Anticancer Properties of Platinum Nanoparticles and Retinoic Acid: Combination Therapy for the Treatment of Human Neuroblastoma Cancer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
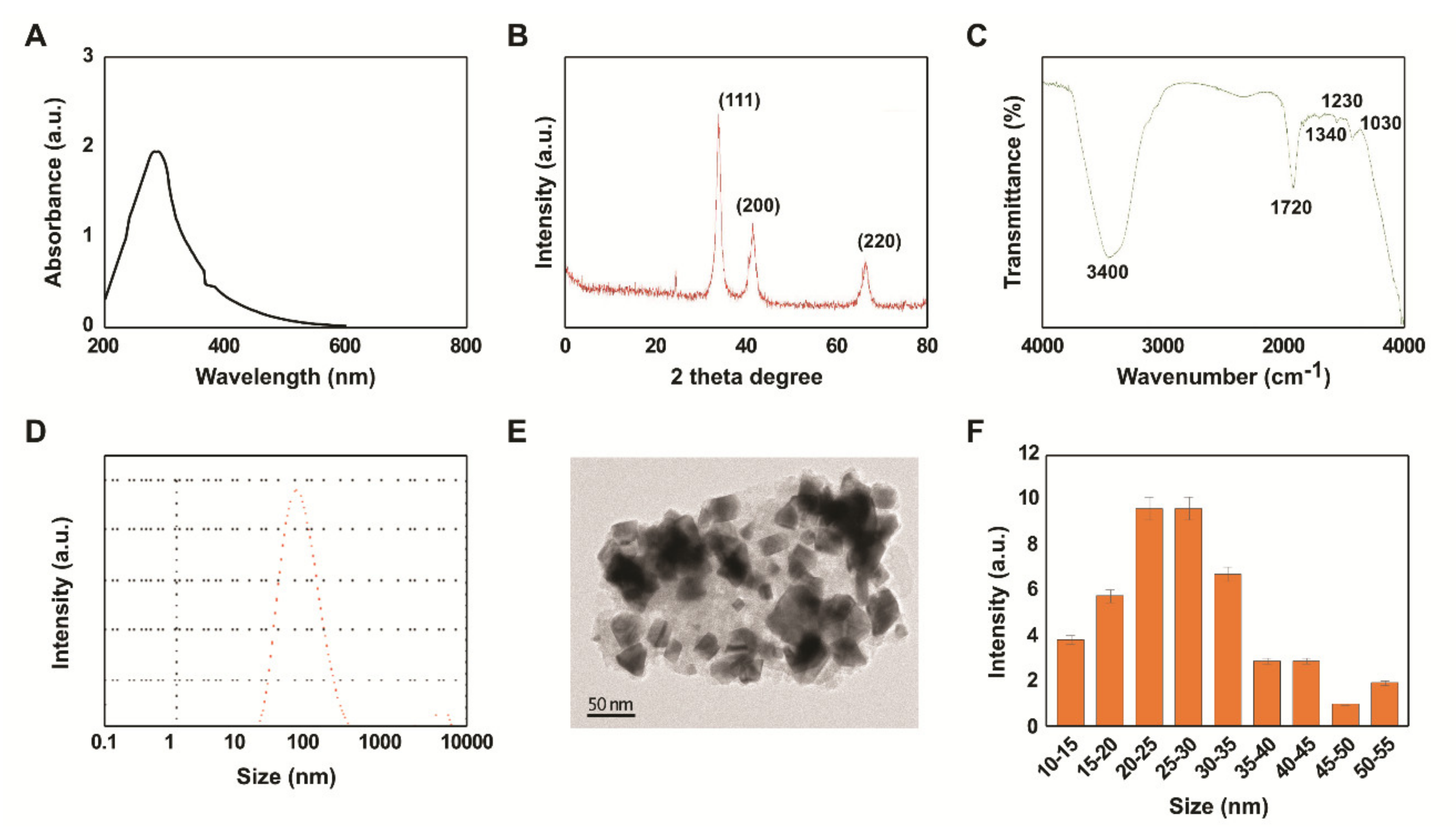
2.1. Synthesis and Characterization of PtNPs Using Beta Carotene
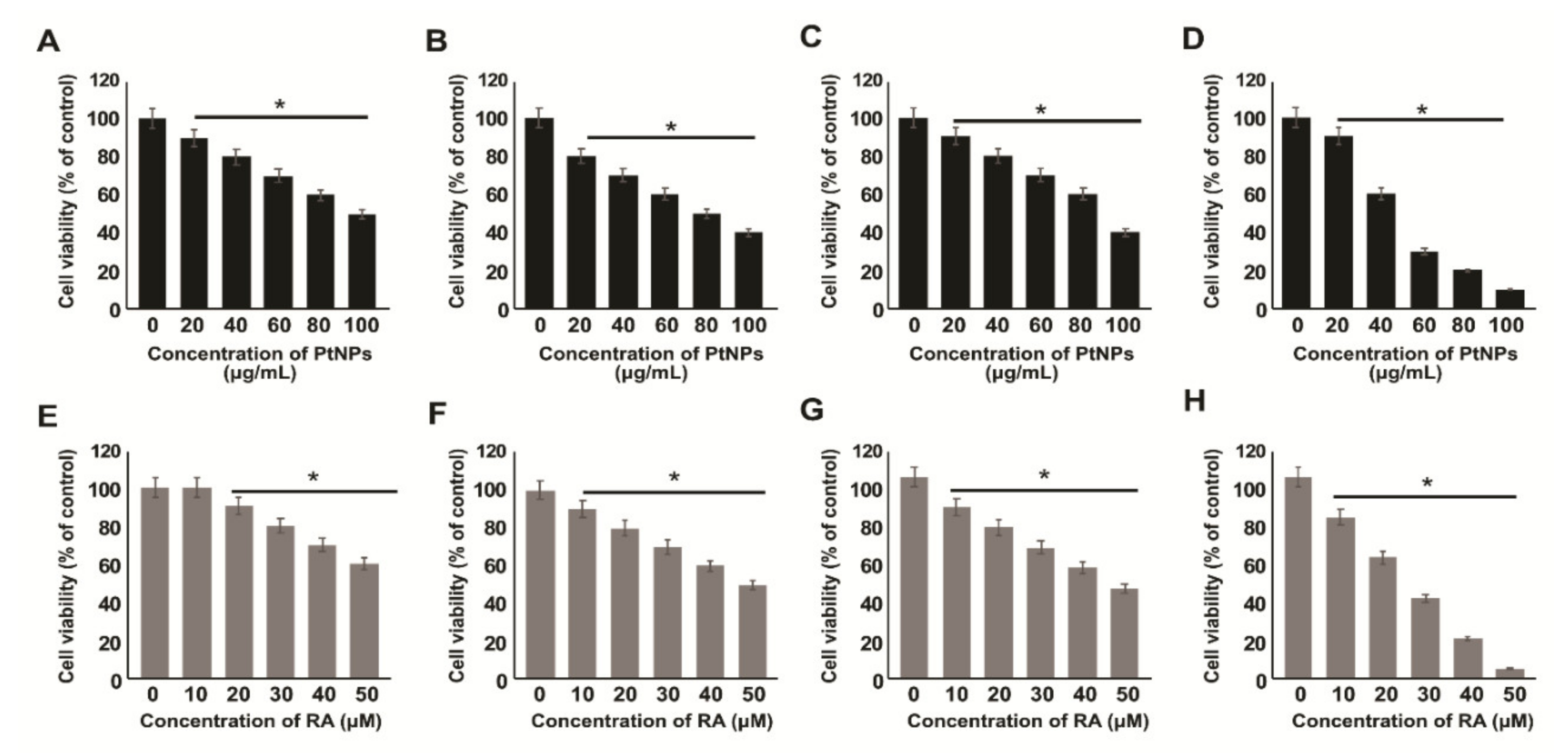
2.2. Effect of PtNPs and RA on the Viability of Various Types of Cancer Cells
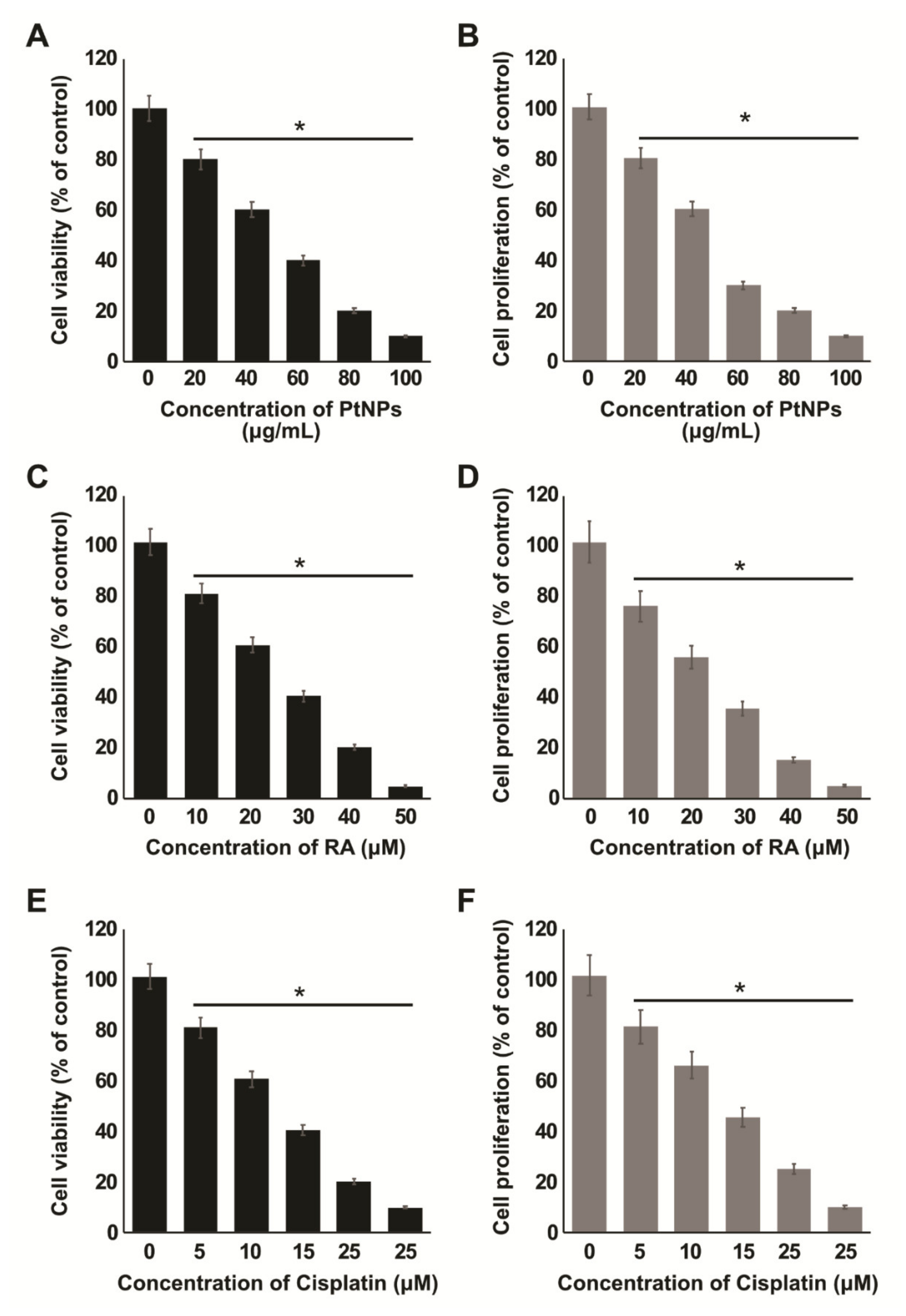
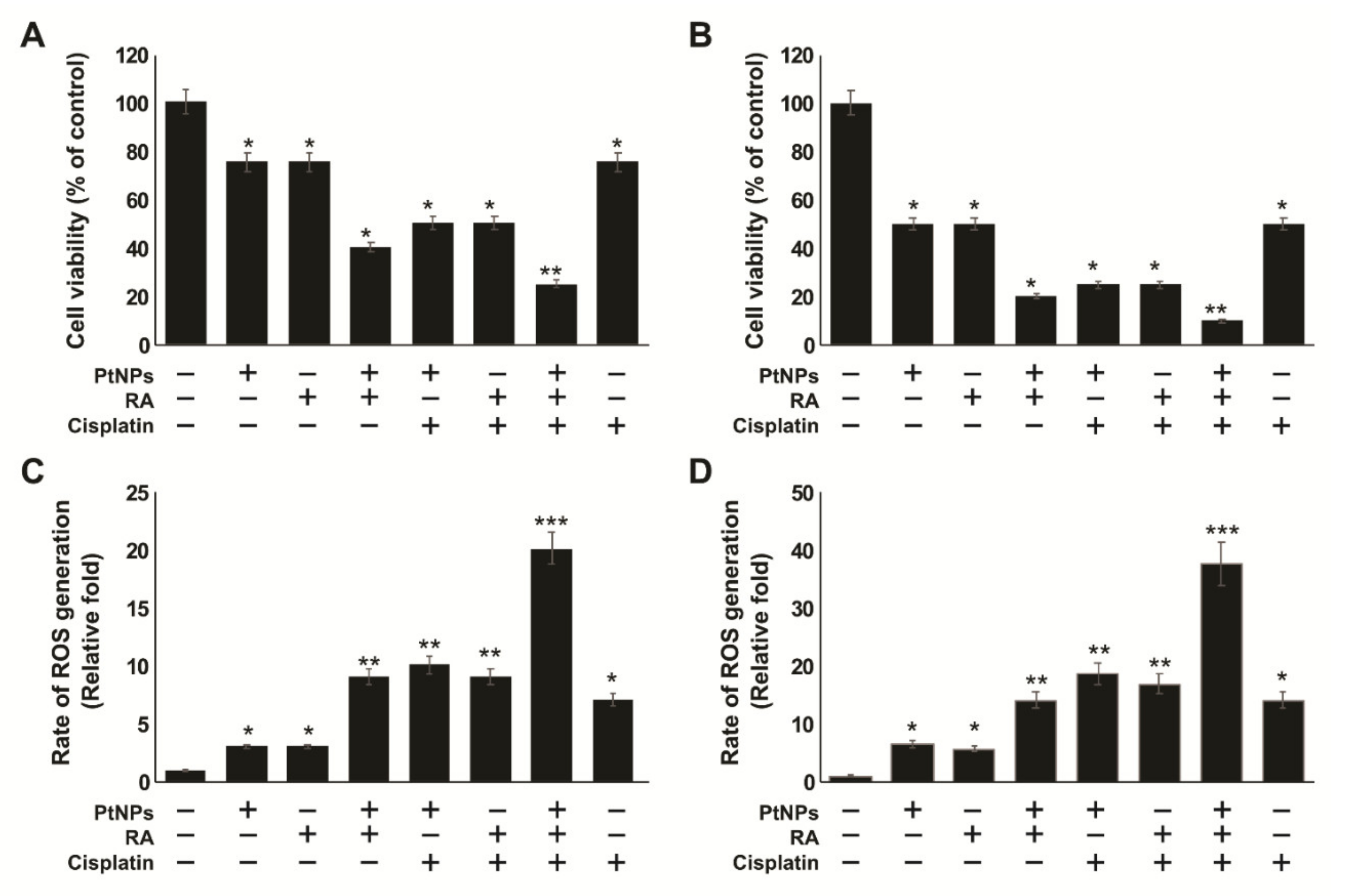
2.3. Dose-Dependent Effects of PtNPs, RA, and Cisplatin on the Viability and Proliferation of SH-SY5Y Cells
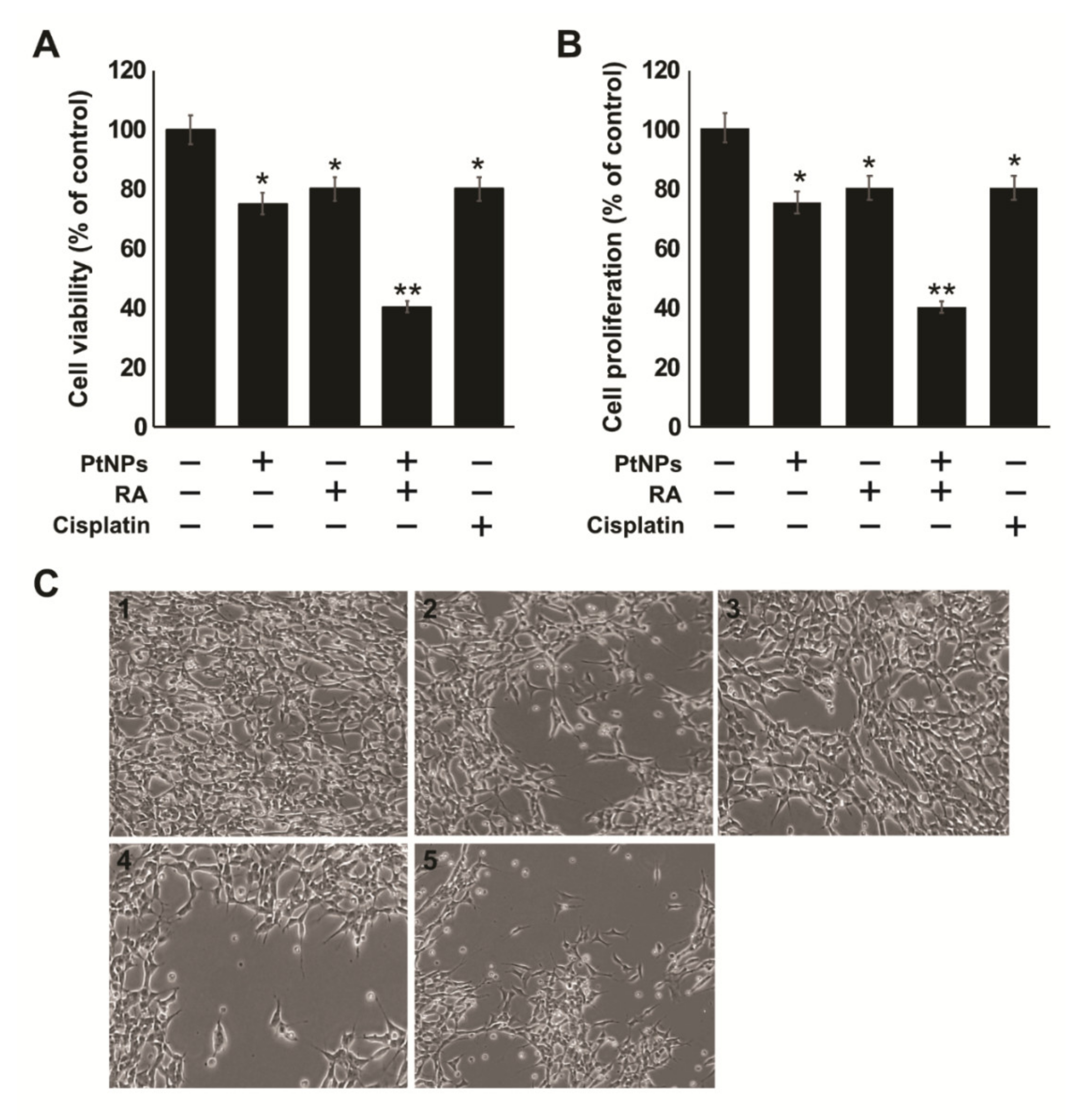
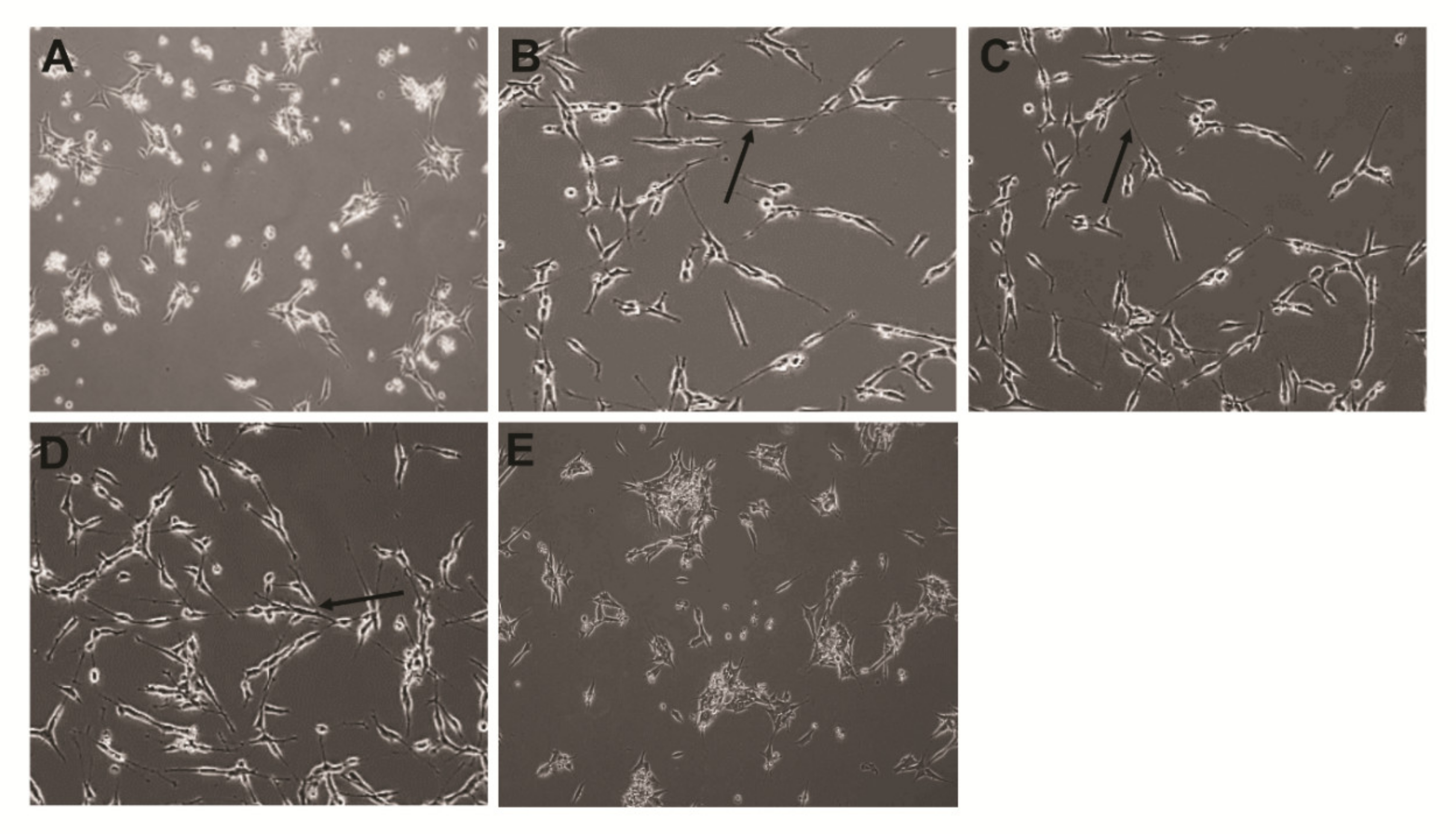
2.4. Effect of the Combination of PtNPs and RA on the Viability, Proliferation, and Morphology of SH-SY5Y Cells
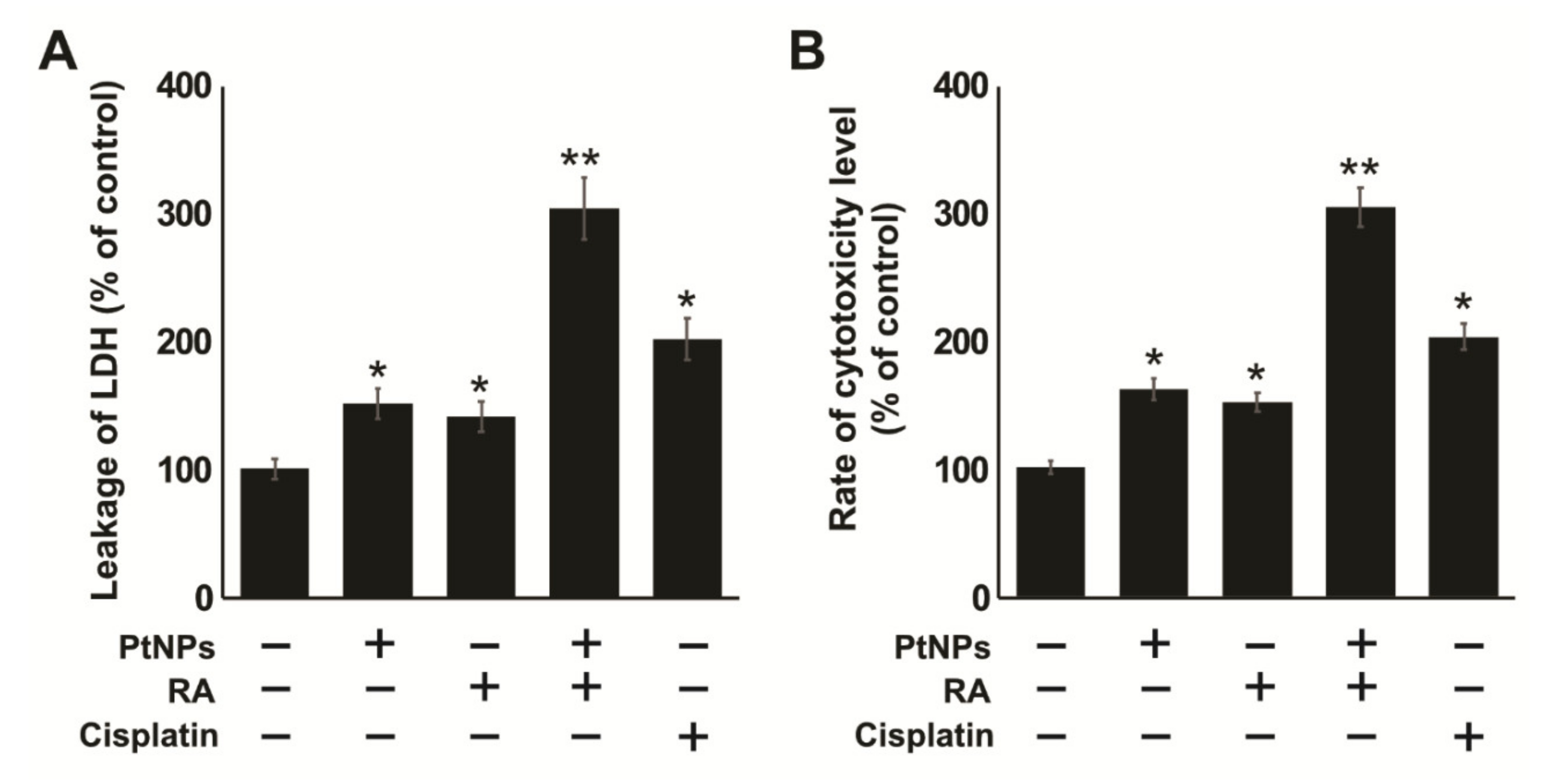
2.5. Combination of PtNPs and RA Induces Lactate Dehydrogenase (LDH) Leakage and Intracellular Protease
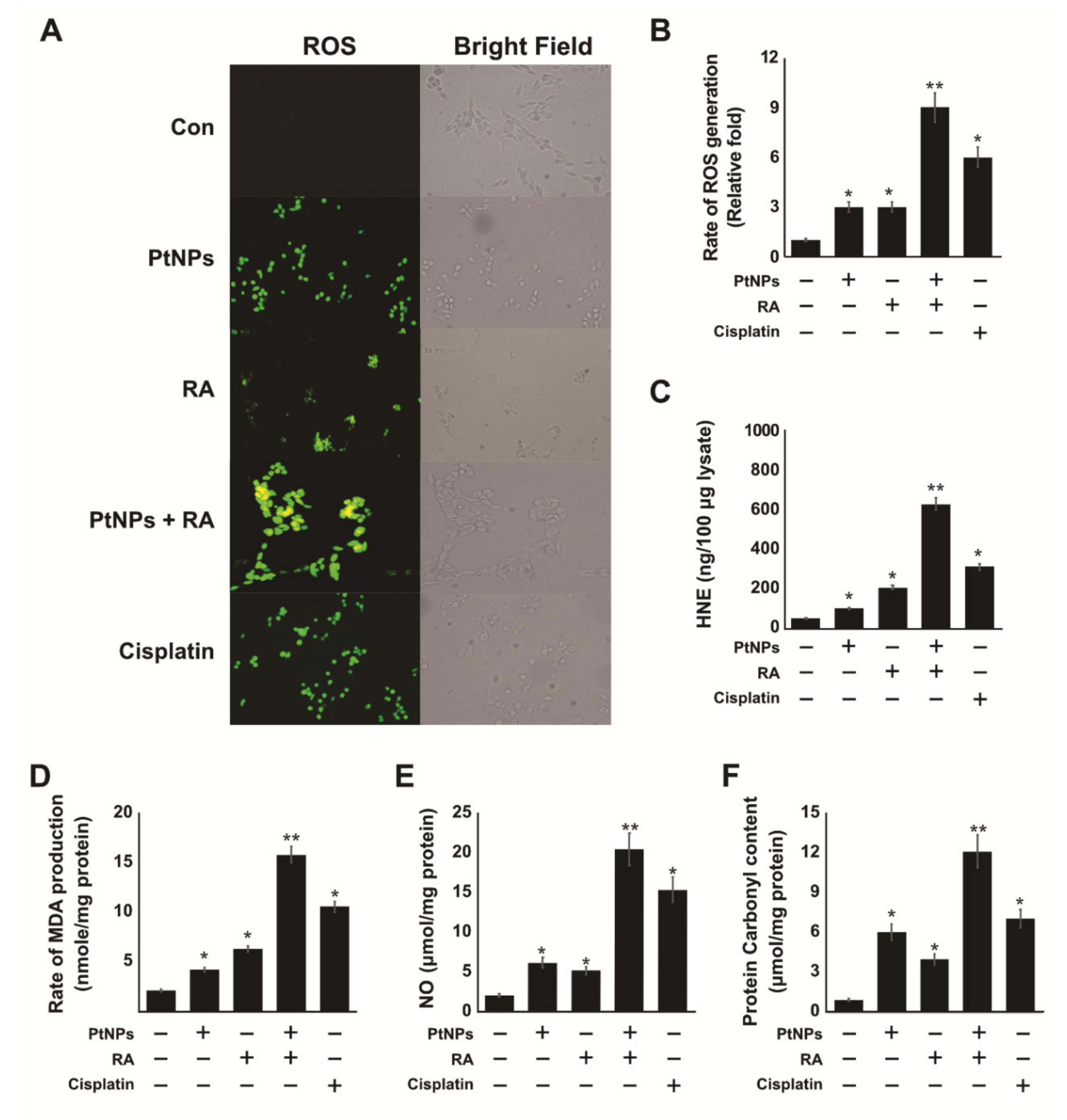
2.6. PtNPs and RA Enhance the Production of Reactive Oxygen Species (ROS), 4-hydroxynonenal (4-HNE), Malondialdehyde (MDA), Nitric Oxide (NO), and Protein Carbonyl Content (PCC)
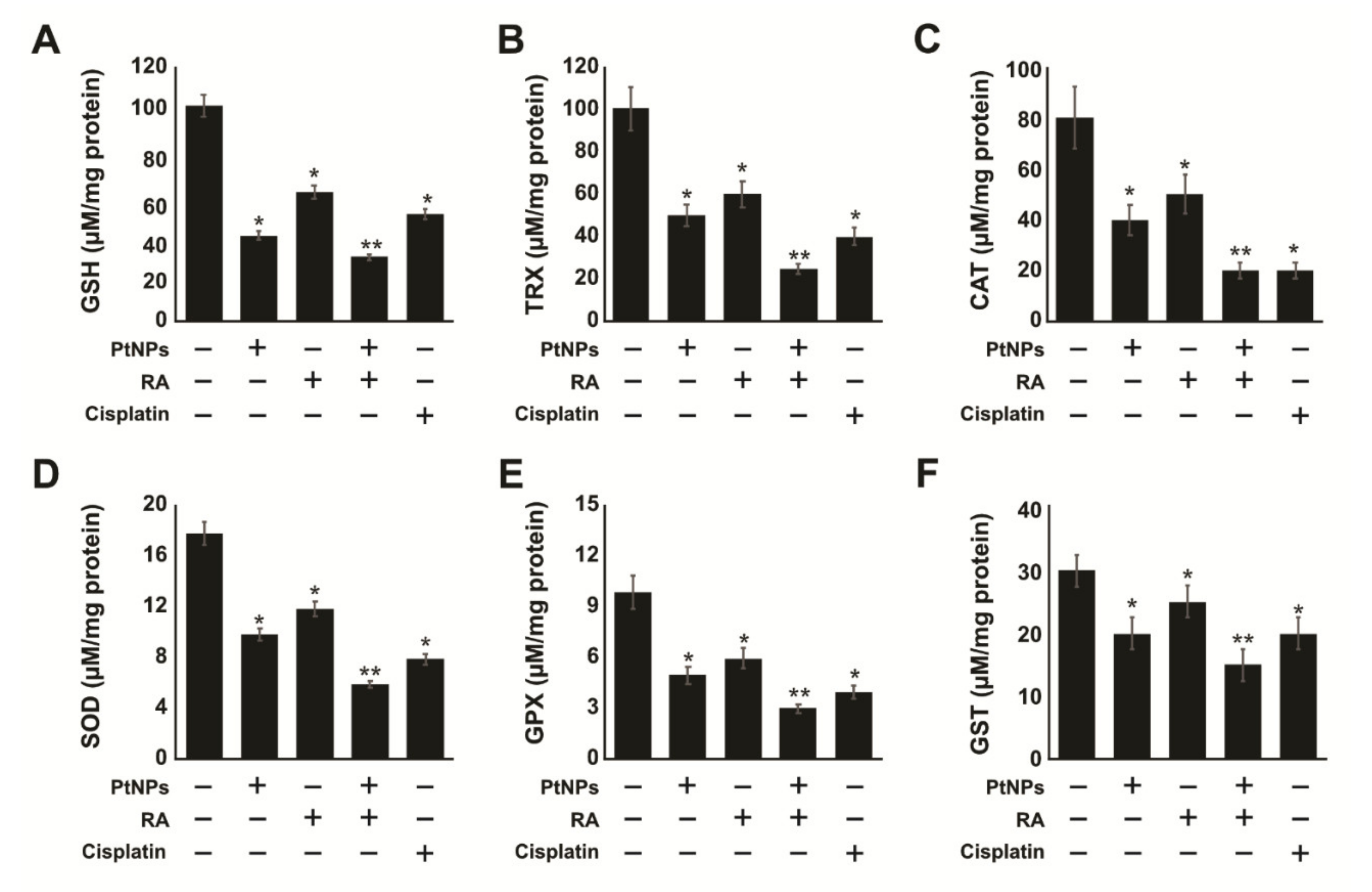
2.7. PtNPs and RA Decrease Antioxidants Levels
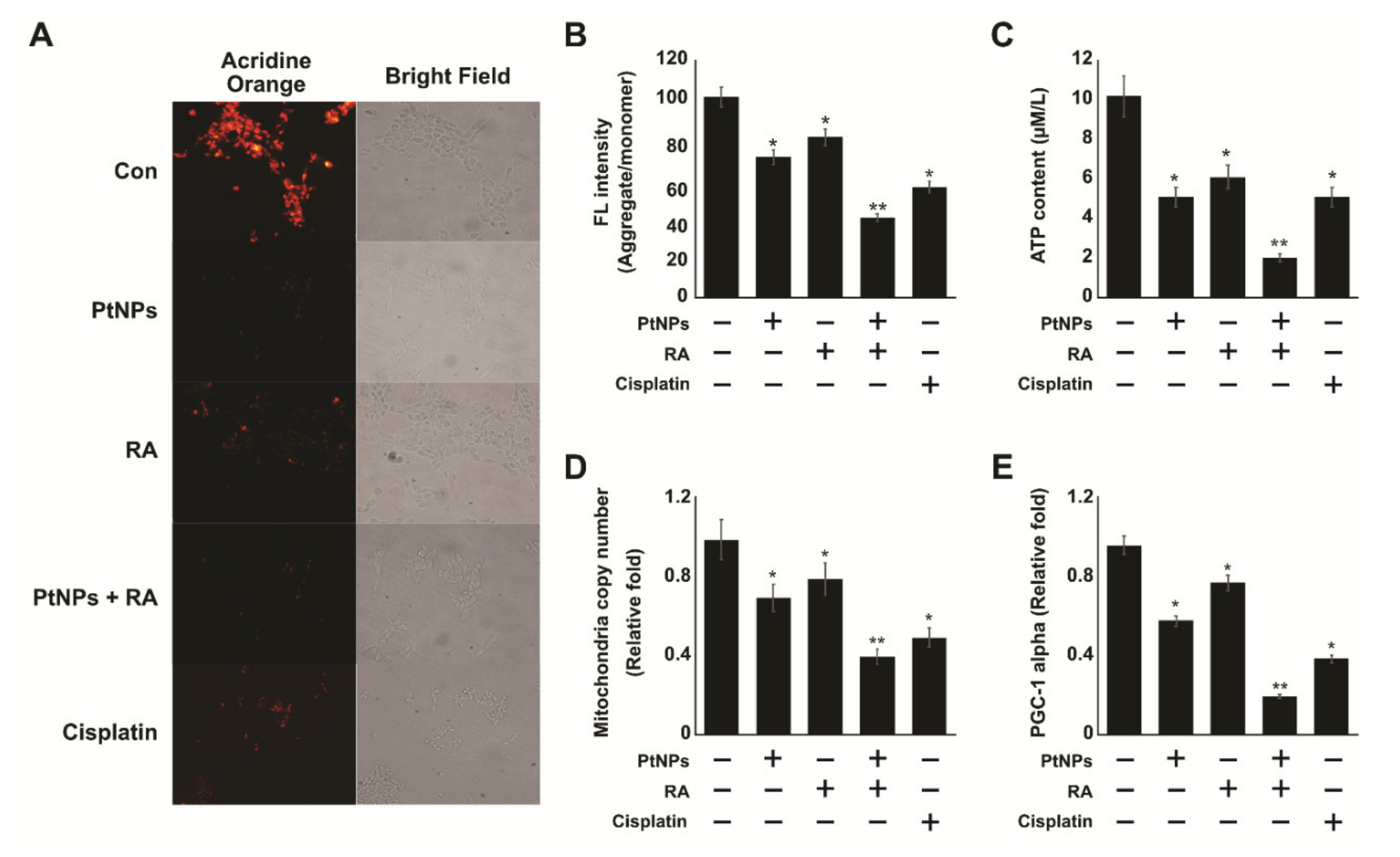
2.8. PtNPs and RA Induce Mitochondrial Dysfunction
2.9. PtNPs and RA Induce ERS and Apoptosis
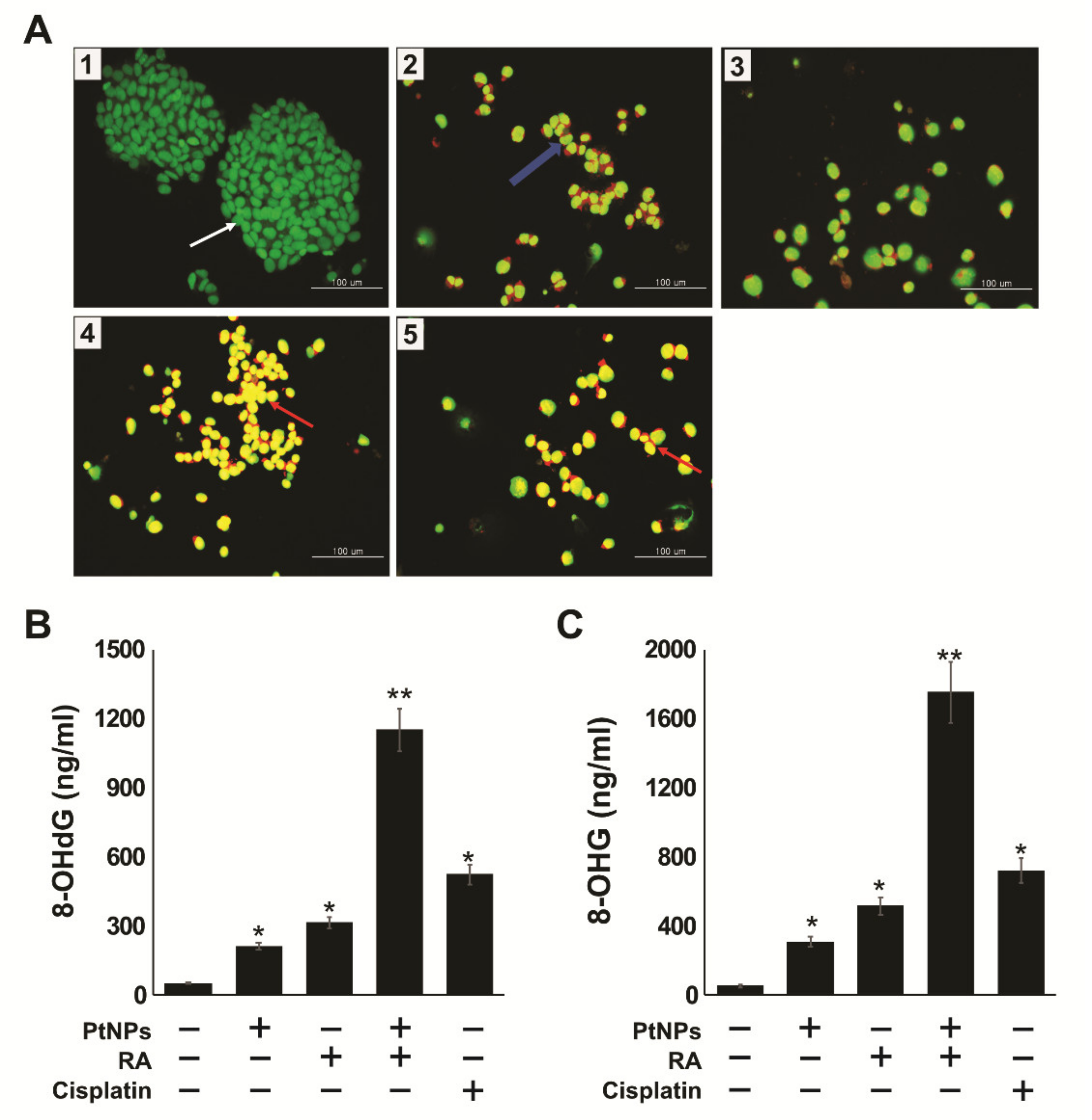
2.10. PtNPs and RA Induce Apoptosis and Oxidative DNA Damage
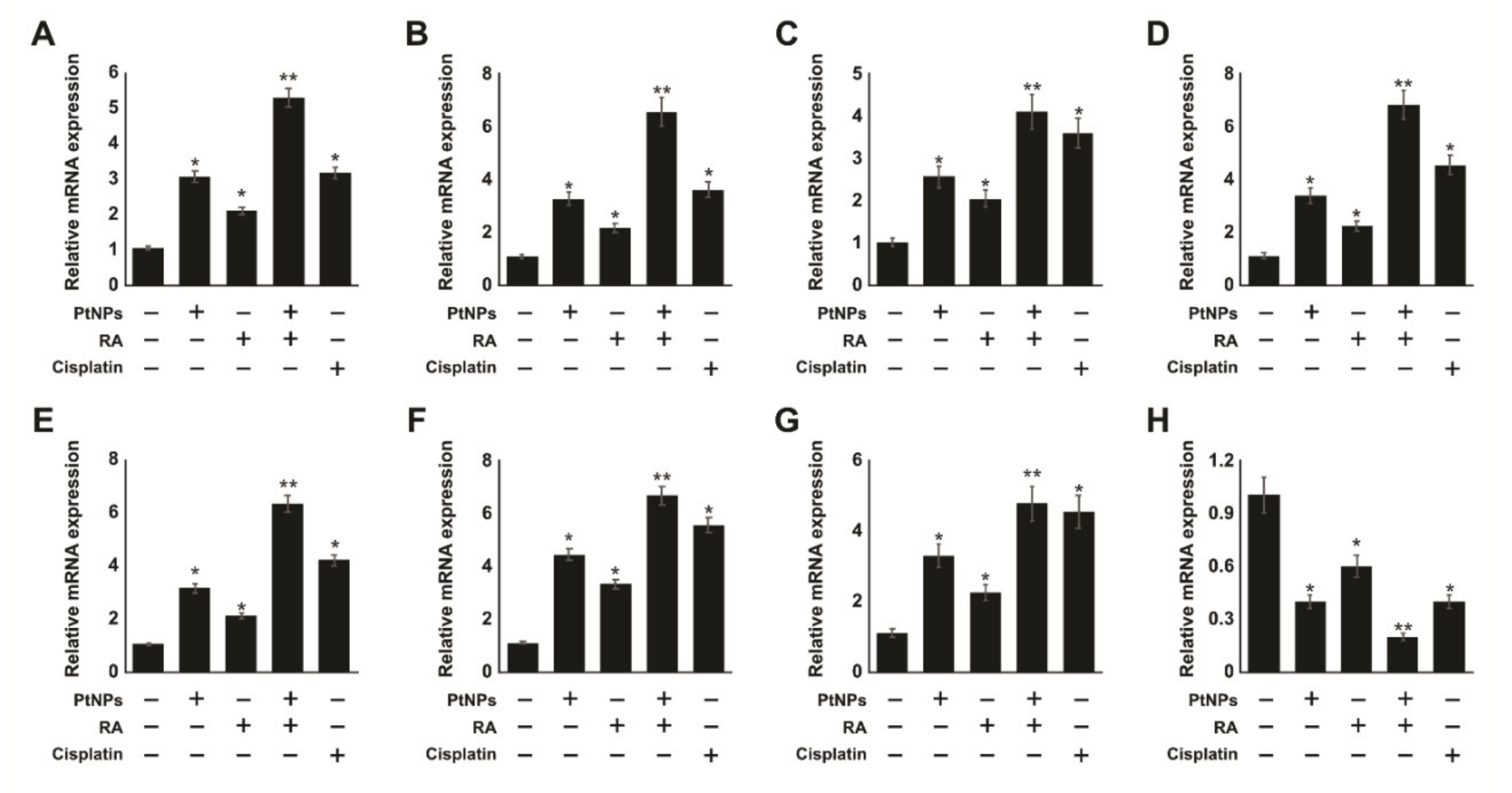
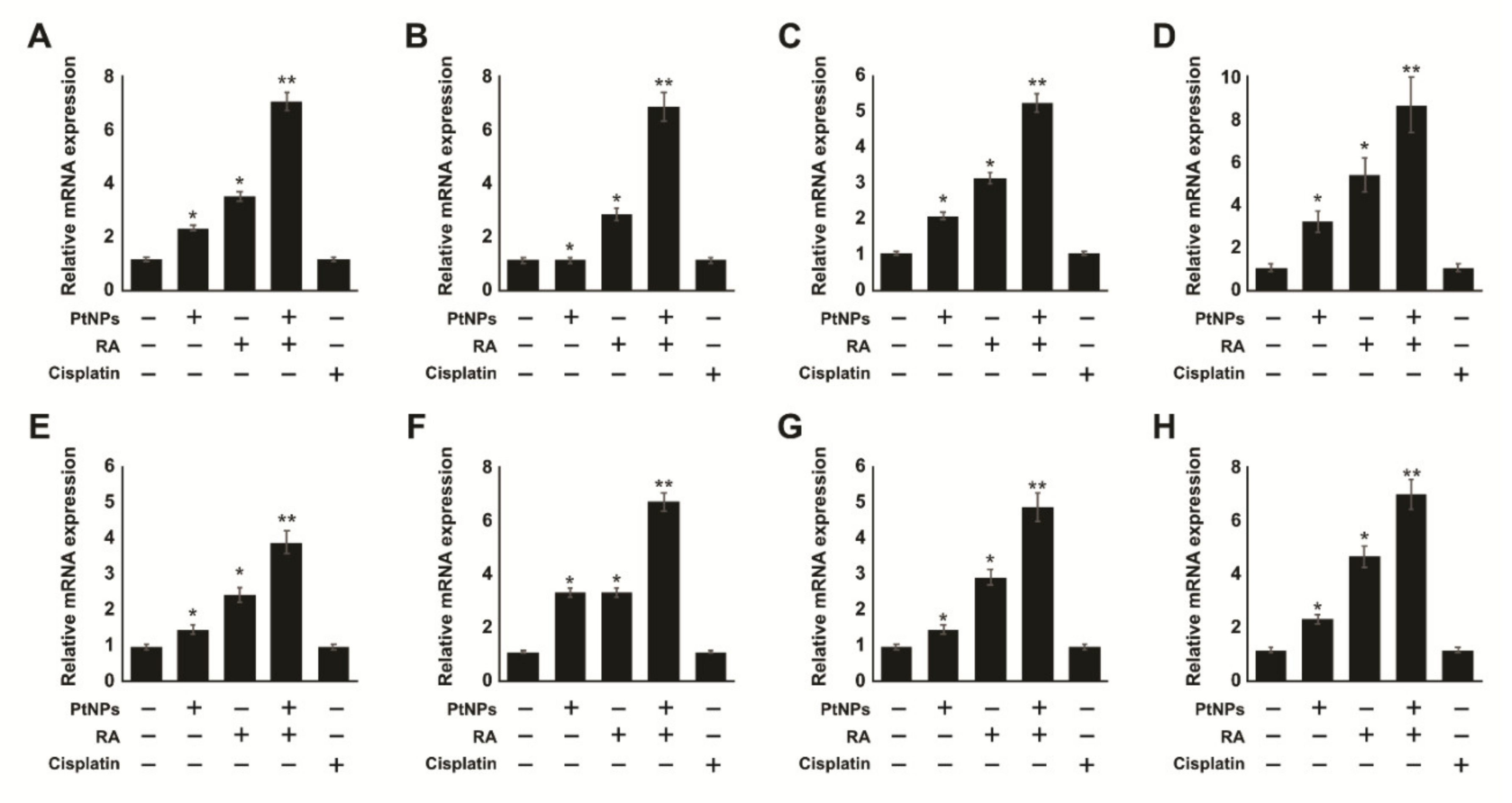
2.11. Combination of PtNPs and RA Increases Differentiation and the Expression of Differentiation Markers in SH-SY5Y Cells
2.12. Effect of Cisplatin on Undifferentiated and Differentiated SH-SY5Y Cells
3. Materials and Methods
3.1. Synthesis and Characterization of PtNPs
3.2. Cell Viability and Cell Proliferation Assay
3.3. Cell Morphology Analysis
3.4. Cytotoxicity Assays
3.5. Determination of the Levels of Reactive Oxygen Species (ROS), Malondialdehyde (MDA), Nitric Oxide (NO), and Protein Carbonyl Content (PCC)
3.6. ELISA
3.7. Measurement of the Levels of Anti-Oxidative Markers
3.8. Determination of Mitochondrial Dysfunctions
3.9. Determination of Apoptosis Using AO/EB Staining
3.10. RT-qPCR
3.11. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fitzmaurice, C.; Allen, C.; Barber, R.M.; Barregard, L.; Bhutta, Z.A.; Brenner, H.; Dicker, D.J.; Chimed-Orchir, O.; Dandona, R.; Dandona, L.; et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-years for 32 Cancer Groups, 1990 to 2015: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2017, 3, 524–548. [Google Scholar] [CrossRef] [PubMed]
- International Agency for Research on Cancer, Cancer Fact Sheets. Available online: http://globocan.iarc.fr/Pages/fact_sheets_cancer.aspx (accessed on 1 September 2019).
- Loo, C.; Lowery, A.; Halas, N.; West, J.; Drezek, R. Immunotargeted nanoshells for integrated cancer imaging and therapy. Nano Lett. 2005, 5, 709–711. [Google Scholar] [PubMed]
- Lal, S.; Clare, S.E.; Halas, N.J. Nanoshell-enabled photothermal cancer therapy: Impending clinical impact. Acc. Chem. Res. 2008, 41, 1842–1851. [Google Scholar] [CrossRef] [PubMed]
- Bardhan, R.; Chen, W.; Perez-Torres, C.; Bartels, M.; Huschka, R.M.; Zhao, L.L.; Morosan, E.; Pautler, R.G.; Joshi, A.; Halas, N.J. Nanoshells with Targeted Simultaneous Enhancement of Magnetic and Optical Imaging and Photothermal Therapeutic Response. Adv. Funct. Mater. 2009, 19, 3901–3909. [Google Scholar] [CrossRef]
- Jørgensen, J.T.; Norregaard, K.; Tian, P.; Bendix, P.M.; Kjaer, A.; Oddershede, L.B. Single Particle and PET-based Platform for Identifying Optimal Plasmonic Nano-Heaters for Photothermal Cancer Therapy. Sci. Rep. 2016, 6, 30076. [Google Scholar] [PubMed]
- Maeda, H.; Wu, J.; Sawa, T.; Matsumura, Y.; Hori, K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: A review. J. Control. Release 2000, 65, 271–284. [Google Scholar] [CrossRef]
- Jaque, D.; Martinez Maestro, L.; del Rosal, B.; Haro-Gonzalez, P.; Benayas, A.; Plaza, J.L.; Martin Rodriguez, E.; Garcia Sole, J. Nanoparticles for photothermal therapies. Nanoscale 2014, 6, 9494–9530. [Google Scholar] [CrossRef]
- Svoboda, K.; Block, S.M. Optical trapping of metallic Rayleigh particles. Opt. Lett. 1994, 19, 930–932. [Google Scholar] [CrossRef]
- Samadi, A.; Klingberg, H.; Jauffred, L.; Kjær, A.; Bendix, P.M.; Oddershede, L.B. Platinum nanoparticles: A non-toxic, effective and thermally stable alternative plasmonic material for cancer therapy and bioengineering. Nanoscale 2018, 10, 9097–9107. [Google Scholar] [CrossRef]
- Linic, S.; Aslam, U.; Boerigter, C.; Morabito, M. Photochemical transformations on plasmonic metal nanoparticles. Nat. Mater. 2015, 14, 567–576. [Google Scholar] [CrossRef]
- Li, D.; Ma, Y.; Du, J.; Tao, W.; Du, X.; Yang, X.; Wang, J. Tumor Acidity/NIR Controlled Interaction of Transformable Nanoparticle with Biological Systems for Cancer Therapy. Nano Lett. 2017, 17, 2871–2878. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ding, J.; Wang, Y.; Cheng, J.; Ji, S.; Zhuang, X.; Chen, X. Sequentially Responsive Shell-Stacked Nanoparticles for Deep Penetration into Solid Tumors. Adv. Mater. (Deerfield BeachFla.) 2017, 29. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, J.; Xu, W.; Xiao, G.; Ding, J.; Chen, X. Tumor microenvironment-labile polymer-doxorubicin conjugate thermogel combined with docetaxel for in situ synergistic chemotherapy of hepatoma. Acta Biomater. 2018, 77, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Bang, Y.J.; Van Cutsem, E.; Feyereislova, A.; Chung, H.C.; Shen, L.; Sawaki, A.; Lordick, F.; Ohtsu, A.; Omuro, Y.; Satoh, T.; et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): A phase 3, open-label, randomised controlled trial. Lancet (Lond. Engl.) 2010, 376, 687–697. [Google Scholar] [CrossRef]
- Gurunathan, S.; Kang, M.H.; Qasim, M.; Kim, J.H. Nanoparticle-Mediated Combination Therapy: Two-in-One Approach for Cancer. Int. J. Mol. Sci. 2018, 19, 3264. [Google Scholar] [CrossRef] [Green Version]
- Cheung, N.K.; Kushner, B.H.; LaQuaglia, M.P.; Kramer, K.; Ambros, P.; Ambros, I.; Ladanyi, M.; Eddy, J.; Bonilla, M.A.; Gerald, W. Survival from non-stage 4 neuroblastoma without cytotoxic therapy: An analysis of clinical and biological markers. Eur. J. Cancer (Oxf. Engl. 1990) 1997, 33, 2117–2120. [Google Scholar] [CrossRef]
- London, W.B.; Castel, V.; Monclair, T.; Ambros, P.F.; Pearson, A.D.; Cohn, S.L.; Berthold, F.; Nakagawara, A.; Ladenstein, R.L.; Iehara, T. Clinical and biologic features predictive of survival after relapse of neuroblastoma: A report from the International Neuroblastoma Risk Group project. J. Clin. Oncol. 2011, 29, 3286. [Google Scholar] [CrossRef] [Green Version]
- Cheung, N.-K.V.; Dyer, M.A. Neuroblastoma: Developmental biology, cancer genomics and immunotherapy. Nat. Rev. Cancer. 2013, 13, 397–411. [Google Scholar] [CrossRef] [Green Version]
- Maris, J.M.; Hogarty, M.D.; Bagatell, R.; Cohn, S.L. Neuroblastoma. Lancet 2007, 369, 2106–2120. [Google Scholar] [CrossRef]
- Matthay, K.K.; Villablanca, J.G.; Seeger, R.C.; Stram, D.O.; Harris, R.E.; Ramsay, N.K.; Swift, P.; Shimada, H.; Black, C.T.; Brodeur, G.M.; et al. Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. Children’s Cancer Group. N. Engl. J. Med. 1999, 341, 1165–1173. [Google Scholar] [CrossRef]
- Matthay, K.K.; Atkinson, J.B.; Stram, D.O.; Selch, M.; Reynolds, C.P.; Seeger, R.C. Patterns of relapse after autologous purged bone marrow transplantation for neuroblastoma: A Childrens Cancer Group pilot study. J. Clin. Oncol. 1993, 11, 2226–2233. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Wurmbach, E.; Waxman, S.; Jing, Y. Upregulation of Bfl-1/A1 in leukemia cells undergoing differentiation by all-trans retinoic acid treatment attenuates chemotherapeutic agent-induced apoptosis. Leukemia 2006, 20, 1009. [Google Scholar] [CrossRef] [PubMed]
- Nehme, A.; Varadarajan, P.; Sellakumar, G.; Gerhold, M.; Niedner, H.; Zhang, Q.; Lin, X.; Christen, R. Modulation of docetaxel-induced apoptosis and cell cycle arrest by all-trans retinoic acid in prostate cancer cells. Br. J. Cancer 2001, 84, 1571. [Google Scholar] [CrossRef]
- Merino, R.; Hurlé, J.M. The molecular basis of retinoid action in tumors. Trends Mol. Med. 2003, 9, 509–511. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.-B.; Minden, M.; McCulloch, E. Phosphorylation of BCL-2 after exposure of human leukemic cells to retinoic acid. Blood 1998, 92, 1768–1775. [Google Scholar] [CrossRef]
- Siddikuzzaman, G.C.; Berlin Grace, V. All trans retinoic acid and cancer. Immunopharmacol. Immunotoxicol. 2011, 33, 241–249. [Google Scholar] [CrossRef]
- Zhu, W.Y.; Jones, C.S.; Kiss, A.; Matsukuma, K.; Amin, S.; De Luca, L.M. Retinoic acid inhibition of cell cycle progression in MCF-7 human breast cancer cells. Exp. Cell Res. 1997, 234, 293–299. [Google Scholar] [CrossRef]
- Liu, H.-W.; Hu, S.-H.; Chen, Y.-W.; Chen, S.-Y. Characterization and drug release behavior of highly responsive chip-like electrically modulated reduced graphene oxide–poly (vinyl alcohol) membranes. J. Mater. Chem. 2012, 22, 17311–17320. [Google Scholar] [CrossRef]
- Yao, J.; Zhang, L.; Zhou, J.; Liu, H.; Zhang, Q. Efficient simultaneous tumor targeting delivery of all-trans retinoid acid and Paclitaxel based on hyaluronic acid-based multifunctional nanocarrier. Mol. Pharm. 2013, 10, 1080–1091. [Google Scholar] [CrossRef]
- Sun, R.; Liu, Y.; Li, S.Y.; Shen, S.; Du, X.J.; Xu, C.F.; Cao, Z.T.; Bao, Y.; Zhu, Y.H.; Li, Y.P.; et al. Co-delivery of all-trans-retinoic acid and doxorubicin for cancer therapy with synergistic inhibition of cancer stem cells. Biomaterials 2015, 37, 405–414. [Google Scholar] [CrossRef]
- Melino, G.; Bernassola, F.; Knight, R.A.; Corasaniti, M.T.; Nistic, G.; Finazzi-Agr, A. S-nitrosylation regulates apoptosis. Nature 1997, 388, 432–433. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.F.; Gurunathan, S. Combination of salinomycin and silver nanoparticles enhances apoptosis and autophagy in human ovarian cancer cells: An effective anticancer therapy. Int. J. Nanomed. 2016, 11, 3655–3675. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.G.; Peng, Q.L.; Gurunathan, S. Combination of palladium nanoparticles and tubastatin-A potentiates apoptosis in human breast cancer cells: A novel therapeutic approach for cancer. Int. J. Nanomed. 2017, 12, 6503–6520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De los Santos, M.; Zambrano, A.; Aranda, A. Combined effects of retinoic acid and histone deacetylase inhibitors on human neuroblastoma SH-SY5Y cells. Mol. Cancer Ther. 2007, 6, 1425–1432. [Google Scholar] [PubMed] [Green Version]
- Kato, Y.; Salumbides, B.C.; Wang, X.-F.; Qian, D.Z.; Williams, S.; Wei, Y.; Sanni, T.B.; Atadja, P.; Pili, R. Antitumor effect of the histone deacetylase inhibitor LAQ824 in combination with 13-cis-retinoic acid in human malignant melanoma. Mol. Cancer Ther. 2007, 6, 70–81. [Google Scholar] [PubMed] [Green Version]
- Spiller, S.E.; Ditzler, S.H.; Pullar, B.J.; Olson, J.M. Response of preclinical medulloblastoma models to combination therapy with 13-cis retinoic acid and suberoylanilide hydroxamic acid (SAHA). J. Neuro-Oncol. 2008, 87, 133–141. [Google Scholar] [CrossRef]
- Witcher, M.; Ross, D.T.; Rousseau, C.; Deluca, L.; Miller, W.H. Synergy between all-trans retinoic acid and tumor necrosis factor pathways in acute leukemia cells. Blood 2003, 102, 237–245. [Google Scholar]
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751–760. [Google Scholar] [CrossRef]
- Davis, M.E.; Chen, Z.G.; Shin, D.M. Nanoparticle therapeutics: An emerging treatment modality for cancer. Nat. Rev. Drug Discov. 2008, 7, 771–782. [Google Scholar] [CrossRef]
- Doane, T.L.; Burda, C. The unique role of nanoparticles in nanomedicine: Imaging, drug delivery and therapy. Chem. Soc. Rev. 2012, 41, 2885–2911. [Google Scholar] [CrossRef]
- Yuan, Y.Y.; Mao, C.Q.; Du, X.J.; Du, J.Z.; Wang, F.; Wang, J. Surface charge switchable nanoparticles based on zwitterionic polymer for enhanced drug delivery to tumor. Adv. Mater. (Deerfield BeachFla.) 2012, 24, 5476–5480. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E.; Zuckerman, J.E.; Choi, C.H.; Seligson, D.; Tolcher, A.; Alabi, C.A.; Yen, Y.; Heidel, J.D.; Ribas, A. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature 2010, 464, 1067–1070. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Liong, M.; Xia, T.; Li, Z.; Ji, Z.; Zink, J.I.; Nel, A.E. Engineered design of mesoporous silica nanoparticles to deliver doxorubicin and P-glycoprotein siRNA to overcome drug resistance in a cancer cell line. ACS Nano 2010, 4, 4539–4550. [Google Scholar] [CrossRef]
- Sarfati, G.; Dvir, T.; Elkabets, M.; Apte, R.N.; Cohen, S. Targeting of polymeric nanoparticles to lung metastases by surface-attachment of YIGSR peptide from laminin. Biomaterials 2011, 32, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.Z.; Du, X.J.; Liu, Y.; Zhu, Y.H.; Liu, Y.Z.; Li, Y.P.; Wang, J. Rational design of polyion complex nanoparticles to overcome cisplatin resistance in cancer therapy. Adv. Mater. (Deerfield BeachFla.) 2014, 26, 931–936. [Google Scholar] [CrossRef] [PubMed]
- Al-Radadi, N.S. Green synthesis of platinum nanoparticles using Saudi’s Dates extract and their usage on the cancer cell treatment. Arab. J. Chem. 2019, 12, 330–349. [Google Scholar] [CrossRef]
- Gurunathan, S.; Jeyaraj, M.; Kang, M.H.; Kim, J.H. The Effects of Apigenin-Biosynthesized Ultra-Small Platinum Nanoparticles on the Human Monocytic THP-1 Cell Line. Cells 2019, 8, 444. [Google Scholar] [CrossRef] [Green Version]
- Gurunathan, S.; Jeyaraj, M.; Kang, M.H.; Kim, J.H. Tangeretin-Assisted Platinum Nanoparticles Enhance the Apoptotic Properties of Doxorubicin: Combination Therapy for Osteosarcoma Treatment. Nanomaterials 2019, 9, 1089. [Google Scholar] [CrossRef] [Green Version]
- Karthik, R.; Sasikumar, R.; Chen, S.-M.; Govindasamy, M.; Kumar, J.V.; Muthuraj, V. Green synthesis of platinum nanoparticles using quercus glauca extract and its electrochemical oxidation of hydrazine in water samples. Int. J. Electrochem. Sci. 2016, 11, 8245–8255. [Google Scholar] [CrossRef]
- Huang, X.; Wu, H.; Liao, X.; Shi, B. One-step, size-controlled synthesis of gold nanoparticles at room temperature using plant tannin. Green Chem. 2010, 12, 395–399. [Google Scholar] [CrossRef]
- Hoshyar, N.; Gray, S.; Han, H.; Bao, G. The effect of nanoparticle size on in vivo pharmacokinetics and cellular interaction. Nanomedicine 2016, 11, 673–692. [Google Scholar]
- Bendale, Y.; Bendale, V.; Paul, S. Evaluation of cytotoxic activity of platinum nanoparticles against normal and cancer cells and its anticancer potential through induction of apoptosis. Integr. Med. Res. 2017, 6, 141–148. [Google Scholar]
- Kutwin, M.; Sawosz, E.; Jaworski, S.; Hinzmann, M.; Wierzbicki, M.; Hotowy, A.; Grodzik, M.; Winnicka, A.; Chwalibog, A. Investigation of platinum nanoparticle properties against U87 glioblastoma multiforme. Arch. Med. Sci. 2017, 13, 1322. [Google Scholar] [PubMed]
- Almeer, R.S.; Ali, D.; Alarifi, S.; Alkahtani, S.; Almansour, M. Green Platinum Nanoparticles Interaction With HEK293 Cells: Cellular Toxicity, Apoptosis, and Genetic Damage. Dose-Response 2018, 16, 1559325818807382. [Google Scholar] [CrossRef] [PubMed]
- Gurunathan, S.; Jeyaraj, M.; Kang, M.H.; Kim, J.H. Graphene Oxide(-)Platinum Nanoparticle Nanocomposites: A Suitable Biocompatible Therapeutic Agent for Prostate Cancer. Polymers 2019, 11, 733. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Kang, Y.; Yin, S.; Song, B.; Wei, L.; Chen, L.; Shao, L. Zinc oxide nanoparticles induce toxic responses in human neuroblastoma SHSY5Y cells in a size-dependent manner. Int. J. Nanomed. 2017, 12, 8085–8099. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.-H.; Ye, N.; Tang, X.-F.; Khan, M.I.; Liu, H.-L.; Shi, N.; Hang, L.-F. Synergistic Effect of Retinoic Acid Polymeric Micelles and Prodrug for the Pharmacodynamic Evaluation of Tumor Suppression. Front Pharm. 2019, 10, 447. [Google Scholar] [CrossRef]
- Sun, X.J.; Zhang, P.; Li, H.H.; Jiang, Z.W.; Jiang, C.C.; Liu, H. Cisplatin combined with metformin inhibits migration and invasion of human nasopharyngeal carcinoma cells by regulating E-cadherin and MMP-9. Asian Pac. J. Cancer Prev. Apjcp 2014, 15, 4019–4023. [Google Scholar] [CrossRef] [Green Version]
- Hong, G.-Y.; Jeong, Y.-I.; Lee, S.J.; Lee, E.; Oh, J.S.; Lee, H.C. Combination of paclitaxel-and retinoic acid-incorporated nanoparticles for the treatment of CT-26 colon carcinoma. Arch. Pharmacal Res. 2011, 34, 407–417. [Google Scholar]
- Redova, M.; Chlapek, P.; Loja, T.; Zitterbart, K.; Hermanova, M.; Sterba, J.; Veselska, R. Influence of LOX/COX inhibitors on cell differentiation induced by all-trans retinoic acid in neuroblastoma cell lines. Int. J. Mol. Med. 2010, 25, 271–280. [Google Scholar]
- Ibrahim, A.B.; Zaki, H.F.; Ibrahim, W.W.; Omran, M.M.; Shouman, S.A. Evaluation of tamoxifen and simvastatin as the combination therapy for the treatment of hormonal dependent breast cancer cells. Toxicol. Rep. 2019, 6, 1114–1126. [Google Scholar] [CrossRef] [PubMed]
- Gurunathan, S.; Kang, M.H.; Kim, J.H. Combination Effect of Silver Nanoparticles and Histone Deacetylases Inhibitor in Human Alveolar Basal Epithelial Cells. Molecules 2018, 23, 2046. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.G.; Peng, Q.L.; Gurunathan, S. Silver nanoparticles enhance the apoptotic potential of gemcitabine in human ovarian cancer cells: Combination therapy for effective cancer treatment. Int. J. Nanomed. 2017, 12, 6487–6502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.-F.; Huang, F.-H.; Zhang, G.-L.; Bai, D.-P.; Massimo, D.F.; Huang, Y.-F.; Gurunathan, S. Novel biomolecule lycopene-reduced graphene oxide-silver nanoparticle enhances apoptotic potential of trichostatin A in human ovarian cancer cells (SKOV3). Int. J. Nanomed. 2017, 12, 7551–7575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurunathan, S.; Kim, J.-H. Biocompatible Gold Nanoparticles Ameliorate Retinoic Acid-Induced Cell Death and Induce Differentiation in F9 Teratocarcinoma Stem Cells. Nanomaterials 2018, 8, 396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiny, P.; Mukherjee, A.; Chandrasekaran, N. DNA damage and mitochondria-mediated apoptosis of A549 lung carcinoma cells induced by biosynthesised silver and platinum nanoparticles. RSC Adv. 2016, 6, 27775–27787. [Google Scholar]
- Forman, H.J. Redox signaling: An evolution from free radicals to aging. Free Radic. Biol. Med. 2016, 97, 398–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentile, F.; Arcaro, A.; Pizzimenti, S.; Daga, M.; Cetrangolo, G.P.; Dianzani, C.; Lepore, A.; Graf, M.; Ames, P.R.; Barrera, G. DNA damage by lipid peroxidation products: Implications in cancer, inflammation and autoimmunity. AIMS Genet. 2017, 4, 103. [Google Scholar] [CrossRef]
- Fruehauf, J.P.; Meyskens, F.L., Jr. Reactive oxygen species: A breath of life or death? Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2007, 13, 789–794. [Google Scholar] [CrossRef] [Green Version]
- Zanetti, D.; Poli, G.; Vizio, B.; Zingaro, B.; Chiarpotto, E.; Biasi, F. 4-hydroxynonenal and transforming growth factor-beta1 expression in colon cancer. Mol. Asp. Med. 2003, 24, 273–280. [Google Scholar] [CrossRef]
- Cerbone, A.; Toaldo, C.; Laurora, S.; Briatore, F.; Pizzimenti, S.; Dianzani, M.U.; Ferretti, C.; Barrera, G. 4-Hydroxynonenal and PPARγ ligands affect proliferation, differentiation, and apoptosis in colon cancer cells. Free Radic. Biol. Med. 2007, 42, 1661–1670. [Google Scholar] [CrossRef] [PubMed]
- Albright, C.D.; Klem, E.; Shah, A.A.; Gallagher, P. Breast cancer cell-targeted oxidative stress: Enhancement of cancer cell uptake of conjugated linoleic acid, activation of p53, and inhibition of proliferation. Exp. Mol. Pathol. 2005, 79, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Corbalan, J.J.; Medina, C.; Jacoby, A.; Malinski, T.; Radomski, M.W. Amorphous silica nanoparticles trigger nitric oxide/peroxynitrite imbalance in human endothelial cells: Inflammatory and cytotoxic effects. Int. J. Nanomed. 2011, 6, 2821. [Google Scholar]
- Uehara, T.; Kikuchi, Y.; Nomura, Y. Caspase activation accompanying cytochrome c release from mitochondria is possibly involved in nitric oxide-induced neuronal apoptosis in SH-SY5Y cells. J. Neurochem. 1999, 72, 196–205. [Google Scholar] [CrossRef]
- Bolaños, J.P.; Almeida, A.; Stewart, V.; Peuchen, S.; Land, J.M.; Clark, J.B.; Heales, S.J. Nitric oxide-mediated mitochondrial damage in the brain: Mechanisms and implications for neurodegenerative diseases. J. Neurochem. 1997, 68, 2227–2240. [Google Scholar] [CrossRef]
- Clementi, E.; Brown, G.C.; Feelisch, M.; Moncada, S. Persistent inhibition of cell respiration by nitric oxide: Crucial role of S-nitrosylation of mitochondrial complex I and protective action of glutathione. Proc. Natl. Acad. Sci. USA 1998, 95, 7631–7636. [Google Scholar] [CrossRef] [Green Version]
- Gurunathan, S.; Kim, J.-H. Graphene Oxide-Silver Nanoparticles Nanocomposite Stimulates Differentiation in Human Neuroblastoma Cancer Cells (SH-SY5Y). Int. J. Mol. Sci. 2017, 18, 2549. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, Y.J.; Carini, M.; Butterfield, D.A. Protein carbonylation. Antioxid. Redox Signal. 2010, 12, 323–325. [Google Scholar] [CrossRef]
- Thanan, R.; Oikawa, S.; Hiraku, Y.; Ohnishi, S.; Ma, N.; Pinlaor, S.; Yongvanit, P.; Kawanishi, S.; Murata, M. Oxidative stress and its significant roles in neurodegenerative diseases and cancer. Int. J. Mol. Sci. 2015, 16, 193–217. [Google Scholar] [CrossRef] [Green Version]
- Petrache Voicu, S.; Dinu, D.; Sima, C.; Hermenean, A.; Ardelean, A.; Codrici, E.; Stan, M.; Zărnescu, O.; Dinischiotu, A. Silica nanoparticles induce oxidative stress and autophagy but not apoptosis in the MRC-5 cell line. Int. J. Mol. Sci. 2015, 16, 29398–29416. [Google Scholar] [CrossRef] [Green Version]
- Driessen, M.D.; Mues, S.; Vennemann, A.; Hellack, B.; Bannuscher, A.; Vimalakanthan, V.; Riebeling, C.; Ossig, R.; Wiemann, M.; Schnekenburger, J. Proteomic analysis of protein carbonylation: A useful tool to unravel nanoparticle toxicity mechanisms. Part. Fibre Toxicol. 2015, 12, 36. [Google Scholar]
- Butterfield, D.A.; Perluigi, M.; Reed, T.; Muharib, T.; Hughes, C.P.; Robinson, R.A.; Sultana, R. Redox proteomics in selected neurodegenerative disorders: From its infancy to future applications. Antioxid. Redox Signal. 2012, 17, 1610–1655. [Google Scholar] [PubMed] [Green Version]
- Dalle-Donne, I.; Aldini, G.; Carini, M.; Colombo, R.; Rossi, R.; Milzani, A. Protein carbonylation, cellular dysfunction, and disease progression. J. Cell. Mol. Med. 2006, 10, 389–406. [Google Scholar] [PubMed]
- Ergin, V.; Hariry, R.E.; Karasu, Ç. Carbonyl stress in aging process: Role of vitamins and phytochemicals as redox regulators. Aging Dis. 2013, 4, 276. [Google Scholar] [PubMed]
- Cabiscol, E.; Tamarit, J.; Ros, J. Protein carbonylation: Proteomics, specificity and relevance to aging. Mass Spectrom. Rev. 2014, 33, 21–48. [Google Scholar] [PubMed]
- Pham-Huy, L.A.; He, H.; Pham-Huy, C. Free radicals, antioxidants in disease and health. Int. J. Biomed. Sci. Ijbs 2008, 4, 89–96. [Google Scholar]
- Ren, X.; Zou, L.; Zhang, X.; Branco, V.; Wang, J.; Carvalho, C.; Holmgren, A.; Lu, J. Redox signaling mediated by thioredoxin and glutathione systems in the central nervous system. Antioxid. Redox Signal. 2017, 27, 989–1010. [Google Scholar] [PubMed]
- Holmgren, A.; Sengupta, R. The use of thiols by ribonucleotide reductase. Free Radic. Biol. Med. 2010, 49, 1617–1628. [Google Scholar] [CrossRef]
- Mitsui, A.; Hamuro, J.; Nakamura, H.; Kondo, N.; Hirabayashi, Y.; Ishizaki-Koizumi, S.; Hirakawa, T.; Inoue, T.; Yodoi, J. Overexpression of human thioredoxin in transgenic mice controls oxidative stress and life span. Antioxid. Redox Signal. 2002, 4, 693–696. [Google Scholar] [CrossRef]
- Trachootham, D.; Zhou, Y.; Zhang, H.; Demizu, Y.; Chen, Z.; Pelicano, H.; Chiao, P.J.; Achanta, G.; Arlinghaus, R.B.; Liu, J.; et al. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell 2006, 10, 241–252. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.S.; Gupta Vallur, P.; Phaeton, R.; Mythreye, K.; Hempel, N. Insights into the Dichotomous Regulation of SOD2 in Cancer. Antioxidants 2017, 6, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatesan, B.; Mahimainathan, L.; Das, F.; Ghosh-Choudhury, N.; Ghosh Choudhury, G. Downregulation of catalase by reactive oxygen species via PI 3 kinase/Akt signaling in mesangial cells. J. Cell. Physiol. 2007, 211, 457–467. [Google Scholar] [CrossRef]
- Fan, J.J.; Hsu, W.H.; Hung, H.H.; Zhang, W.J.; Lee, Y.L.A.; Chen, K.C.; Chu, C.Y.; Ko, T.P.; Lee, M.T.; Lin, C.W. Reduction in MnSOD promotes the migration and invasion of squamous carcinoma cells. Int. J. Oncol. 2019, 54, 1639–1650. [Google Scholar] [CrossRef] [Green Version]
- Roy, K.; Wu, Y.; Meitzler, J.L.; Juhasz, A.; Liu, H.; Jiang, G.; Lu, J.; Antony, S.; Doroshow, J.H. NADPH oxidases and cancer. Clin. Sci. 2015, 128, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Gurunathan, S.; Choi, Y.-J.; Han, J.W.; Song, H.; Kim, J.-H. Silver nanoparticles suppresses brain-derived neurotrophic factor-induced cell survival in the human neuroblastoma cell line SH-SY5Y. J. Ind. Eng. Chem. 2017, 47, 62–73. [Google Scholar] [CrossRef]
- Ventura-Clapier, R.; Garnier, A.; Veksler, V. Transcriptional control of mitochondrial biogenesis: The central role of PGC-1α. Cardiovasc. Res. 2008, 79, 208–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roussel, B.D.; Kruppa, A.J.; Miranda, E.; Crowther, D.C.; Lomas, D.A.; Marciniak, S.J. Endoplasmic reticulum dysfunction in neurological disease. Lancet Neurol. 2013, 12, 105–118. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Shao, H.; Liu, W.; Gu, W.; Shu, X.; Mo, Y.; Chen, X.; Zhang, Q.; Jiang, M. Endoplasmic reticulum stress and oxidative stress are involved in ZnO nanoparticle-induced hepatotoxicity. Toxicol. Lett. 2015, 234, 40–49. [Google Scholar] [CrossRef] [Green Version]
- Yu, K.-N.; Chang, S.-H.; Park, S.J.; Lim, J.; Lee, J.; Yoon, T.-J.; Kim, J.-S.; Cho, M.-H. Titanium dioxide nanoparticles induce endoplasmic reticulum stress-mediated autophagic cell death via mitochondria-associated endoplasmic reticulum membrane disruption in normal lung cells. PLoS ONE 2015, 10, e0131208. [Google Scholar] [CrossRef] [Green Version]
- Simard, J.-C.; Durocher, I.; Girard, D. Silver nanoparticles induce irremediable endoplasmic reticulum stress leading to unfolded protein response dependent apoptosis in breast cancer cells. Apoptosis 2016, 21, 1279–1290. [Google Scholar] [CrossRef]
- Christen, V.; Fent, K. Silica nanoparticles induce endoplasmic reticulum stress response and activate mitogen activated kinase (MAPK) signalling. Toxicol. Rep. 2016, 3, 832–840. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Zhang, Y.; Wang, Z.; Liu, R.; Gong, X. Bufalin induces the interplay between apoptosis and autophagy in glioma cells through endoplasmic reticulum stress. Int. J. Biol. Sci. 2014, 10, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niizuma, H.; Nakamura, Y.; Ozaki, T.; Nakanishi, H.; Ohira, M.; Isogai, E.; Kageyama, H.; Imaizumi, M.; Nakagawara, A. Bcl-2 is a key regulator for the retinoic acid-induced apoptotic cell death in neuroblastoma. Oncogene 2006, 25, 5046. [Google Scholar] [CrossRef] [Green Version]
- Giorgi, C.; Bonora, M.; Sorrentino, G.; Missiroli, S.; Poletti, F.; Suski, J.M.; Ramirez, F.G.; Rizzuto, R.; Di Virgilio, F.; Zito, E. p53 at the endoplasmic reticulum regulates apoptosis in a Ca2+-dependent manner. Proc. Natl. Acad. Sci. USA 2015, 112, 1779–1784. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Wan, L.; Lu, H.; Li, X. High expression of active ATF6 aggravates endoplasmic reticulum stress-induced vascular endothelial cell apoptosis through the mitochondrial apoptotic pathway. Mol. Med. Rep. 2018, 17, 6483–6489. [Google Scholar] [CrossRef]
- Gurunathan, S.; Han, J.W.; Eppakayala, V.; Jeyaraj, M.; Kim, J.H. Cytotoxicity of biologically synthesized silver nanoparticles in MDA-MB-231 human breast cancer cells. Biomed. Res. Int. 2013, 2013, 535796. [Google Scholar] [CrossRef] [Green Version]
- Gurunathan, S.; Han, J.W.; Park, J.H.; Kim, E.; Choi, Y.J.; Kwon, D.N.; Kim, J.H. Reduced graphene oxide-silver nanoparticle nanocomposite: A potential anticancer nanotherapy. Int. J. Nanomed. 2015, 10, 6257–6276. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.-G.; Wang, Y.-H.; Xing, H.-H.; Gurunathan, S. Quercetin-mediated synthesis of graphene oxide-silver nanoparticle nanocomposites: A suitable alternative nanotherapy for neuroblastoma. Int. J. Nanomed. 2017, 12, 5819–5839. [Google Scholar] [CrossRef] [Green Version]
- Nakabeppu, Y. Cellular levels of 8-oxoguanine in either DNA or the nucleotide pool play pivotal roles in carcinogenesis and survival of cancer cells. Int. J. Mol. Sci. 2014, 15, 12543–12557. [Google Scholar] [CrossRef] [Green Version]
- Guglielmo, C.D.; Lapuente, J.D.; Porredon, C.; Ramos-López, D.; Sendra, J.; Borràs, M. In vitro safety toxicology data for evaluation of gold nanoparticles–chronic cytotoxicity, genotoxicity and uptake. J. Nanosci. Nanotechnol. 2012, 12, 6185–6191. [Google Scholar] [CrossRef] [PubMed]
- Meena, R.; Rani, M.; Pal, R.; Rajamani, P. Nano-TiO 2-induced apoptosis by oxidative stress-mediated DNA damage and activation of p53 in human embryonic kidney cells. Appl. Biochem. Biotechnol. 2012, 167, 791–808. [Google Scholar] [CrossRef] [PubMed]
- Guan, R.; Kang, T.; Lu, F.; Zhang, Z.; Shen, H.; Liu, M. Cytotoxicity, oxidative stress, and genotoxicity in human hepatocyte and embryonic kidney cells exposed to ZnO nanoparticles. Nanoscale Res. Lett. 2012, 7, 602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, C.; Tao, G.; Yang, L.; Liu, J.; He, H.; Zhuang, Z. The role of reactive oxygen species in silicon dioxide nanoparticle-induced cytotoxicity and DNA damage in HaCaT cells. Mol. Biol. Rep. 2012, 39, 4915–4925. [Google Scholar] [CrossRef] [PubMed]
- Ammer, H.; Schulz, R. Retinoic acid-induced differentiation of human neuroblastoma SH-SY5Y cells is associated with changes in the abundance of G proteins. J. Neurochem. 1994, 62, 1310–1318. [Google Scholar] [CrossRef]
- Encinas, M.; Iglesias, M.; Liu, Y.; Wang, H.; Muhaisen, A.; Cena, V.; Gallego, C.; Comella, J.X. Sequential treatment of SH-SY5Y cells with retinoic acid and brain-derived neurotrophic factor gives rise to fully differentiated, neurotrophic factor-dependent, human neuron-like cells. J. Neurochem. 2000, 75, 991–1003. [Google Scholar] [CrossRef]
- Teppola, H.; Sarkanen, J.-R.; Jalonen, T.O.; Linne, M.-L. Morphological differentiation towards neuronal phenotype of SH-SY5Y neuroblastoma cells by estradiol, retinoic acid and cholesterol. Neurochem. Res. 2016, 41, 731–747. [Google Scholar]
- Dayem, A.A.; Kim, B.; Gurunathan, S.; Choi, H.Y.; Yang, G.; Saha, S.K.; Han, D.; Han, J.; Kim, K.; Kim, J.H.; et al. Biologically synthesized silver nanoparticles induce neuronal differentiation of SH-SY5Y cells via modulation of reactive oxygen species, phosphatases, and kinase signaling pathways. Biotechnol. J. 2014, 9, 934–943. [Google Scholar] [CrossRef]
- Han, J.W.; Gurunathan, S.; Jeong, J.-K.; Choi, Y.-J.; Kwon, D.-N.; Park, J.-K.; Kim, J.-H. Oxidative stress mediated cytotoxicity of biologically synthesized silver nanoparticles in human lung epithelial adenocarcinoma cell line. Nanoscale Res. Lett. 2014, 9, 459. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Li, Z.; Xu, X.; Chen, C.; Wei, W.; Fan, M.; Chen, X.; Li, J.J.; Wang, Y.; Huang, J. All-trans retinoic acids induce differentiation and sensitize a radioresistant breast cancer cells to chemotherapy. BMC Complementary Altern. Med. 2016, 16, 113. [Google Scholar] [CrossRef] [Green Version]
- Park, S.Y.; Park, J.; Sim, S.H.; Sung, M.G.; Kim, K.S.; Hong, B.H.; Hong, S. Enhanced differentiation of human neural stem cells into neurons on graphene. Adv. Mater. (Deerfield BeachFla.) 2011, 23, H263–H267. [Google Scholar] [CrossRef]
- Han, J.W.; Gurunathan, S.; Choi, Y.-J.; Kim, J.-H. Dual functions of silver nanoparticles in F9 teratocarcinoma stem cells, a suitable model for evaluating cytotoxicity- and differentiation-mediated cancer therapy. Int. J. Nanomed. 2017, 12, 7529–7549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Guan, D.X.; Shi, J.; Gao, H.; Li, J.J.; Zhao, J.S.; Qiu, L.; Liu, J.; Li, N.; Guo, W.X.; et al. All-trans retinoic acid potentiates the chemotherapeutic effect of cisplatin by inducing differentiation of tumor initiating cells in liver cancer. J. Hepatol. 2013, 59, 1255–1263. [Google Scholar] [CrossRef]
- Erlejman, A.G.; Oteiza, P.I. The oxidant defense system in human neuroblastoma IMR-32 cells predifferentiation and postdifferentiation to neuronal phenotypes. Neurochem. Res. 2002, 27, 1499–1506. [Google Scholar] [CrossRef] [PubMed]
- Yanes, O.; Clark, J.; Wong, D.M.; Patti, G.J.; Sanchez-Ruiz, A.; Benton, H.P.; Trauger, S.A.; Desponts, C.; Ding, S.; Siuzdak, G. Metabolic oxidation regulates embryonic stem cell differentiation. Nat. Chem. Biol. 2010, 6, 411–417. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Khvorostov, I.; Hong, J.S.; Oktay, Y.; Vergnes, L.; Nuebel, E.; Wahjudi, P.N.; Setoguchi, K.; Wang, G.; Do, A.; et al. UCP2 regulates energy metabolism and differentiation potential of human pluripotent stem cells. EMBO J. 2011, 30, 4860–4873. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, M.; Miyoshi, M.; Sakai, S.; Nishiokada, A.; Aoyama-Ishikawa, M.; Maeshige, N.; Usami, Y.; Hamada, Y.; Takahashi, M.; Usami, M. Lard-based high-fat diet increases secretory leukocyte protease inhibitor expression and attenuates the inflammatory response of acute lung injury in endotoxemic rats. Clin. Nutr. 2015, 34, 997–1009. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.J.; Gurunathan, S.; Kim, J.H. Graphene Oxide-Silver Nanocomposite Enhances Cytotoxic and Apoptotic Potential of Salinomycin in Human Ovarian Cancer Stem Cells (OvCSCs): A Novel Approach for Cancer Therapy. Int. J. Mol. Sci. 2018, 19, 710. [Google Scholar] [CrossRef] [PubMed] [Green Version]













© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gurunathan, S.; Jeyaraj, M.; Kang, M.-H.; Kim, J.-H. Anticancer Properties of Platinum Nanoparticles and Retinoic Acid: Combination Therapy for the Treatment of Human Neuroblastoma Cancer. Int. J. Mol. Sci. 2020, 21, 6792. https://doi.org/10.3390/ijms21186792
Gurunathan S, Jeyaraj M, Kang M-H, Kim J-H. Anticancer Properties of Platinum Nanoparticles and Retinoic Acid: Combination Therapy for the Treatment of Human Neuroblastoma Cancer. International Journal of Molecular Sciences. 2020; 21(18):6792. https://doi.org/10.3390/ijms21186792
Chicago/Turabian StyleGurunathan, Sangiliyandi, Muniyandi Jeyaraj, Min-Hee Kang, and Jin-Hoi Kim. 2020. "Anticancer Properties of Platinum Nanoparticles and Retinoic Acid: Combination Therapy for the Treatment of Human Neuroblastoma Cancer" International Journal of Molecular Sciences 21, no. 18: 6792. https://doi.org/10.3390/ijms21186792
APA StyleGurunathan, S., Jeyaraj, M., Kang, M. -H., & Kim, J. -H. (2020). Anticancer Properties of Platinum Nanoparticles and Retinoic Acid: Combination Therapy for the Treatment of Human Neuroblastoma Cancer. International Journal of Molecular Sciences, 21(18), 6792. https://doi.org/10.3390/ijms21186792