Genetic and Methylome Variation in Turkish Brachypodium Distachyon Accessions Differentiate Two Geographically Distinct Subpopulations

 , , , ,
, , , ,  ,
,  , , , ,
, , , ,  and
and 
Abstract
:1. Introduction
2. Results
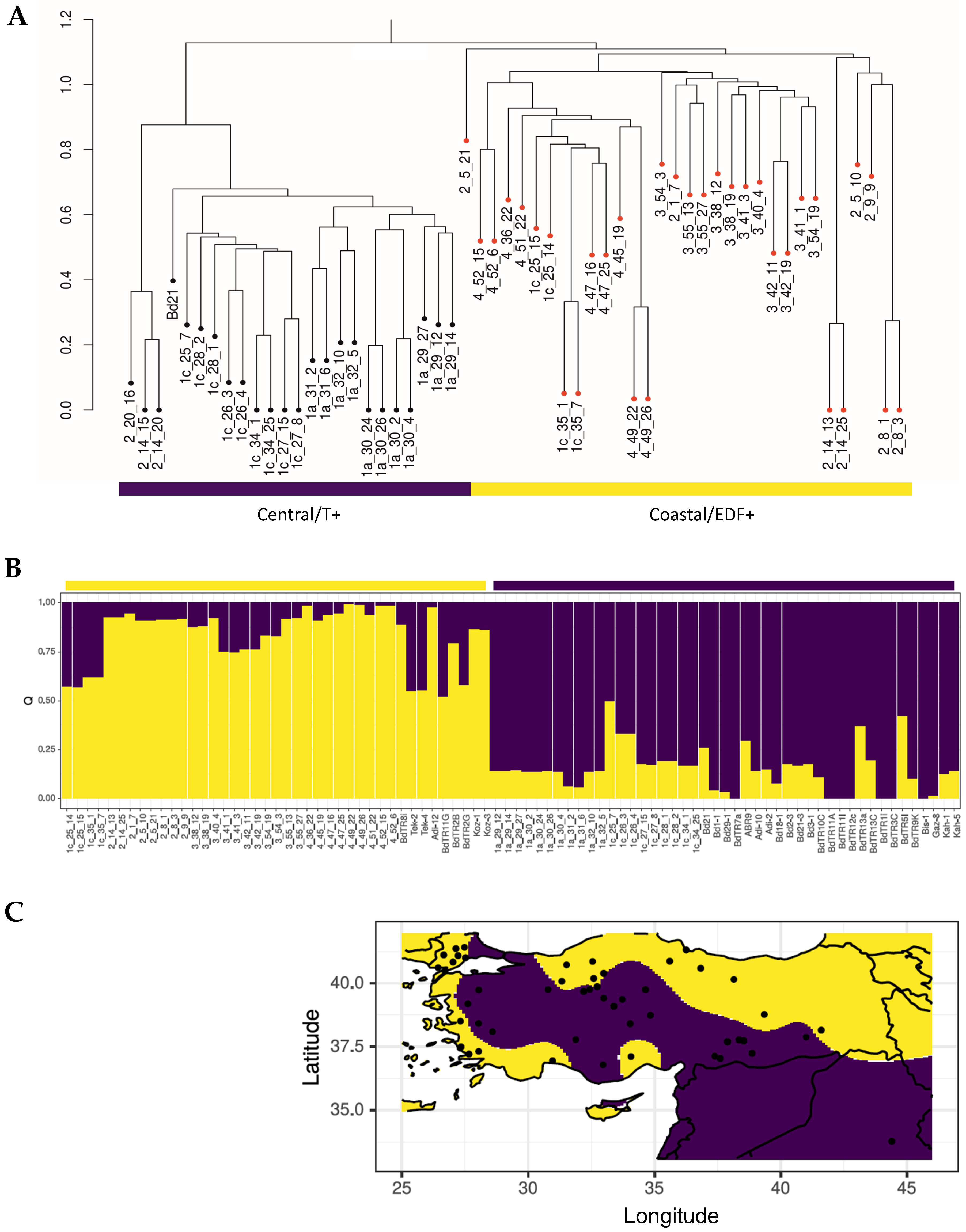
2.1. Genomic Diversity Reveals Two Subpopulations in Turkey
2.2. Whole Genome Methylation Assessments Also Indicate Two Subpopulations in the Turkish Population of Brachypodium
2.3. Phenomic Assessment of the Turkish Brachypodium Collection
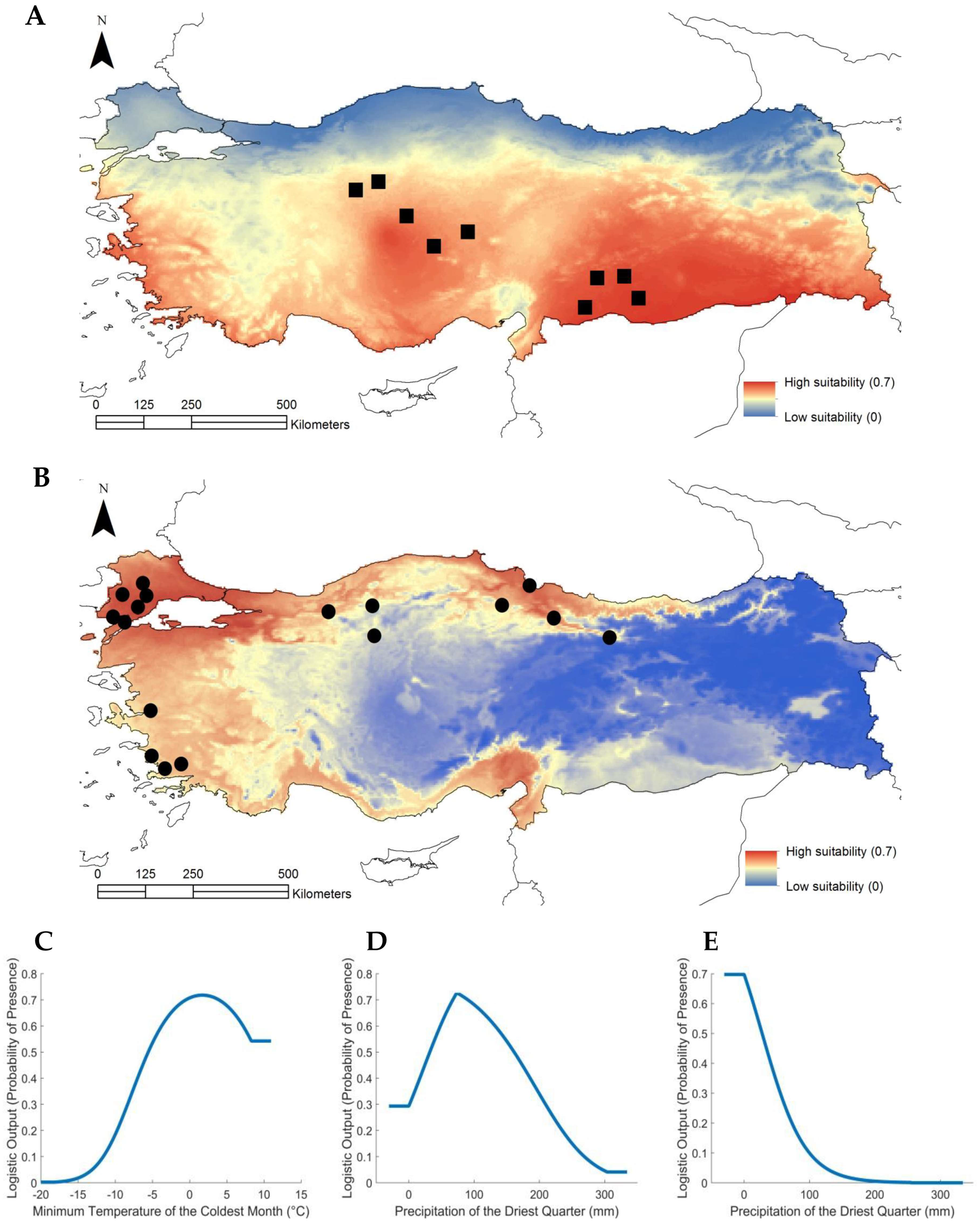
2.4. Relating Climatic Niches to the Two Subpopulations in the Turkish Population of Brachypodium
3. Discussion
4. Materials and Methods
4.1. Derivation of Turkish Lines
4.2. Whole Genome Sequencing and Single Nucleotide Polymorphism Calling
4.3. Bisulfite Sequencing and Data Analysis
4.4. Phenomic Experiments
4.5. Climate Modelling
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- IBI (The International Brachypodium Initiative). Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature 2010, 463, 763–768. [Google Scholar] [CrossRef]
- Mur, L.A.; Allainguillaume, J.; Catalan, P.; Hasterok, R.; Jenkins, G.; Lesniewska, K.; Thomas, I.; Vogel, J. Exploiting the Brachypodium tool box in cereal and grass research. New Phytol. 2011, 191, 334–347. [Google Scholar] [CrossRef] [PubMed]
- Scholthof, K.B.G.; Irigoyen, S.; Catalan, P.; Mandadi, K.K. Brachypodium: A monocot grass model genus for plant biology. Plant. Cell 2018, 30, 1673–1694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rancour, D.M.; Marita, J.M.; Hatfield, R.D. Cell wall composition throughout development for the model grass Brachypodium distachyon. Front. Plant. Sci. 2012, 3, 266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woods, D.P.; Ream, T.S.; Bouche, F.; Lee, J.; Thrower, N.; Wilkerson, C.; Amasino, R.M. Establishment of a vernalization requirement in Brachypodium distachyon requires repressor of vernalization1. Proc. Natl. Acad. Sci. USA 2017, 114, 6623–6628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, L.H.; Han, J.; Corke, F.M.; Akinyemi, A.; Didion, T.; Nielsen, K.K.; Doonan, J.H.; Mur, L.A.; Bosch, M. Linking dynamic phenotyping with metabolite analysis to study natural variation in drought responses of Brachypodium distachyon. Front. Plant. Sci. 2016, 7, 1751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Priest, H.D.; Fox, S.E.; Rowley, E.R.; Murray, J.R.; Michael, T.P.; Mockler, T.C. Analysis of global gene expression in Brachypodium distachyon reveals extensive network plasticity in response to abiotic stress. PLoS ONE 2014, 9, e87499. [Google Scholar] [CrossRef]
- Dell’Acqua, M.; Zuccolo, A.; Tuna, M.; Gianfranceschi, L.; Pe, M.E. Targeting environmental adaptation in the monocot model Brachypodium distachyon: A multi-faceted approach. BMC Genom. 2014, 15, 801. [Google Scholar] [CrossRef] [Green Version]
- Wilson, P.; Streich, J.; Borevitz, J. Genomic diversity and climate adaptation in Brachypodium. bioRxiv 2015. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.P.; Contreras-Moreira, B.; Woods, D.P.; Des Marais, D.L.; Burgess, D.; Shu, S.; Stritt, C.; Roulin, A.C.; Schackwitz, W.; Tyler, L.; et al. Extensive gene content variation in the Brachypodium distachyon pan-genome correlates with population structure. Nat. Commun. 2017, 8, 2184. [Google Scholar] [CrossRef]
- Finnegan, E.J.; Genger, R.K.; Peacock, W.J.; Dennis, E.S. DNA methylation in plants. Annu. Rev. Plant. Physiol. Plant. Mol. Biol. 1998, 49, 223–247. [Google Scholar] [CrossRef] [PubMed]
- Mitchell-Olds, T.; Willis, J.H.; Goldstein, D.B. Which evolutionary processes influence natural genetic variation for phenotypic traits? Nat. Rev. Genet. 2007, 8, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Foust, C.M.; Preite, V.; Schrey, A.W.; Alvarez, M.; Robertson, M.H.; Verhoeven, K.J.; Richards, C.L. Genetic and epigenetic differences associated with environmental gradients in replicate populations of two salt marsh perennials. Mol. Ecol. 2016, 25, 1639–1652. [Google Scholar] [CrossRef] [PubMed]
- Herrera, C.M.; Bazaga, P. Genetic and epigenetic divergence between disturbed and undisturbed subpopulations of a Mediterranean shrub: A 20-year field experiment. Ecol. Evol. 2016, 6, 3832–3847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrera, C.M.; Medrano, M.; Bazaga, P. Comparative spatial genetics and epigenetics of plant populations: Heuristic value and a proof of concept. Mol. Ecol. 2016, 25, 1653–1664. [Google Scholar] [CrossRef] [PubMed]
- Stassen, J.H.M.; Lopez, A.; Jain, R.; Pascual-Pardo, D.; Luna, E.; Smith, L.M.; Ton, J. The relationship between transgenerational acquired resistance and global DNA methylation in Arabidopsis. Sci. Rep. 2018, 8, 14761. [Google Scholar] [CrossRef] [Green Version]
- Dubin, M.J.; Zhang, P.; Meng, D.; Remigereau, M.S.; Osborne, E.J.; Paolo Casale, F.; Drewe, P.; Kahles, A.; Jean, G.; Vilhjalmsson, B.; et al. DNA methylation in Arabidopsis has a genetic basis and shows evidence of local adaptation. Elife 2015, 4, e05255. [Google Scholar] [CrossRef]
- Kawakatsu, T.; Huang, S.C.; Jupe, F.; Sasaki, E.; Schmitz, R.J.; Urich, M.A.; Castanon, R.; Nery, J.R.; Barragan, C.; He, Y.; et al. Epigenomic diversity in a global collection of Arabidopsis thaliana accessions. Cell 2016, 166, 492–505. [Google Scholar] [CrossRef] [Green Version]
- Schmid, M.W.; Heichinger, C.; Coman Schmid, D.; Guthorl, D.; Gagliardini, V.; Bruggmann, R.; Aluri, S.; Aquino, C.; Schmid, B.; Turnbull, L.A.; et al. Contribution of epigenetic variation to adaptation in Arabidopsis. Nat. Commun. 2018, 9, 4446. [Google Scholar] [CrossRef] [Green Version]
- Aller, E.S.T.; Jagd, L.M.; Kliebenstein, D.J.; Burow, M. Comparison of the relative potential for epigenetic and genetic variation to contribute to trait stability. G3 2018, 8, 1733–1746. [Google Scholar] [CrossRef] [Green Version]
- Eichten, S.R.; Stuart, T.; Srivastava, A.; Lister, R.; Borevitz, J.O. DNA methylation profiles of diverse Brachypodium distachyon align with underlying genetic diversity. Genome Res. 2016, 26, 1520–1531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, P.B.; Streich, J.C.; Murray, K.D.; Eichten, S.R.; Cheng, R.; Aitken, N.C.; Spokas, K.; Warthmann, N.; Gordon, S.P.; Vogel, J.P.; et al. Global diversity of the Brachypodium species complex as a resource for genome-wide association studies demonstrated for agronomic traits in response to climate. Genetics 2019, 211, 317–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eichten, S.R.; Srivastava, A.; Reddiex, A.J.; Ganguly, D.R.; Heussler, A.; Streich, J.C.; Wilson, P.B.; Borevitz, J.O. Extending the genotype in Brachypodium by including DNA methylation reveals a joint contribution with genetics on adaptive traits. G3 2020, 10, 1629–1637. [Google Scholar] [CrossRef] [Green Version]
- Roessler, K.; Takuno, S.; Gaut, B.S. CG methylation covaries with differential gene expression between leaf and floral bud tissues of Brachypodium distachyon. PLoS ONE 2016, 11, e0150002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKnight, T.L.; Hess, D. Physical Geography: A Landscape Appreciation, 7th ed.; Prentice Hall: Upper Saddle River, NJ, USA, 2002. [Google Scholar]
- Vogel, J.P.; Tuna, M.; Budak, H.; Huo, N.; Gu, Y.Q.; Steinwand, M.A. Development of SSR markers and analysis of diversity in Turkish populations of Brachypodium distachyon. BMC Plant. Biol 2009, 9, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neilson, E.H.; Edwards, A.M.; Blomstedt, C.K.; Berger, B.; Moller, B.L.; Gleadow, R.M. Utilization of a high-throughput shoot imaging system to examine the dynamic phenotypic responses of a C4 cereal crop plant to nitrogen and water deficiency over time. J. Exp. Bot. 2015, 66, 1817–1832. [Google Scholar] [CrossRef]
- Phillips, S.J.; Anderson, R.P.; Schapire, R.E. Maximum entropy modeling of species geographic distributions. Ecol. Model. 2006, 190, 231–259. [Google Scholar] [CrossRef] [Green Version]
- Baulcombe, D.C.; Dean, C. Epigenetic regulation in plant responses to the environment. Cold Spring Harb. Perspect Biol. 2014, 6, a019471. [Google Scholar] [CrossRef]
- Schmitz, R.J.; Ecker, J.R. Epigenetic and epigenomic variation in Arabidopsis thaliana. Trends Plant. Sci. 2012, 17, 149–154. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Geng, Y.; Li, B.; Chen, J.; Yang, J. Genome-wide DNA methylation alterations of Alternanthera philoxeroides in natural and manipulated habitats: Implications for epigenetic regulation of rapid responses to environmental fluctuation and phenotypic variation. Plant. Cell Environ. 2010, 33, 1820–1827. [Google Scholar] [CrossRef]
- Lira-Medeiros, C.F.; Parisod, C.; Fernandes, R.A.; Mata, C.S.; Cardoso, M.A.; Ferreira, P.C. Epigenetic variation in mangrove plants occurring in contrasting natural environment. PLoS ONE 2010, 5, e10326. [Google Scholar] [CrossRef]
- Schmitz, R.J.; Schultz, M.D.; Lewsey, M.G.; O’Malley, R.C.; Urich, M.A.; Libiger, O.; Schork, N.J.; Ecker, J.R. Transgenerational epigenetic instability is a source of novel methylation variants. Science 2011, 334, 369–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Secco, D.; Wang, C.; Shou, H.; Schultz, M.D.; Chiarenza, S.; Nussaume, L.; Ecker, J.R.; Whelan, J.; Lister, R. Stress induced gene expression drives transient DNA methylation changes at adjacent repetitive elements. Elife 2015, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharbel, T.F.; Haubold, B.; Mitchell-Olds, T. Genetic isolation by distance in Arabidopsis thaliana: Biogeography and postglacial colonization of Europe. Mol. Ecol. 2000, 9, 2109–2118. [Google Scholar] [CrossRef] [PubMed]
- Slotkin, R.K.; Martienssen, R. Transposable elements and the epigenetic regulation of the genome. Nat. Rev. Genet. 2007, 8, 272–285. [Google Scholar] [CrossRef]
- Zemach, A.; Kim, M.Y.; Hsieh, P.H.; Coleman-Derr, D.; Eshed-Williams, L.; Thao, K.; Harmer, S.L.; Zilberman, D. The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin. Cell 2013, 153, 193–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matzke, M.A.; Mosher, R.A. RNA-directed DNA methylation: An epigenetic pathway of increasing complexity. Nat. Rev. Genet. 2014, 15, 394–408. [Google Scholar] [CrossRef]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef]
- Stroud, H.; Do, T.; Du, J.; Zhong, X.; Feng, S.; Johnson, L.; Patel, D.J.; Jacobsen, S.E. Non-CG methylation patterns shape the epigenetic landscape in Arabidopsis. Nat. Struct. Mol. Biol. 2014, 21, 64–72. [Google Scholar] [CrossRef] [Green Version]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 2008, 452, 215–219. [Google Scholar] [CrossRef] [Green Version]
- Lister, R.; O’Malley, R.C.; Tonti-Filippini, J.; Gregory, B.D.; Berry, C.C.; Millar, A.H.; Ecker, J.R. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 2008, 133, 523–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiebaut, F.; Hemerly, A.S.; Ferreira, P.C.G. A role for epigenetic regulation in the adaptation and stress responses of non-model plants. Front. Plant. Sci. 2019, 10, 246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stritt, C.; Gordon, S.P.; Wicker, T.; Vogel, J.P.; Roulin, A.C. Recent activity in expanding populations and purifying selection have shaped transposable element landscapes across natural accessions of the mediterranean grass Brachypodium distachyon. Genome Biol. Evol. 2018, 10, 304–318. [Google Scholar] [CrossRef] [PubMed]
- Wyler, M.; Stritt, C.; Walser, J.-C.; Baroux, C.; Roulin, A.C. Impact of transposable elements on methylation and gene expression across natural accessions of Brachypodium distachyon. Genome Biol. Evol. 2020, evaa180. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J. DNA protocols for plants. In Molecular Techniques in Taxonomy; Hewitt, G.M., Johnston, A.W.B., Young, J.P.W., Eds.; Springer: Berlin/Heidelberg, Germany, 1991; pp. 283–293. [Google Scholar]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Tarasov, A.; Vilella, A.J.; Cuppen, E.; Nijman, I.J.; Prins, P. Sambamba: Fast processing of NGS alignment formats. Bioinformatics 2015, 31, 2032–2034. [Google Scholar] [CrossRef]
- Zheng, X.; Levine, D.; Shen, J.; Gogarten, S.M.; Laurie, C.; Weir, B.S. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 2012, 28, 3326–3328. [Google Scholar] [CrossRef] [Green Version]
- Caye, K.; Deist, T.M.; Martins, H.; Michel, O.; François, O. TESS3: Fast inference of spatial population structure and genome scans for selection. Mol. Ecol. Resour. 2016, 16, 540–548. [Google Scholar] [CrossRef]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef]
- Akalin, A.; Kormaksson, M.; Li, S.; Garrett-Bakelman, F.E.; Figueroa, M.E.; Melnick, A.; Mason, C.E. methylKit: A comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012, 13, R87. [Google Scholar] [CrossRef] [Green Version]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warren, D.L.; Glor, R.E.; Turelli, M. ENMTools: A toolbox for comparative studies of environmental niche models. Ecography 2010, 33, 607–611. [Google Scholar] [CrossRef]
- Fick, S.E.; Hijmans, R.J. WorldClim 2: New 1-km spatial resolution climate surfaces for global land areas. Int. J. Climatol. 2017, 37, 4302–4315. [Google Scholar] [CrossRef]
- Beale, C.M.; Brewer, M.J.; Lennon, J.J. A new statistical framework for the quantification of covariate associations with species distributions. Methods Ecol. Evol. 2014, 5, 421–432. [Google Scholar] [CrossRef]
- Beale, C.M.; Lennon, J.J.; Gimona, A. Opening the climate envelope reveals no macroscale associations with climate in European birds. Proc. Natl. Acad. Sci. USA 2008, 105, 14908–14912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, H.W.; Cross, D.E.; Crump, H.L.; Drost, C.J.; Thomas, C.J. Climate suitability for European ticks: Assessing species distribution models against null models and projection under AR5 climate. Parasites Vectors 2015, 8, 440. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | Station | Collecting Date | Latitude | Longitude | Altitude (m) * |
|---|---|---|---|---|---|
| 2 | 1 | 22-May-2016 | 38.5055 | 27.31671667 | 349 |
| 2 | 5 | 23-May-2016 | 37.49243333 | 27.3395 | 67 |
| 2 | 8 | 24-May-2016 | 37.20506667 | 27.65306667 | 40 |
| 2 | 9 | 24-May-2016 | 37.31233333 | 28.03705 | 626 |
| 2 | 14 | 25-May-2016 | 36.94201667 | 30.96305 | 10 |
| 2 | 20 | 27-May-2016 | 36.95461667 | 34.7507 | 161 |
| 1c | 25 | 28-June-2016 | 39.86911667 | 32.7329 | 1042 |
| 1c | 26 | 28-June-2016 | 39.68008333 | 32.19811667 | 879 |
| 1c | 27 | 29-June-2016 | 38.40685 | 34.03873333 | 1122 |
| 1c | 28 | 30-June-2016 | 38.738 | 34.83881667 | 1063 |
| 1a | 29 | 1-July-2016 | 37.73385 | 38.53376667 | 668 |
| 1a | 30 | 1-July-2016 | 37.69656667 | 37.89476667 | 696 |
| 1a | 31 | 2-July-2016 | 37.03268333 | 37.60995 | 735 |
| 1a | 32 | 2-July-2016 | 37.23601667 | 38.87008333 | 605 |
| 1c | 34 | 3-July-2016 | 39.09433333 | 33.39311667 | 933 |
| 1c | 35 | 3-July-2016 | 40.19286667 | 32.59326667 | 1059 |
| 4 | 36 | 3-July-2016 | 40.73106667 | 31.51755 | 865 |
| 3 | 38 | 7-July-2016 | 41.12035 | 26.65313333 | 58 |
| 3 | 40 | 23-July-2016 | 40.61361667 | 26.43273333 | 63 |
| 3 | 41 | 27-July-2016 | 41.0926 | 27.22096667 | 97 |
| 3 | 42 | 8-August-2016 | 41.3691 | 27.13661667 | 50 |
| 4 | 45 | 15-August-2016 | 40.86231667 | 32.54991667 | 1242 |
| 4 | 47 | 16-July-2016 | 40.87441667 | 35.60698333 | 605 |
| 4 | 49 | 16-July-2016 | 40.59275 | 36.83505 | 283 |
| 4 | 51 | 16-July-2016 | 40.15375 | 38.14713333 | 920 |
| 4 | 52 | 19-July-2016 | 41.32165 | 36.25826667 | 128 |
| 3 | 54 | 29-July-2016 | 40.83846667 | 27.02001667 | 205 |
| 3 | 55 | 29-July-2016 | 40.5003 | 26.70376667 | 88 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skalska, A.; Stritt, C.; Wyler, M.; Williams, H.W.; Vickers, M.; Han, J.; Tuna, M.; Savas Tuna, G.; Susek, K.; Swain, M.; et al. Genetic and Methylome Variation in Turkish Brachypodium Distachyon Accessions Differentiate Two Geographically Distinct Subpopulations. Int. J. Mol. Sci. 2020, 21, 6700. https://doi.org/10.3390/ijms21186700
Skalska A, Stritt C, Wyler M, Williams HW, Vickers M, Han J, Tuna M, Savas Tuna G, Susek K, Swain M, et al. Genetic and Methylome Variation in Turkish Brachypodium Distachyon Accessions Differentiate Two Geographically Distinct Subpopulations. International Journal of Molecular Sciences. 2020; 21(18):6700. https://doi.org/10.3390/ijms21186700
Chicago/Turabian StyleSkalska, Aleksandra, Christoph Stritt, Michele Wyler, Hefin W. Williams, Martin Vickers, Jiwan Han, Metin Tuna, Gulsemin Savas Tuna, Karolina Susek, Martin Swain, and et al. 2020. "Genetic and Methylome Variation in Turkish Brachypodium Distachyon Accessions Differentiate Two Geographically Distinct Subpopulations" International Journal of Molecular Sciences 21, no. 18: 6700. https://doi.org/10.3390/ijms21186700
APA StyleSkalska, A., Stritt, C., Wyler, M., Williams, H. W., Vickers, M., Han, J., Tuna, M., Savas Tuna, G., Susek, K., Swain, M., Wóycicki, R. K., Chaudhary, S., Corke, F., Doonan, J. H., Roulin, A. C., Hasterok, R., & Mur, L. A. J. (2020). Genetic and Methylome Variation in Turkish Brachypodium Distachyon Accessions Differentiate Two Geographically Distinct Subpopulations. International Journal of Molecular Sciences, 21(18), 6700. https://doi.org/10.3390/ijms21186700