, M.; Wühr, M.; Coughlin, M.; Lemaitre, R.; Gygi, S.P.; Alberti, S.; Drechsel, D.; Hyman, A.A.; Mitchison, T.J. Amyloid-like self-assembly of a cellular compartment. Cell 2016, 166, 637–650. [Google Scholar] [CrossRef] [Green Version]
- Pepling, M.E.; Wilhelm, J.E.; Hara, A.L.O.; Gephardt, G.W.; Spradling, A.C. Mouse oocytes within germ cell cysts and primordial follicles contain a Balbiani body. Proc. Natl. Acad. Sci. USA 2007, 104, 187–192. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.; Yin, M.; Chen, J.; Li, X. Assembly and substrate recognition of curli biogenesis system. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Perov, S.; Lidor, O.; Salinas, N.; Golan, N.; Tayeb-Fligelman, E.; Deshmukh, M.; Willbold, D.; Landau, M. Structural insights into curli CsgA cross-β fibril architecture inspire repurposing of anti-amyloid compounds as anti-biofilm agents. PLoS Pathog. 2019, 15, PMC6748439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipke, P.N.; Garcia, M.C.; Alsteens, D.; Ramsook, C.B.; Klotz, S.A.; Dufrêne, Y.F. Strengthening relationships: Amyloids create adhesion nanodomains in yeasts. Trends Microbiol. 2012, 20, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberti, S.; Halfmann, R.; King, O.; Kapila, A.; Lindquist, S. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell 2009, 137, 146–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majumdar, A.; Cesario, W.C.; White-Grindley, E.; Jiang, H.; Ren, F.; Khan, M.R.; Li, L.; Choi, E.M.-L.; Kannan, K.; Guo, F.; et al. Critical role of amyloid-like oligomers of Drosophila Orb2 in the persistence of memory. Cell 2012, 148, 515–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grignaschi, E.; Cereghetti, G.; Grigolato, F.; Kopp, M.R.G.; Caimi, S.; Faltova, L.; Saad, S.; Peter, M.; Arosio, P. A hydrophobic low-complexity region regulates aggregation of the yeast pyruvate kinase Cdc19 into amyloid-like aggregates in vitro. J. Biol. Chem. 2018, 293, 11424–11432. [Google Scholar] [CrossRef] [Green Version]
- Hervas, R.; Rau, M.J.; Park, Y.; Zhang, W.; Murzin, A.G.; Fitzpatrick, J.A.J.; Scheres, S.H.W.; Si, K. Cryo-EM structure of a neuronal functional amyloid implicated in memory persistence in Drosophila. Science (80-.) 2020, 367, 1230–1234. [Google Scholar] [CrossRef]
- Ulamec, S.M.; Radford, S.E. Spot the Difference: Function versus Toxicity in Amyloid Fibrils. Trends Biochem. Sci. 2020, 45, 635–636. [Google Scholar] [CrossRef]
- Stefani, M. Biochemical and biophysical features of both oligomer/fibril and cell membrane in amyloid cytotoxicity. FEBS J. 2010, 277, 4602–4613. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, U.; Nilson, A.N.; Kayed, R. The role of amyloid-β oligomers in toxicity, propagation, and immunotherapy. EBioMedicine 2016, 6, 42–49. [Google Scholar] [CrossRef] [Green Version]
- Demuro, A.; Mina, E.; Kayed, R.; Milton, S.C.; Parker, I.; Glabe, C.G. Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J. Biol. Chem. 2005, 280, 17294–17300. [Google Scholar] [CrossRef] [Green Version]
- Kayed, R.; Sokolov, Y.; Edmonds, B.; McIntire, T.M.; Milton, S.C.; Hall, J.E.; Glabe, C.G. Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J. Biol. Chem. 2004, 279, 46363–46366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, M.P.; Hewitt, E.W. Why are functional amyloids non-toxic in humans? Biomolecules 2017, 7, 7655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watt, B.; Tenza, D.; Lemmon, M.A.; Kerje, S.; Raposo, G.; Andersson, L.; Marks, M.S. Mutations in or near the transmembrane domain alter PMEL amyloid formation from functional to pathogenic. PLoS Genet. 2011, 7, e1002286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubén , H.; Li, L.; Majumdar, A.; Fernández-Ramírez, M.D.C.; Unruh, J.R.; Slaughter, B.D.; Galera-Prat, A.; Santana, E.; Suzuki, M.; Nagai, Y.; et al. Molecular basis of Orb2 amyloidogenesis and blockade of memory consolidation. PLoS Biol. 2016, 14, e1002361. [Google Scholar]
- Fowler, D.M.; Koulov, A.V.; Alory-jost, C.; Marks, M.S.; Balch, W.E.; Kelly, J.W. Functional amyloid formation within mammalian tissue. PLoS Biol. 2006, 4, 0100–0107. [Google Scholar] [CrossRef] [PubMed]
- Watt, B.; Van Niel, G.; Fowler, D.M.; Hurbain, I.; Luk, K.C.; Stayrook, S.E.; Lemmon, M.A.; Raposo, G.; Shorter, J.; Kelly, J.W.; et al. N-terminal domains elicit formation of functional Pmel17 amyloid fibrils. J. Biol. Chem. 2009, 284, 35543–35555. [Google Scholar] [CrossRef] [Green Version]
- Sleutel, M.; Van Den Broeck, I.; Van Gerven, N.; Feuillie, C.; Jonckheere, W.; Valotteau, C.; Dufrêne, Y.F.; Remaut, H. Nucleation and growth of a bacterial functional amyloid at single-fiber resolution. Nat. Chem. Biol. 2017, 13, 902–910. [Google Scholar] [CrossRef] [Green Version]
- Roberts, R.G. Good amyloids, bad amyloids-what’s the difference? PLoS Biol. 2016, 14, e1002362. [Google Scholar] [CrossRef] [Green Version]
- Narayanaswamy, R.; Levy, M.; Tsechansky, M.; Stovall, G.M.; O’Connell, J.D.; Mirrielees, J.; Ellington, A.D.; Marcotte, E.M. Widespread reorganization of metabolic enzymes into reversible assemblies upon nutrient starvation. Proc. Natl. Acad. Sci. USA 2009, 106, 10147–10152. [Google Scholar] [CrossRef] [Green Version]
- Wallace, E.W.J.; Kear-scott, J.L.; Pilipenko, E.V.; Schwartz, M.H.; Laskowski, P.R.; Rojek, A.E.; Katanski, C.D.; Riback, J.A.; Dion, M.F.; Franks, A.M.; et al. Reversible, specific, active aggregates of endogenous proteins assemble upon heat stress. Cell 2015, 162, 1286–1298. [Google Scholar] [CrossRef] [Green Version]
- Munder, M.C.; Midtvedt, D.; Franzmann, T.; Nüske, E.; Otto, O.; Herbig, M.; Ulbricht, E.; Müller, P.; Taubenberger, A.; Maharana, S.; et al. A pH-driven transition of the cytoplasm from a fluid- to a solid-like state promotes entry into dormancy. eLife 2016, 5, e09347. [Google Scholar] [CrossRef] [PubMed]
- Guyonnet, B.; Egge, N.; Cornwall, G.A. Functional amyloids in the mouse sperm acrosome. Mol. Cell. Biol. 2014, 34, 2624–2634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dannies, P.S. Prolactin and growth hormone aggregates in secretory granules: The need to understand the structure of the aggregate. Endocr. Rev. 2012, 33, 254–270. [Google Scholar] [CrossRef] [Green Version]
- Sun, D.; Wu, R.; Li, P.; Yu, L. Phase Separation in Regulation of Aggrephagy. J. Mol. Biol. 2020, 432, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Chuang, E.; Hori, A.M.; Hesketh, C.D.; Shorter, J. Amyloid assembly and disassembly. J. Cell Sci. 2018, 131, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Hipp, M.S.; Park, S.; Hartl, F.U. Proteostasis impairment in protein misfolding and aggregation diseases. Trends Cell Biol. 2014, 24, 506–514. [Google Scholar] [CrossRef]
- Jackson, M.P.; Hewitt, E.W. Cellular proteostasis: Degradation of misfolded proteins by lysosomes. Essays Biochem. 2016, 60, 173–180. [Google Scholar]
- Barmada, S.J.; Serio, A.; Arjun, A.; Bilican, B.; Daub, A.; Ando, D.M.; Tsvetkov, A.; Pleiss, M.; Li, X.; Peisach, D.; et al. Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat. Chem. Biol. 2014, 10, 677–685. [Google Scholar] [CrossRef] [Green Version]
- Webb, J.L.; Ravikumar, B.; Atkins, J.; Skepper, J.N.; Rubinsztein, D.C. Alpha-synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 2003, 278, 25009–25013. [Google Scholar] [CrossRef] [Green Version]
- Falcon, B.; Noad, J.; Mcmahon, H.; Randow, F.; Goedert, M. Galectin-8-mediated autophagy and seeded tau aggregation protects against seeded tau aggregation. J. Biol. Chem. 2018, 293, 2438–2451. [Google Scholar] [CrossRef] [Green Version]
- Cho, M.-H.; Cho, K.; Kang, H.-J.; Jeon, E.-Y.; Kim, H.-S.; Kwon, H.-J.; Kim, H.-M.; Kim, D.-H.; Yoon, S.-Y. Autophagy in microglia degrades extracellular β-amyloid fibrils and regulates the NLRP3 inflammasome. Autophagy 2014, 10, 1761–1775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zare-shahabadi, A.; Masliah, E.; Johnson, G.V.W.; Rezaei, N. Autophagy in Alzheimer’s disease. Rev. Neurosci. 2016, 26, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.A.; Rhim, H. Therapeutic implication of autophagy in neurodegenerative diseases. BMB Rep. 2017, 50, 345–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldberg, A.L. Protein degradation and protection against misfolded or damaged proteins. Nature 2003, 426, 895–899. [Google Scholar] [CrossRef] [PubMed]
- Myeku, N.; Clelland, C.L.; Emrani, S.; Kukushkin, N.V.; Yu, W.H.; Goldberg, A.L.; Duff, K.E. Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early by activating cAMP-PKA signaling. Nat. Med. 2016, 22, 46–53. [Google Scholar] [CrossRef]
- Kroschwald, S.; Munder, M.C.; Maharana, S.; Franzmann, T.M.; Richter, D.; Ruer, M.; Hyman, A.A.; Alberti, S. Different material states of Pub1 condensates define distinct modes of stress adaptation and recovery. Cell Rep. 2018, 23, 3327–3339. [Google Scholar] [CrossRef]
- den Brave, F.; Cairo, L.V.; Jagadeesan, C.; Ruger-Herreros, C.; Mogk, A.; Bukau, B.; Jentsch, S. Chaperone-Mediated Protein Disaggregation Triggers Proteolytic Clearance of Intra-nuclear Protein Inclusions. Cell Rep. 2020, 31, 107680. [Google Scholar] [CrossRef]
- Burmann, B.M.; Gerez, J.A.; Matečko-Burmann, I.; Campioni, S.; Kumari, P.; Ghosh, D.; Mazur, A.; Aspholm, E.E.; Šulskis, D.; Wawrzyniuk, M.; et al. Regulation of α-synuclein by chaperones in mammalian cells. Nature 2020, 577, 127–132. [Google Scholar] [CrossRef]
- Mack, K.L.; Shorter, J. Engineering and evolution of molecular chaperones and protein disaggregases with enhanced activity. Front. Mol. Biosci. 2016, 3, 8. [Google Scholar] [CrossRef] [Green Version]
- Lindberg, I.; Shorter, J.; Wiseman, R.L.; Chiti, F.; Dickey, C.A.; Mclean, X.J. Chaperones in neurodegeneration. J. Neurosci. 2015, 35, 13853–13859. [Google Scholar] [CrossRef] [Green Version]
- Weickert, S.; Wawrzyniuk, M.; John, L.H.; Rüdiger, S.G.D.; Drescher, M. The mechanism of Hsp90-induced oligomerizaton of Tau. Sci. Adv. 2020, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desantis, M.E.; Leung, E.H.; Sweeny, E.A.; Jackrel, M.E.; Cushman-nick, M.; Neuhaus-follini, A.; Vashist, S.; Sochor, M.A.; Knight, M.N.; Shorter, J. Operational plasticity enables Hsp104 to disaggregate diverse amyloid and nonamyloid clients. Cell 2012, 151, 778–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desantis, M.E.; Shorter, J. Hsp104 drives “Protein-Only” positive selection of Sup35 prion strains encoding strong [PSI+]. Chem. Biol. 2012, 19, 1400–1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackrel, M.E.; Desantis, M.E.; Martinez, B.A.; Castellano, L.M.; Stewart, R.M.; Caldwell, K.A.; Caldwell, G.A.; Shorter, J. Potentiated Hsp104 variants antagonize diverse proteotoxic misfolding events. Cell 2015, 156, 170–182. [Google Scholar] [CrossRef] [Green Version]
- Torrente, M.P.; Chuang, E.; Noll, M.M.; Jackrel, M.E.; Go, M.S.; Shorter, J. Mechanistic insights into Hsp104 potentiation. J. Biol. Chem. 2016, 291, 5101–5115. [Google Scholar] [CrossRef] [Green Version]
- Mcglinchey, R.P.; Lee, J.C. Why study functional amyloids? Lessons from the repeat domain of Pmel17. J. Mol. Biol. 2018, 430, 3696–3706. [Google Scholar] [CrossRef]
- Langner, M.; Kubica, K. The electrostatics of lipid surfaces. Chem. Phys. Lipids 1999, 101, 3–35. [Google Scholar] [CrossRef]
- Kinnunen, P.K.J. Amyloid formation on lipid membrane surfaces. Open Biol. J. 2009, 2, 163–175. [Google Scholar] [CrossRef]
- Iannuzzi, C.; Irace, G.; Sirangelo, I. The effect of glycosaminoglycans (GAGs) on amyloid aggregation and toxicity. Molecules 2015, 20, 2510–2528. [Google Scholar] [CrossRef] [Green Version]
- Solomon, J.P.; Bourgault, S.; Powers, E.T.; Kelly, J.W. Heparin binds 8 kDa gelsolin cross-β-sheet oligomers and accelerates amyloidogenesis by hastening fibril extension. Biochemistry 2011, 50, 2486–2498. [Google Scholar] [CrossRef] [Green Version]
- Bourgault, S.; Solomon, J.P.; Reixach, N.; Kelly, J.W. Sulfated glycosaminoglycans accelerate transthyretin amyloidogenesis by quaternary structural conversion. Biochemistry 2011, 50, 1001–1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Zhang, Y. Nucleic acid-mediated protein aggregation and assembly. Adv. Protein Chem. Struct. Biol. 2011, 84, 1–40. [Google Scholar]
- Sasahara, K.; Yamaguchi, K.; So, M.; Goto, Y. Polyphosphates diminish solubility of a globular protein and thereby promote amyloid aggregation. J. Biol. Chem. 2019, 294, 15318–15329. [Google Scholar] [CrossRef] [PubMed]
- Murphy, R.M. Kinetics of amyloid formation and membrane interaction with amyloidogenic proteins. Biochim. Biophys. Acta 2007, 1768, 1923–1934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fantini, J.; Yahi, N. Molecular insights into amyloid regulation by membrane cholesterol and sphingolipids: Common mechanisms in neurodegenerative diseases. Expert Rev. Mol. Med. 2019, 12, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Soria, M.A.; Cervantes, S.A.; Bajakian, T.H.; Siemer, A.B. The functional amyloid Orb2A binds to lipid membranes. Biophys. J. 2017, 113, 37–47. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.; Lee, J.C. Lysophospholipid-containing membranes modulate the fibril formation of the repeat domain of a human functional amyloid, Pmel17. J. Mol. Biol. 2014, 426, 4074–4086. [Google Scholar] [CrossRef] [Green Version]
- Malishev, R.; Abbasi, R.; Jelinek, R.; Chai, L. Bacterial model membranes reshape fibrillation of a functional amyloid protein. Biochemistry 2018, 57, 5230–5238. [Google Scholar] [CrossRef]
- Relini, A.; Marano, N.; Gliozzi, A. Probing the interplay between amyloidogenic proteins and membranes using lipid monolayers and bilayers. Adv. Colloid Interface Sci. 2014, 207, 81–92. [Google Scholar] [CrossRef]
- Dulce, P.-G.; Christophe, M.; Minh Bao, H.; Fernando, S.; Ludmilla, S.; Diaz Julia Elisa, S.; Rita, R.-V. Glycosaminoglycans, protein aggregation and neurodegeneration. Curr. Protein Pept. Sci. 2011, 999, 1–11. [Google Scholar] [CrossRef]
- Dharmadana, D.; Reynolds, N.P.; Conn, C.E.; Valéry, C. Molecular interactions of amyloid nanofibrils with biological aggregation modifiers: Implications for cytotoxicity mechanisms and biomaterial design. Interface Focus 2017, 7, 20160160. [Google Scholar] [CrossRef] [PubMed]
- De Carufel, C.A.; Nguyen, P.T.; Sahnouni, S.; Bourgault, S. New insights into the roles of sulfated glycosaminoglycans in islet amyloid polypeptide amyloidogenesis and cytotoxicity. Biopolymers 2013, 100, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Vieira, T.C.R.G.; Cordeiro, Y.; Caughey, B.; Silva, J.L. Heparin binding confers prion stability and impairs its aggregation. FASEB J. 2014, 28, 2667–2676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehra, S.; Ghosh, D.; Kumar, R.; Mondal, M.; Gadhe, L.G.; Das, S.; Anoop, A.; Jha, N.N.; Jacob, R.S.; Chatterjee, D.; et al. Glycosaminoglycans have variable effects on α-synuclein aggregation and differentially affect the activities of the resulting amyloid fibrils. J. Biol. Chem. 2018, 293, 12975–12991. [Google Scholar] [CrossRef] [Green Version]
- Saridaki, T.; Zampagni, M.; Mannini, B.; Evangelisti, E.; Taddei, N.; Cecchi, C.; Chiti, F. Glycosaminoglycans (GAGs) suppress the toxicity of HypF-N prefibrillar aggregates. J. Mol. Biol. 2012, 421, 616–630. [Google Scholar] [CrossRef]
- Chen, Z.; Cheng, W. Reversible aggregation of HIV-1 Gag proteins mediated by nucleic acids. Biochem. Biophys. Res. Commun. 2017, 482, 1437–1442. [Google Scholar] [CrossRef] [Green Version]
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef]
- Brangwynne, C.P.; Mitchison, T.J.; Hyman, A.A. Active liquid-like behavior of nucleoli determines their size and shape in Xenopus laevis oocytes. Proc. Natl. Acad. Sci. USA 2011, 108, 4334–4339. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.; Lee, H.O.; Jawerth, L.; Maharana, S.; Jahnel, M.; Hein, M.Y.; Stoynov, S.; Mahamid, J.; Saha, S.; Franzmann, T.M.; et al. A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 2015, 162, 1066–1077. [Google Scholar] [CrossRef] [Green Version]
- Saha, S.; Weber, C.A.; Nousch, M.; Adame-Arana, O.; Hoege, C.; Hein, M.Y.; Osborne-Nishimura, E.; Mahamid, J.; Jahnel, M.; Jawerth, L.; et al. Polar positioning of phase-separated liquid compartments in cells regulated by an mRNA competition mechanism. Cell 2016, 166, 1572–1584. [Google Scholar] [CrossRef] [Green Version]
- Mao, Y.S.; Zhang, B.; Spector, D.L. Biogenesis and function of nuclear bodies. Trends Genet. 2011, 27, 295–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H. Higher-order assemblies in a new paradigm of signal transduction. Cell 2013, 153, 287–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decker, C.J.; Parker, R. P-bodies and stress granules: Possible roles in the control of translation and mRNA degradation. Cold Spring Harb. Perspect. Biol. 2012, 4, a012286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberti, S.; Dormann, D. Liquid–liquid phase separation in disease. Annu. Rev. Genet. 2019, 53, 171–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stochaj, U.; Weber, S.C. Nucleolar Organization and Functions in Health and Disease. Cells 2020, 9, 526. [Google Scholar] [CrossRef] [Green Version]
- Dundr, M.; Hebert, M.D.; Karpova, T.S.; Stanek, D.; Xu, H.; Shpargel, K.B.; Meier, U.T.; Neugebauer, K.M.; Matera, A.G.; Misteli, T. In vivo kinetics of Cajal body components. J. Cell Biol. 2004, 164, 831–842. [Google Scholar] [CrossRef] [Green Version]
- Weidtkamp-peters, S.; Lenser, T.; Negorev, D.; Gerstner, N.; Hofmann, T.G.; Schwanitz, G.; Hoischen, C.; Maul, G.; Dittrich, P.; Hemmerich, P. Dynamics of component exchange at PML nuclear bodies. J. Cell Sci. 2008, 121, 2731–2743. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.R.W.; Kornberg, A. Inorganic polyphosphate in the origin and survival of species. Proc. Natl. Acad. Sci. USA 2004, 101, 16085–16087. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Jakob, U. Inorganic polyphosphate, a multifunctional polyanionic protein scaffold. J. Biol. Chem. 2019, 294, 2180–2190. [Google Scholar] [CrossRef] [Green Version]
- Cremers, C.M.; Knoefler, D.; Gates, S.; Martin, N.; Dahl, J.; Lempart, J.; Xie, L.; Chapman, M.R.; Galvan, V.; Southworth, D.R.; et al. Polyphosphate: A conserved modifier of amyloidogenic processes. Mol. Cell 2016, 63, 768–780. [Google Scholar] [CrossRef] [Green Version]
- Lempart, J.; Tse, E.; Lauer, J.A.; Ivanova, M.I.; Sutter, A.; Yoo, N.; Huettemann, P.; Southworth, D.; Jakob, U. Mechanistic insights into the protective roles of polyphosphate against amyloid cytotoxicity. Life Sci. Alliance 2019, 2, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leal, S.S.; Botelho, H.M.; Gomes, C.M. Metal ions as modulators of protein conformation and misfolding in neurodegeneration. Coord. Chem. Rev. 2012, 256, 2253–2270. [Google Scholar] [CrossRef]
- Cicero, C.E.; Mostile, G.; Vasta, R.; Rapisarda, V.; Signorelli, S.S.; Ferrante, M.; Zappia, M.; Nicoletti, A. Metals and neurodegenerative diseases. A systematic review. Environ. Res. 2017, 159, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yin, Y.L.; Liu, X.Z.; Shen, P.; Zheng, Y.G.; Lan, X.R.; Lu, C.B.; Wang, J.Z. Current understanding of metal ions in the pathogenesis of Alzheimer’s disease. Transl. Neurodegener. 2020, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Yu, L.; Sun, Y.; Dong, X. Kinetic insights into Zn2+-induced amyloid β-protein aggregation revealed by stopped-flow fluorescence spectroscopy. J. Phys. Chem. 2017, 121, 3909–3917. [Google Scholar] [CrossRef]
- Hu, J.; Zhang, D.; Liu, X.; Li, X.; Cheng, X.; Chen, J.; Du, H.; Liang, Y. Pathological concentration of zinc dramatically accelerates abnormal aggregation of full-length human Tau and thereby significantly increases Tau toxicity in neuronal cells. Biochim. Biophys. Acta 2017, 1863, 414–427. [Google Scholar] [CrossRef]
- Moroz, O.V.; Burkitt, W.; Wittkowski, H.; He, W.; Ianoul, A.; Novitskaya, V.; Xie, J.; Polyakova, O.; Lednev, I.K.; Shekhtman, A.; et al. Both Ca2+ and Zn2+ are essential for S100A12 protein oligomerization and function. BMC Biochem. 2009, 10, 11. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Aleshintsev, A.; Zhuang, J.; Brenowitz, M.; Gupta, R. Ca(II) and Zn(II) cooperate to modulate the structure and self-assembly of S100A12. Biochemistry 2019, 58, 2269–2281. [Google Scholar] [CrossRef]
- Liu, Z.; Song, F.; Ma, Z.L.; Xiong, Q.; Wang, J.; Guo, D.; Sun, G. Bivalent copper ions promote fibrillar aggregation of KCTD1 and induce cytotoxicity. Sci. Rep. 2016, 6, 32658. [Google Scholar] [CrossRef] [Green Version]
- Funk, K.E.; Thomas, S.N.; Schafer, K.N.; Cooper, G.L.; Clark, D.J.; Yang, A.J.; Kuret, J. Lysine methylation is an endogenous post-translational modification of tau protein in human brain and a modulator of aggregation propensity. Biochem. J. 2015, 462, 77–88. [Google Scholar] [CrossRef] [Green Version]
- Schedin-weiss, S.; Winblad, B.; Tjernberg, L.O. The role of protein glycosylation in Alzheimer disease. FEBS J. 2014, 281, 46–62. [Google Scholar] [CrossRef] [PubMed]
- Cohen, T.J.; Hwang, A.W.; Restrepo, C.R.; Yuan, C.; John, Q.; Lee, V.M.Y. An acetylation switch controls TDP-43 function and aggregation propensity. Nat. Commun. 2015, 6, 5845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdolvahabi, A.; Shi, Y.; Rhodes, N.R.; Cook, N.P.; Martı, A.A.; Shaw, B.F. Arresting amyloid with coulomb’s Law: Acetylation of ALS-Linked SOD1 by aspirin impedes aggregation. Biophys. J. 2015, 108, 1199–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gal, J.; Chen, J.; Barnett, K.R.; Yang, L.; Brumley, E.; Zhu, H. HDAC6 regulates mutant SOD1 aggregation through two SMIR motifs and tubulin acetylation. J. Biol. Chem. 2013, 288, 15035–15045. [Google Scholar] [CrossRef] [Green Version]
- DiMauro, M.A.; Nandi, S.K.; Raghavan, C.T.; Kar, R.K.; Wang, B.; Bhunia, A.; Nagaraj, R.H.; Biswas, A. Acetylation of Gly1 and Lys2 Promotes Aggregation of Human γD- Crystallin. Biochemistry 2014, 53, 7269–7282. [Google Scholar] [CrossRef]
- Johnson, G.V.W.; Stoothoff, W.H. Tau phosphorylation in neuronal cell function and dysfunction. J. Cell Sci. 2004, 117, 5721–5729. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Martin, T.; Cuchillo-ibáñez, I.; Noble, W.; Nyenya, F.; Anderton, B.H.; Hanger, D.P. Tau phosphorylation affects its axonal transport and degradation. Neurobiol. Aging. 2013, 34, 2146–2157. [Google Scholar] [CrossRef] [Green Version]
- Tenreiro, S.; Reimão-Pinto, M.M.; Antas, P.; Rino, J.; Wawrzycka, D.; Macedo, D.; Rosado-Ramos, R.; Amen, T.; Waiss, M.; Magalhães, F.; et al. Phosphorylation modulates clearance of alpha-synuclein inclusions in a yeast model of Parkinson’s disease. PLoS Genet. 2014, 10, e1004302. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Xie, J.; Xia, Y.; Yu, S.; Gu, Z.; Feng, R.; Luo, G.; Wang, D.; Wang, K.; Jiang, M.; et al. LK6/Mnk2a is a new kinase of alpha synuclein phosphorylation mediating neurodegeneration. Sci. Rep. 2015, 5, 12564. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Yeh, P.; Chiu, H.; Tang, C.; Tu, B.P. Hyperphosphorylation as a defense mechanism to reduce TDP-43 aggregation. PLoS ONE 2011, 6, e23075. [Google Scholar] [CrossRef] [Green Version]
- Nonaka, T.; Suzuki, G.; Tanaka, Y.; Kametani, F.; Hirai, S.; Okado, H.; Miyashita, T.; Saitoe, M.; Akiyama, H.; Masai, H.; et al. Phosphorylation of TAR DNA-binding protein of 43 kDa (TDP-43) by trucated casein kinase 1δ triggers mislocalization and accumulation of TDP-43. J. Biol. Chem. 2016, 291, 5473–5483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhoads, S.N.; Monahan, Z.T.; Yee, D.S.; Shewmaker, F.P. The role of post-translational modifications on prion-like aggregation and liquid-phase separation of FUS. Int. J. Mol. Sci. 2018, 19, 886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gould, N.; Doulias, P.T.; Tenopoulou, M.; Raju, K.; Ischiropoulos, H. Regulation of protein function and signaling by reversible cysteine s-nitrosylation. J. Biol. Chem. 2013, 288, 26473–26479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marinelli, P.; Navarro, S.; Graña-Montes, R.; Bañó-Polo, M.; Fernández, M.R.; Papaleo, E.; Ventura, S. A single cysteine post-translational oxidation suffices to compromise globular proteins kinetic stability and promote amyloid formation. Redox Biol. 2018, 14, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Wineman-Fisher, V.; Tudorachi, L.; Nissim, E.; Miller, Y. The removal of disulfide bonds in amylin oligomers leads to the conformational change of the “native” amylin oligomers. Phys. Chem. Chem. Phys. 2016, 18, 12438–12442. [Google Scholar] [CrossRef] [Green Version]
- Anoop, A.; Ranganathan, S.; Dhaked, B.D.; Jha, N.N.; Pratihar, S.; Ghosh, S.; Sahay, S.; Kumar, S.; Das, S.; Kombrabail, M.; et al. Elucidating the role of disulfide bond on amyloid formation and fibril reversibility of somatostatin-14: Relevance to its storage and secretion. J. Biol. Chem. 2014, 289, 16884–16903. [Google Scholar] [CrossRef] [Green Version]
- Chattopadhyay, M.; Nwadibia, E.; Strong, C.D.; Gralla, E.B.; Valentine, J.S.; Whitelegge, J.P. The disulfide bond, but not zinc or dimerization, controls initiation and seeded growth in amyotrophic lateral sclerosis-linked Cu, Zn superoxide dismutase (SOD1) fibrillation. J. Biol. Chem. 2015, 290, 30624–30636. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Yan, J.; Zhang, X.; Huang, K. Disulfide bonds in amyloidogenesis diseases related proteins. Proteins 2013, 81, 1862–1873. [Google Scholar] [CrossRef]
- Nakajima, H.; Amano, W.; Fujita, A.; Fukuhara, A.; Azuma, Y.-T.; Hata, F.; Inui, T.; Takeuchi, T. The active site cysteine of the proapoptotic protein glyceraldehyde-3-phosphate dehydrogenase is essential in oxidative stress-induced aggregation and cell death. J. Biol. Chem. 2007, 282, 26562–26574. [Google Scholar] [CrossRef]
- Göbl, C.; Morris, V.K.; van Dam, L.; Visscher, M.; Polderman, P.E.; Hartlmüller, C.; de Ruiter, H.; Hora, M.; Liesinger, L.; Birner-Gruenberger, R.; et al. Cysteine oxidation triggers amyloid fibril formation of the tumor suppressor p16INK4A. bioRxiv 2020, 28, 101316. [Google Scholar] [CrossRef]
- Carlomagno, Y.; Chung, D.C.; Yue, M.; Castanedes-casey, M.; Madden, B.J.; Dunmore, J.; Tong, J.; DeTure, M.; Dickson, D.W.; Petrucelli, L.; et al. An acetylation–phosphorylation switch that regulates tau aggregation propensity and function. J. Biol. Chem. 2017, 292, 15277–15286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiki, A.; Deguire, S.M.; Ruggeri, F.S.; Sanfelice, D.; Ansaloni, A.; Wang, Z.; Cendrowska, U.; Burai, R.; Vieweg, S.; Pastore, A.; et al. Mutant exon1 huntingtin aggregation is regulated by T3 phosphorylation-induced structural changes and crosstalk between T3 phosphorylation and acetylation at K6. Angew. Chem. Int. Ed. Engl. 2017, 56, 5202–5207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaffert, L.N.; Carter, W.G. Do post-translational modifications influence protein aggregation in neurodegenerative diseases: A systematic review. Brain Sci. 2020, 10, 232. [Google Scholar] [CrossRef]
- Bechtel, T.J.; Weerapana, E. From structure to redox: The diverse functional roles of disulfides and implications in disease. Proteomics 2017, 17, 1–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Z.; Yan, L.-J. Protein oxidative modifications: Beneficial roles in disease and health. J. Biochem. Pharmacol. Res. 2013, 1, 15–26. [Google Scholar]
- Poole, L.B. The basics of thiols and cysteines in redox biology and chemistry. Free Radic. Biol. Med. 2015, 80, 148–157. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Liang, J.; Li, C.; He, Z.; Yuan, H.; Huang, B.; Liu, X.; Tang, B.; Pang, D.; Du, H.; et al. Pathological hydrogen peroxide triggers the fibrillization of wild-type SOD1 via sulfenic acid modification of Cys-111. Cell Death Dis. 2018, 9, 67. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.J.; Kulkarni, S.; Pawson, T. FF domains of CA150 bind transcription and splicing factors through multiple weak interactions. Mol. Cell. Biol. 2014, 24, 9274–9285. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Sordella, R.; Chen, G.; Hakre, S.; Roy, A.L.; Settleman, J. An FF domain-dependent protein interaction mediates a signaling pathway for growth factor-induced gene expression. Mol. Cell 2005, 17, 23–35. [Google Scholar] [CrossRef]
- Chiu, J.; Hogg, P.J. Allosteric disulfides: Sophisticated molecular structures enabling flexible protein regulation. J. Biol. Chem. 2019, 294, 2949–2960. [Google Scholar] [CrossRef] [Green Version]
- Trivedi, M.; Laurence, J.; Siahaan, T. The role of thiols and disulfides on protein stability. Curr. Protein Pept. Sci. 2009, 10, 614–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridgway, Z.; Zhang, X.; Wong, A.G.; Abedini, A.; Schmidt, A.M.; Raleigh, D.P. Analysis of the role of the conserved disulfide in amyloid formation by human islet amyloid polypeptide in homogeneous and heterogeneous environments. Biochemistry 2018, 57, 3065–3074. [Google Scholar] [CrossRef] [PubMed]
- Honda, R. Role of the disulfide bond in prion protein amyloid formation: A thermodynamic and kinetic analysis. Biophys. J. 2017, 114, 885–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laghaei, R.; Mousseau, N. Effect of the disulfide bond on the monomeric structure of human amylin studied by simulations. J. Phys. Chem. B 2010, 114, 7071–7077. [Google Scholar] [CrossRef] [PubMed]
- Yonemoto, L.T.; Kroon, G.J.A.; Dyson, H.J.; Balch, W.E.; Kelly, J.W. Amylin proprotein processing generates progressively more amyloidogenic peptides that initially sample the helical state. Biochemistry 2009, 47, 9900–9910. [Google Scholar] [CrossRef] [Green Version]
- Vaiana, S.M.; Best, R.B.; Yau, W.; Eaton, W.A.; Hofrichter, J. Evidence for a partially structured state of the amylin monomer. Biophys. J. 2009, 97, 2948–2957. [Google Scholar] [CrossRef] [Green Version]
- Ho, T.; Watt, B.; Spruce, L.A.; Seeholzer, S.H.; Marks, M.S. The Kringle-like domain facilitates post-endoplasmic reticulum changes to premelanosome protein (PMEL) oligomerization and disulfide bond configuration and promotes amyloid formation. J. Biol. Chem. 2016, 291, 3595–3612. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Freed, C.R. Tyrosine-to-cysteine modification of human α-synuclein enhances protein aggregation and cellular toxicity. J. Biol. Chem. 2004, 279, 10128–10135. [Google Scholar] [CrossRef] [Green Version]
- Zha, X.; Wang, R.; Collier, D.M.; Snyder, P.M.; Wemmie, J.A.; Welsh, M.J. Oxidant regulated inter-subunit disulfide bond formation between ASIC1a subunits. Proc. Natl. Acad. Sci. USA 2009, 106, 3573–3578. [Google Scholar] [CrossRef] [Green Version]
- Banci, L.; Bertini, I.; Durazo, A.; Girotto, S.; Gralla, E.B.; Martinelli, M.; Valentine, J.S.; Vieru, M.; Whitelegge, J.P. Metal-free superoxide dismutase forms soluble oligomers under physiological conditions: A possible general mechanism for familial ALS. Proc. Natl. Acad. Sci. USA 2007, 104, 11263–11267. [Google Scholar] [CrossRef] [Green Version]
- Buffone, M.G.; Foster, J.A.; Gerton, G.L. The role of the acrosomal matrix in fertilization. Int. J. Dev. Biol. 2008, 52, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Gautam, S.; Karmakar, S.; Batra, R.; Sharma, P.; Pradhan, P.; Singh, J.; Kundu, B.; Chowhury, P.K. Polyphenols in combination with β-cyclodextrin can inhibit and disaggregate α-synuclein amyloids under cell mimicking conditions: A promising therapeutic alternative. Biochim. Biophys. Acta 2017, 1865, 589–603. [Google Scholar] [CrossRef] [PubMed]
- Velander, P.; Wu, L.; Henderson, F.; Zhang, S.; Bevan, D.R.; Xu, B. Natural product-based amyloid inhibitors. Biochem Pharmacol. 2017, 139, 40–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirangelo, I.; Borriello, M.; Vilasi, S.; Iannuzzi, C. Hydroxytyrosol inhibits protein oligomerization and amyloid aggregation in human insulin. Int. J. Mol. Sci. 2020, 21, 4636. [Google Scholar] [CrossRef]
- Sheng, J.; Olrichs, N.K.; Geerts, W.J.; Kaloyanova, D.V.; Helms, J.B. Metal ions and redox balance regulate distinct amyloid-like aggregation pathways of GAPR-1. Sci. Rep. 2019, 9, 15048. [Google Scholar] [CrossRef] [Green Version]
- Sheng, J.; Olrichs, N.K.; Geerts, W.J.; Li, X.; Rehman, A.U.; Gadella, B.M.; Kaloyanova, D.V.; Helms, J.B. Zinc binding regulates amyloid-like aggregation of GAPR-1. Biosci. Rep. 2019, 39, BSR20182345. [Google Scholar] [CrossRef] [Green Version]
- Olrichs, N.K.; Mahalka, A.K.; Kaloyanova, D.; Kinnunen, P.K.; Helms, B.J. Golgi-Associated plant Pathogenesis Related protein 1 (GAPR-1) forms amyloid-like fibrils by interaction with acidic phospholipids and inhibits Aβ aggregation. Amyloid 2014, 21, 88–96. [Google Scholar] [CrossRef]
- Shoji-Kawata, S.; Sumpter, R.; Leveno, M.; Campbell, G.R.; Zou, Z.; Kinch, L.; Wilkins, A.D.; Sun, Q.; Pallauf, K.; MacDuff, D.; et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature 2013, 494, 201–206. [Google Scholar] [CrossRef] [Green Version]
- Eberle, H.B.; Serrano, R.L.; Füllekrug, J.; Schlosser, A.; Lehmann, W.D.; Lottspeich, F.; Kaloyanova, D.; Wieland, F.T.; Helms, J.B. Identification and characterization of a novel human plant pathogenesis-related protein that localizes to lipid-enriched microdomains in the Golgi complex. J. Cell Sci. 2002, 115, 827–838. [Google Scholar]
- Nah, J.; Pyo, J.-O.; Jung, S.; Yoo, S.-M.; Kam, T.-I.; Chang, J.; Han, J.; A.An, S.S.; Onodera, T.; Jung, Y.-K. BECN1/Beclin 1 is recruited into lipid rafts by prion to activate autophagy in response to amyloid β42. Autophagy 2013, 9, 2009–2021. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhao, Y.; Su, M.; Glover, K.; Chakravarthy, S.; Colbert, C.L.; Levine, B.; Sinha, S.C. Structural insights into the interaction of the conserved mammalian proteins GAPR-1 and Beclin 1, a key autophagy protein. Acta Crystallogr. D Struct. Biol. 2017, 73, 775–792. [Google Scholar] [CrossRef] [PubMed]
- Adi-Harel, S.; Erlich, S.; Schmukler, E.; Cohen-kedar, S.; Segev, O.; Mizrachy, L.; Hirsch, J.A.; Pinkas-kramarski, R. Beclin 1 self-association is independent of autophagy induction by amino acid deprivation and rapamycin treatment. J. Cell. Biochem. 2010, 1271, 1262–1271. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Choi, W.; Hu, W.; Mi, N.; Guo, Q.; Ma, M.; Liu, M. Crystal structure and biochemical analyses reveal Beclin 1 as a novel membrane binding protein. Cell Res. 2012, 22, 473–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnett, L.A.; Xiang, X.; Bieber, A.L.; Chandler, D.E. Crisp proteins and sperm chemotaxis: Discovery in amphibians and explorations in mammals. Int. J. Dev. Biol. 2008, 52, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, H.; Burnett, L.; Xiang, X.; Olson, J.; Willis, S.; Miao, A.M.Y.; Akema, T.; Bieber, A.L.; Chandler, D.E. Purification and multimer formation of allurin, a sperm chemoattractant from Xenopus laevis egg jelly. Mol. Reprod. Dev. 2009, 76, 527–536. [Google Scholar] [CrossRef]
- Maldera, J.A.; Vasen, G.; Ernesto, J.I.; Weigel-Muñoz, M.; Cohen, D.J.; Cuasnicu, P.S. Evidence for the involvement of zinc in the association of CRISP1 with rat sperm during epididymal maturation. Biol. Reprod. 2011, 85, 503–510. [Google Scholar] [CrossRef]
- Wang, Y.-L.; Kuo, J.-H.; Lee, S.-C.; Liu, J.-S.; Hsieh, Y.-C.; Shih, Y.-T.; Chen, C.-J.; Chiu, J.-J.; Wu, W.-G. Cobra CRISP functions as an inflammatory modulator via a novel Zn2+- and heparan sulfate-dependent transcriptional regulation of endothelial cell adhesion molecules. J. Biol. Chem. 2010, 285, 37872–37883. [Google Scholar] [CrossRef] [Green Version]
- Olrichs, N.K.; Helms, J.B. Novel insights into the function of the conserved domain of the CAP superfamily of proteins. AIMS Biophys. 2016, 3, 232–246. [Google Scholar] [CrossRef]
- Lu, S.; Faris, J.D.; Sherwood, R.; Edwards, M.C. Dimerization and protease resistance: New insight into the function of PR-1. J. Plant Physiol. 2013, 170, 105–110. [Google Scholar] [CrossRef]
- Lu, S.; Faris, J.D.; Sherwood, R.; Friesen, T.L.; Edwards, M.C. A dimeric PR-1-type pathogenesis-related protein interacts with ToxA and potentially mediates ToxA-induced necrosis in sensitive wheat. Mol. Plant Pathol. 2014, 15, 650–663. [Google Scholar] [CrossRef]
- Prados-Rosales, R.C.; Roldán-Rodríguez, R.; Serena, C.; López-Berges, M.S.; Guarro, J.; Martínez-del-Pozo, Á.; Di Pietro, A. A PR-1-like protein of Fusarium oxysporum functions in virulence on mammalian hosts. J. Biol. Chem. 2012, 287, 21970–21979. [Google Scholar] [CrossRef] [Green Version]
- Asojo, O.A.; Goud, G.; Dhar, K.; Loukas, A.; Zhan, B.; Deumic, V.; Liu, S.; Borgstahl, G.E.O.; Hotez, P.J. X-ray structure of Na-ASP-2, a pathogenesis-related-1 protein from the nematode parasite, Necator americanus, and a vaccine antigen for human hookworm infection. J. Mol. Biol. 2005, 346, 801–814. [Google Scholar] [CrossRef] [PubMed]
- Borloo, J.; Geldhof, P.; Peelaers, I.; Van Meulder, F.; Ameloot, P.; Callewaert, N.; Vercruysse, J.; Claerebout, E.; Strelkov, S.V.; Weeks, S.D.; et al. Structure of Ostertagia ostertagi ASP-1: Insights into disulfide-mediated cyclization and dimerization. Acta Crystallogr. Sect. D Biol. Crystallogr. 2013, 69, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Asojo, O.A. Structure of a two-CAP-domain protein from the human hookworm parasite Necator americanus. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 455–462. [Google Scholar] [CrossRef] [Green Version]
- Ma, D.; Francischetti, I.M.B.; Ribeiro, J.M.C.; Andersen, J.F. The structure of hookworm platelet inhibitor (HPI), a CAP superfamily member from Ancylostoma caninum. Acta Crystallogr. F Struct. Biol. Commun. 2015, 71, 643–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brangulis, K.; Jaudzems, K.; Petrovskis, I.; Akopjana, I.; Kazaks, A.; Tars, K. Structural and functional analysis of BB0689 from Borrelia burgdorferi, a member of the bacterial CAP superfamily. J. Struct. Biol. 2015, 192, 320–330. [Google Scholar] [CrossRef]
- Wang, F.; Li, H.; Liu, M.-n.; Song, H.; Han, H.-m.; Wang, Q.-l.; Yin, C.-c.; Zhou, Y.-c.; Qi, Z.; Shu, Y.-y.; et al. Structural and functional analysis of natrin, a venom protein that targets various ion channels. Biochem. Biophys. Res. Commun. 2006, 351, 443–448. [Google Scholar] [CrossRef]
- Baroni, R.M.; Luo, Z.; Darwiche, R.; Hudspeth, E.M.; Schneiter, R.; Pereira, G.A.G.; Mondego, J.M.C.; Asojo, O.A. Crystal Structure of MpPR-1i, a SCP/TAPS protein from Moniliophthora perniciosa, the fungus that causes Witches’ Broom Disease of Cacao. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Serrano, R.L.; Kuhn, A.; Hendricks, A.; Helms, J.B.; Sinning, I.; Groves, M.R. Structural analysis of the human Golgi-associated plant pathogenesis related protein GAPR-1 implicates dimerization as a regulatory mechanism. J. Mol. Biol. 2004, 339, 173–183. [Google Scholar] [CrossRef]
- Groves, M.R.; Kuhn, A.; Hendricks, A.; Radke, S.; Serrano, R.L.; Helms, J.B.; Sinning, I. Crystallization of a Golgi-associated PR-1-related protein (GAPR-1) that localizes to lipid-enriched microdomains. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 730–732. [Google Scholar] [CrossRef]
- Gibbs, G.M.; Roelants, K.; O’Bryan, M.K. The CAP superfamily: Cysteine-rich secretory proteins, antigen 5, and pathogenesis-related 1 proteins-roles in reproduction, cancer, and immune defense. Endocr. Rev. 2008, 29, 865–897. [Google Scholar] [CrossRef] [PubMed]
- Darwiche, R.; Kelleher, A.; Hudspeth, E.M.; Schneiter, R.; Asojo, O.A. Structural and functional characterization of the CAP domain of pathogen-related yeast 1 (Pry1) protein. Sci. Rep. 2016, 6, 28838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Galen, J.; Olrichs, N.K.; Schouten, A.; Serrano, R.L.; Nolte-’t Hoen, E.N.M.; Eerland, R.; Kaloyanova, D.; Gros, P.; Helms, J.B. Interaction of GAPR-1 with lipid bilayers is regulated by alternative homodimerization. Biochim. Biophys. Acta 2012, 1818, 2175–2183. [Google Scholar] [CrossRef] [Green Version]
- Fernández, C.; Szyperski, T.; Bruyère, T.; Ramage, P.; Mösinger, E.; Wüthrich, K. NMR solution structure of the pathogenesis-related protein P14a. J. Mol. Biol. 1997, 266, 576–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osredkar, J.; Sustar, N. Copper and zinc, biological role and significance of copper/zinc imbalance. J. Clin. Toxicol. 2011, 3, 1–19. [Google Scholar] [CrossRef] [Green Version]
- De Leon-Rodriguez, L.; Lubag, A.J.M.J.; Sherry, A.D. Imaging free zinc levels in vivo–what can be learned? Inorg. Chim. Acta 2012, 393, 12–23. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, J.H.; Maryon, E.B. How mammalian cells acquire copper: An essential but potentially toxic metal. Biophys. J. 2016, 110, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Torkian, S.; Khanjani, N.; Mahmoodi, M.R.; Khosravi, V. A review of copper concentrations in Iranian populations. Environ. Monit. Assess. 2019, 191, 537. [Google Scholar] [CrossRef]
- Takeda, A. Invovlement of zinc in neuronal death in the hippocapus. Biomed Res. Trace Elem. 2007, 18, 204–210. [Google Scholar]
- de Larminat, M.A.; Cuasnicú, P.S.; Blaquier, J.A. Changes in trophic and functional parameters of the rat epididymis during sexual maturation. Biol. Reprod. 1981, 25, 813–819. [Google Scholar] [CrossRef]
- Saito, S.; Zeitz, L.; Bush, I.M.; Lee, R.; Jr, W.F.W. Zinc uptake in canine or rat spermatozoa. Am. J. Physiol. 1969, 217, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Mawson, C.A.; Fischer, M.I. Zinc and carbonic anhydrase in human semen. Biochem. J. 1953, 55, 696–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelleher, S.L.; McCormick, N.H.; Velasquez, V.; Lopez, V. Zinc in specialized secretory tissues: Roles in the pancreas, prostate, and mammary gland. Adv. Nutr. 2011, 2, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Fallah, A.; Mohammad-Hasani, A.; Colagar, A.H. Zinc is an essential element for male fertility: A review of Zn roles in men’s health, germination, sperm quality, and fertilization. J. Reprod. Infertil. 2018, 19, 69–81. [Google Scholar]
- Yamaguchi, S.; Miura, C.; Kikuchi, K.; Celino, F.T.; Agusa, T.; Tanabe, S.; Miura, T. Zinc is an essential trace element for spermatogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 10859–10864. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Dong, X.; Hu, X.; Long, Z.; Wang, L.; Liu, Q.; Sun, B.; Wang, Q.; Wu, Q.; Li, L. Zinc levels in seminal plasma and their correlation with male infertility: A systematic review and meta-analysis. Sci. Rep. 2016, 6, 22386. [Google Scholar] [CrossRef]
- Zhang, N.; Duncan, F.E.; Que, E.L.; O’Halloran, T.V.; Woodruff, T.K. The fertilization-induced zinc spark is a novel biomarker of mouse embryo quality and early development. Sci. Rep. 2016, 6, 22772. [Google Scholar] [CrossRef] [Green Version]
- Da Ros, V.G.; Muñoz, M.W.; Battistone, M.A.; Brukman, N.; Carvajal, G.; Curci, L.; Gomez-Elias, M.D.; Cohen, D.J.; Cuasnicú, P.S. From the epididymis to the egg: Participation of CRISP proteins in mammalian fertilization. Asian J. Androl. 2015, 17, 711–715. [Google Scholar]
- Cohen, D.J.; Maldera, J.A.; Vasen, G.; Ernesto, J.I.; Munoz, M.W.; Battistone, M.A.; Cuasnicu, P.S. Epididymal protein CRISP1 plays different roles during the fertilization process. J. Androl. 2011, 32, 672–678. [Google Scholar] [CrossRef]
- Cohen, D.J.; Rochwerger, L.; Ellerman, D.A.; Morgenfe, M.M.; Busso, D.; Cuasnicú, P.S. Relationship between the association of rat epididymal protein “DE” with spermatozoa and the behavior and function of the protein. Mol. Reprod. Dev. 2000, 56, 180–188. [Google Scholar]
- Roberts, K.P.; Wamstad, J.A.; Ensrud, K.M.; Hamilton, D.W. Inhibition of capacitation-associated tyrosine phosphorylation signaling in rat sperm by epididymal protein Crisp-1. Biol. Reprod. 2003, 69, 572–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busso, D.; Cohen, D.J.; Maldera, J.A.; Dematteis, A.; Cuasnicu, P.S. A novel function for CRISP1 in rodent fertilization: Involvement in sperm-zona pellucida interaction. Biol. Reprod. 2007, 77, 848–854. [Google Scholar] [CrossRef] [PubMed]
- Bedford, J.M.; Moore, H.D.M.; Franklin, L.E. Significance of the equatorial segment of the acrosome of the spermatozoon in eutherian mammals. Exp. Cell Res. 1979, 119, 119–126. [Google Scholar] [CrossRef]
- Ernesto, J.I.; Muñoz, M.W.; Battistone, M.A.; Vasen, G.; Martínez-López, P.; Orta, G.; Figueiras-Fierro, D.; De la Vega-Beltran, J.L.; Moreno, I.A.; Guidobaldi, H.A.; et al. CRISP1 as a novel CatSper regulator that modulates sperm motility and orientation during fertilization. J. Cell Biol. 2015, 210, 1213–1224. [Google Scholar] [CrossRef] [PubMed]
- Aalberts, M.; van Dissel-Emiliani, F.M.F.; van Adrichem, N.P.H.; van Wijnen, M.; Wauben, M.H.M.; Stout, T.A.E.; Stoorvogel, W. Identification of distinct populations of prostasomes that differentially express prostate stem cell Antigen, Annexin A1, and GLIPR2 in humans. Biol. Reprod. 2012, 86, 82. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, O.; Berg, A.L.; Hanrieder, J.; Arnerup, G.; Lindström, A.-K.; Brittebo, E.B. Intracellular fibril formation, calcification, and enrichment of chaperones, cytoskeletal, and intermediate filament proteins in the adult hippocampus CA1 following neonatal exposure to the nonprotein amino acid BMAA. Arch. Toxicol. 2015, 89, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Baxter, R.M.; Crowell, T.P.; George, J.A.; Getman, M.E.; Gardner, H. The plant pathogenesis related protein GLIPR-2 is highly expressed in fibrotic kidney and promotes epithelial to mesenchymal transition in vitro. Matrix Biol. 2007, 26, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Breen, S.; Williams, S.J.; Winterberg, B.; Kobe, B.; Solomon, P.S. Wheat PR-1 proteins are targeted by necrotrophic pathogen effector proteins. Plant J. 2016, 1, 13–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breen, S.; Williams, S.J.; Outram, M.; Kobe, B.; Solomon, P.S. Emerging insights into the functions of pathogenesis-related protein 1. Trends Plant Sci. 2017, 22, 871–879. [Google Scholar] [CrossRef]
- Ribeiro, F.R.; Paulo, P.; Costa, V.L.; Barros-Silva, J.D.; Ramalho-Carvalho, J.; Jerónimo, C.; Henrique, R.; Lind, G.E.; Skotheim, R.I.; Lothe, R.A.; et al. Cysteine-Rich Secretory Protein-3 ( CRISP3 ) is strongly up-regulated in prostate carcinomas with the TMPRSS2-ERG fusion gene. PLoS ONE 2011, 6, e22317. [Google Scholar] [CrossRef]
- Li, L.; Fattah, E.A.; Cao, G.; Ren, C.; Yang, G.; Goltsov, A.A.; Chinault, A.C.; Cai, W.-W.; Timme, T.L.; Thompson, T.C. Glioma pathogenesis-related protein 1 exerts tumor suppressor activities through proapoptotic reactive oxygen species-c-Jun-NH2 kinase signaling. Cancer Res. 2008, 68, 434–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sheng, J.; Olrichs, N.K.; Gadella, B.M.; Kaloyanova, D.V.; Helms, J.B. Regulation of Functional Protein Aggregation by Multiple Factors: Implications for the Amyloidogenic Behavior of the CAP Superfamily Proteins. Int. J. Mol. Sci. 2020, 21, 6530. https://doi.org/10.3390/ijms21186530
Sheng J, Olrichs NK, Gadella BM, Kaloyanova DV, Helms JB. Regulation of Functional Protein Aggregation by Multiple Factors: Implications for the Amyloidogenic Behavior of the CAP Superfamily Proteins. International Journal of Molecular Sciences. 2020; 21(18):6530. https://doi.org/10.3390/ijms21186530
Chicago/Turabian StyleSheng, Jie, Nick K. Olrichs, Bart M. Gadella, Dora V. Kaloyanova, and J. Bernd Helms. 2020. "Regulation of Functional Protein Aggregation by Multiple Factors: Implications for the Amyloidogenic Behavior of the CAP Superfamily Proteins" International Journal of Molecular Sciences 21, no. 18: 6530. https://doi.org/10.3390/ijms21186530
APA StyleSheng, J., Olrichs, N. K., Gadella, B. M., Kaloyanova, D. V., & Helms, J. B. (2020). Regulation of Functional Protein Aggregation by Multiple Factors: Implications for the Amyloidogenic Behavior of the CAP Superfamily Proteins. International Journal of Molecular Sciences, 21(18), 6530. https://doi.org/10.3390/ijms21186530