The Macrophages-Microbiota Interplay in Colorectal Cancer (CRC)-Related Inflammation: Prognostic and Therapeutic Significance



Abstract
:1. Introduction
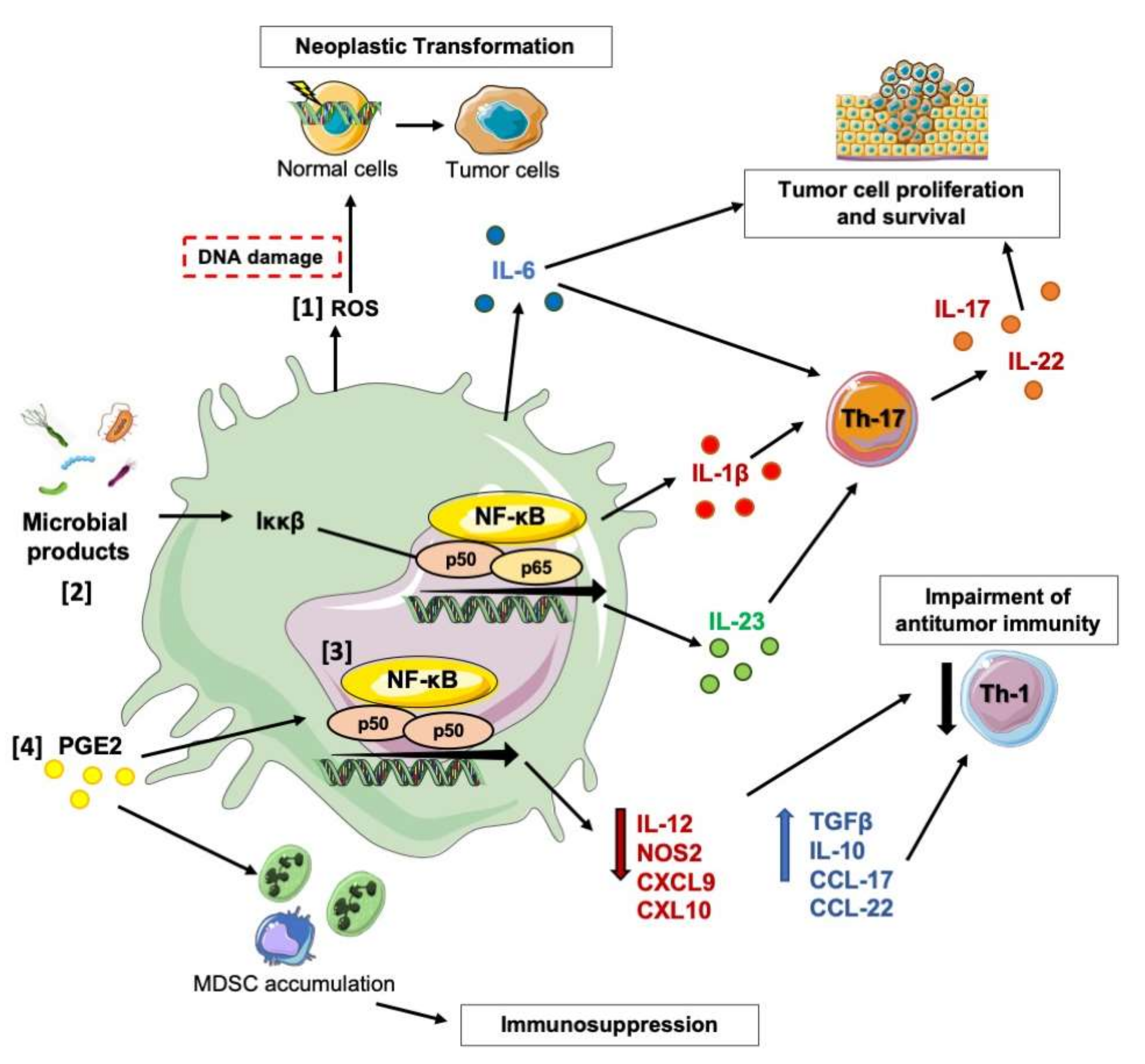
2. Molecular Pathways Underpinning the Pro-Tumor Activity of Macrophages in CRC
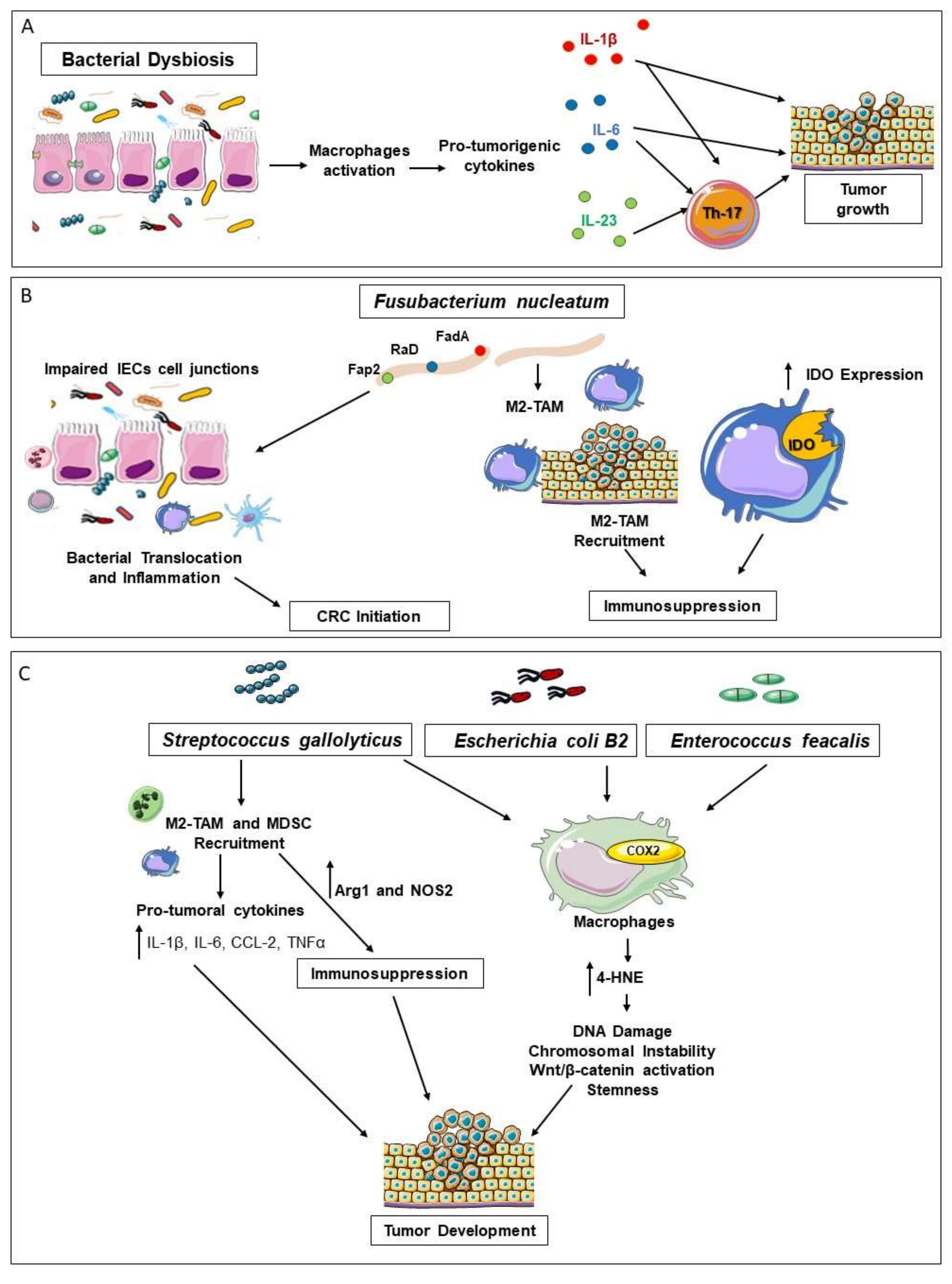
3. Cross-Talk between Gut Microbiota and Macrophages in CRC Development
4. The Interplay between Dietary Habits, Intestinal Microbiota, and Macrophages in CRC
5. Macrophages as Prognostic and Predictive Biomarker in Human CRC
6. The Interplay of Macrophages and Microbiota in Conventional Anti-Cancer Therapies
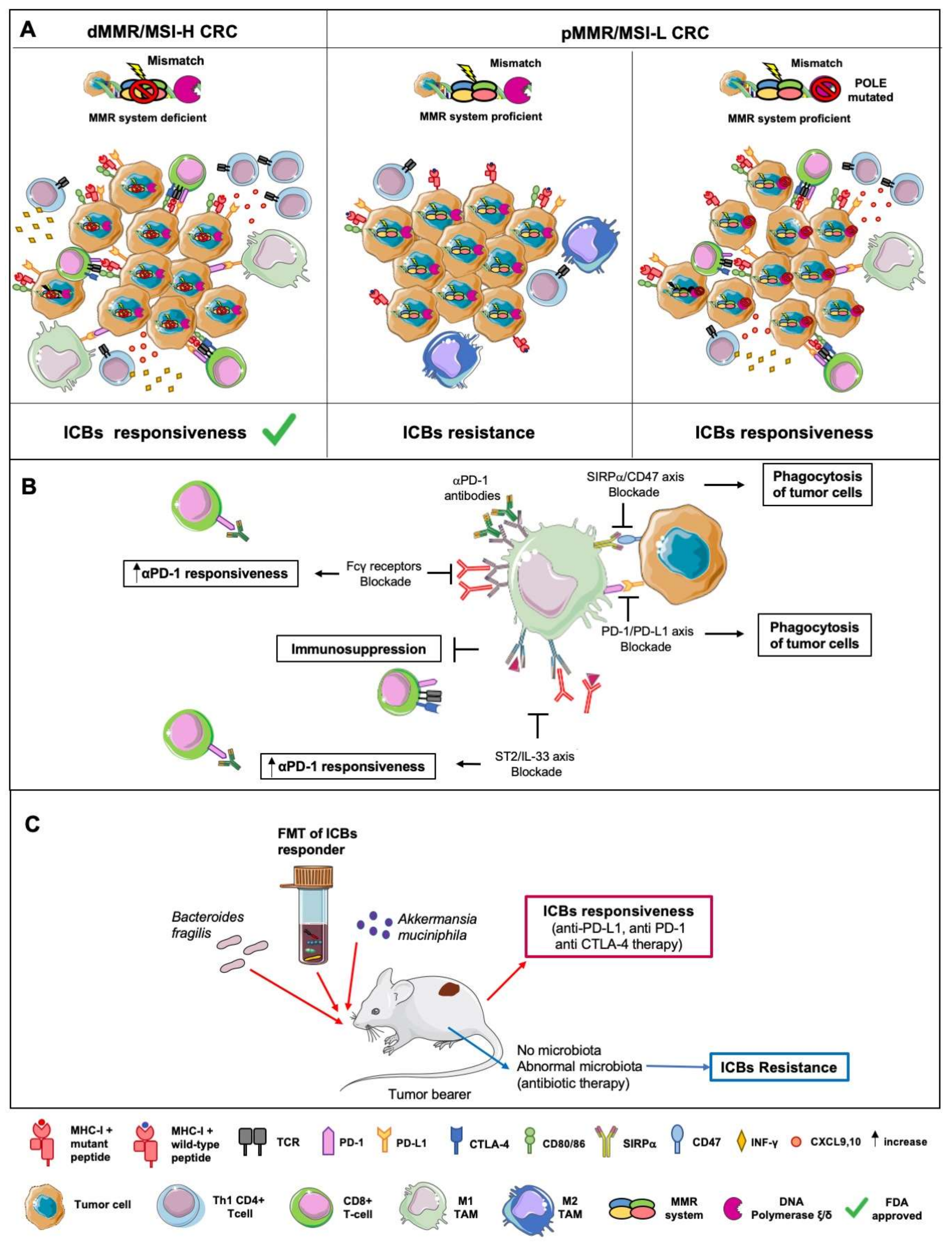
7. The Interplay of Macrophages and Microbiota in ICBs-Based Immunotherapy
8. Conclusions and Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 4-HNE | 4-hydroxy-2-nonenal |
| 5-FU | 5-fluorouracil |
| Arg1 | Arginase 1 |
| CAC | Colitis-associated cancer |
| CCL | Chemokine (C-C motif) ligand |
| CCR | Chemokine (C-C motif) receptor |
| CD | Cluster of Differentiation |
| COX-2 | Cyclooxygenase-2 |
| Cpg-ODN | CpG-oligodeoxynucleotide |
| CRC | Colorectal cancer |
| CSF1R | Colony-stimulating factor 1 receptor |
| CTLA- A | Cytotoxic T-lymphocyte-associated protein 4 |
| CXCL | Chemokine (C-X-C motif) ligand |
| DCLK1 | Doublecortin-like kinase 1 |
| DCs | Dendritic cells |
| dMMR | DNA mismatch-repair |
| DSS | Dextran sulfate sodium |
| EMT | Epithelial-mesenchymal transition |
| EP | Prostaglandin E receptor |
| FadA | Fusubacterium adhesin A |
| Fap | Fatty-acid-binding protein 2 |
| FMT | Faecal microbiota transplantation |
| HLA | Human leukocyte antigen |
| HLA-DR | Major histocompatibility complex, class II, DR |
| IBD | Inflammatory bowel disease |
| ICBs | Immune checkpoint blockers |
| ICD | Immunogenic cell death |
| IDO | Indoleamina 2,3-diossigenasi |
| IFN | Interferon |
| IKK | Nuclear factor NF-kappa-B inhibitor kinase |
| IL | Interleukin |
| IL1R | Interleukin 1 receptor |
| LPS | Lipopolysaccharide |
| MDSCs | Myeloid-derived suppressor cells |
| M-MDSCs | Monocytic myeloid-derived suppressor cells |
| MSI-H | Microsatellite instability-high |
| MSI-L | Microsatellite instability-low |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NK | Natural killer cells |
| NO | Nitric oxide |
| NOS2 | Nitric oxide synthase 2 |
| NOX 2 | NADPH oxidase 2 |
| NSAIDs | Nonsteroidal anti-inflammatory drugs |
| OMVs | Outer membrane vesicles |
| PD-1 | Programmed-death protein 1 |
| PD-L1 | PD-1 ligand |
| PGE2 | Prostaglandin E2 |
| POL | DNA polymerase |
| Rad DRNS | Arginine-inhibitable adhesinReactive nitrogen species |
| ROSSCFA | Reactive oxygen speciesShort-chain fatty acids |
| scRNA-Seq | Single cell RNA-Seq |
| Sirpα | Signal-regulatory protein α |
| Stat3 | Transducer and activator of transcription 3 |
| TAMs | Tumor-associated macrophages |
| TGF | Transforming growth factor |
| Th | T helper |
| TLR | Toll-like receptor |
| TMB | Tumor mutational burden |
| TME | Tumor microenvironment |
| TNF | Tumor necrosis factor |
| TRMs | Tissue-resident macrophages |
References
- Mantovani, A.; Sica, A. Macrophages, innate immunity and cancer: Balance, tolerance, and diversity. Curr. Opin. Immunol. 2010, 22, 231–237. [Google Scholar] [CrossRef]
- Galon, J.; Mlecnik, B.; Bindea, G.; Angell, H.K.; Berger, A.; Lagorce, C.; Lugli, A.; Zlobec, I.; Hartmann, A.; Bifulco, C.; et al. Towards the introduction of the “Immunoscore” in the classification of malignant tumours. J. Pathol. 2014, 232, 199–209. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Pagès, F.; Mlecnik, B.; Marliot, F.; Bindea, G.; Ou, F.-S.; Bifulco, C.; Lugli, A.; Zlobec, I.; Rau, T.T.; Berger, M.D.; et al. International validation of the consensus Immunoscore for the classification of colon cancer: A prognostic and accuracy study. The Lancet 2018, 391, 2128–2139. [Google Scholar] [CrossRef]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Qian, B.-Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Sica, A.; Locati, M. Macrophage polarization comes of age. Immunity 2005, 23, 344–346. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S. Alternative activation of macrophages. Nat. Rev. Immunol. 2003, 3, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Invest. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Sica, A.; Erreni, M.; Allavena, P.; Porta, C. Macrophage polarization in pathology. Cell. Mol. Life Sci. 2015, 72, 4111–4126. [Google Scholar] [CrossRef]
- Ginhoux, F.; Guilliams, M. Tissue-Resident Macrophage Ontogeny and Homeostasis. Immunity 2016, 44, 439–449. [Google Scholar] [CrossRef]
- Perdiguero, E.G.; Geissmann, F. The development and maintenance of resident macrophages. Nat. Immunol. 2016, 17, 2–8. [Google Scholar] [CrossRef] [Green Version]
- Bian, Z.; Gong, Y.; Huang, T.; Lee, C.Z.W.; Bian, L.; Bai, Z.; Shi, H.; Zeng, Y.; Liu, C.; He, J.; et al. Deciphering human macrophage development at single-cell resolution. Nature 2020, 582, 571–576. [Google Scholar] [CrossRef]
- De Schepper, S.; Verheijden, S.; Aguilera-Lizarraga, J.; Viola, M.F.; Boesmans, W.; Stakenborg, N.; Voytyuk, I.; Schmidt, I.; Boeckx, B.; Dierckx de Casterlé, I.; et al. Self-Maintaining Gut Macrophages Are Essential for Intestinal Homeostasis. Cell 2018, 175, 400–415e13. [Google Scholar] [CrossRef] [Green Version]
- Shaw, T.N.; Houston, S.A.; Wemyss, K.; Bridgeman, H.M.; Barbera, T.A.; Zangerle-Murray, T.; Strangward, P.; Ridley, A.J.L.; Wang, P.; Tamoutounour, S.; et al. Tissue-resident macrophages in the intestine are long lived and defined by Tim-4 and CD4 expression. J. Exp. Med. 2018, 215, 1507–1518. [Google Scholar] [CrossRef]
- Bujko, A.; Atlasy, N.; Landsverk, O.J.B.; Richter, L.; Yaqub, S.; Horneland, R.; Øyen, O.; Aandahl, E.M.; Aabakken, L.; Stunnenberg, H.G.; et al. Transcriptional and functional profiling defines human small intestinal macrophage subsets. J. Exp. Med. 2018, 215, 441–458. [Google Scholar] [CrossRef] [Green Version]
- Bain, C.C.; Bravo-Blas, A.; Scott, C.L.; Perdiguero, E.G.; Geissmann, F.; Henri, S.; Malissen, B.; Osborne, L.C.; Artis, D.; Mowat, A.M. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat. Immunol. 2014, 15, 929–937. [Google Scholar] [CrossRef] [Green Version]
- Bernardo, D.; Marin, A.C.; Fernández-Tomé, S.; Montalban-Arques, A.; Carrasco, A.; Tristán, E.; Ortega-Moreno, L.; Mora-Gutiérrez, I.; Díaz-Guerra, A.; Caminero-Fernández, R.; et al. Human intestinal pro-inflammatory CD11chighCCR2+CX3CR1+ macrophages, but not their tolerogenic CD11c-CCR2-CX3CR1- counterparts, are expanded in inflammatory bowel disease. Mucosal Immunol 2018, 11, 1114–1126. [Google Scholar] [CrossRef]
- Lavin, Y.; Winter, D.; Blecher-Gonen, R.; David, E.; Keren-Shaul, H.; Merad, M.; Jung, S.; Amit, I. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 2014, 159, 1312–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, S.; Plüddemann, A. Tissue macrophages: Heterogeneity and functions. BMC Biol. 2017, 15, 53. [Google Scholar] [CrossRef] [PubMed]
- Locati, M.; Curtale, G.; Mantovani, A. Diversity, Mechanisms, and Significance of Macrophage Plasticity. Annu. Rev. Pathol. 2020, 15, 123–147. [Google Scholar] [CrossRef] [Green Version]
- Bowman, R.L.; Klemm, F.; Akkari, L.; Pyonteck, S.M.; Sevenich, L.; Quail, D.F.; Dhara, S.; Simpson, K.; Gardner, E.E.; Iacobuzio-Donahue, C.A.; et al. Macrophage Ontogeny Underlies Differences in Tumor-Specific Education in Brain Malignancies. Cell Rep. 2016, 17, 2445–2459. [Google Scholar] [CrossRef] [Green Version]
- Loyher, P.-L.; Hamon, P.; Laviron, M.; Meghraoui-Kheddar, A.; Goncalves, E.; Deng, Z.; Torstensson, S.; Bercovici, N.; Baudesson de Chanville, C.; Combadière, B.; et al. Macrophages of distinct origins contribute to tumor development in the lung. J. Exp. Med. 2018, 215, 2536–2553. [Google Scholar] [CrossRef]
- Gubin, M.M.; Esaulova, E.; Ward, J.P.; Malkova, O.N.; Runci, D.; Wong, P.; Noguchi, T.; Arthur, C.D.; Meng, W.; Alspach, E.; et al. High-Dimensional Analysis Delineates Myeloid and Lymphoid Compartment Remodeling during Successful Immune-Checkpoint Cancer Therapy. Cell 2018, 175, 1014–1030e19. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Herndon, J.M.; Sojka, D.K.; Kim, K.-W.; Knolhoff, B.L.; Zuo, C.; Cullinan, D.R.; Luo, J.; Bearden, A.R.; Lavine, K.J.; et al. Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity 2017, 47, 323–338e6. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin Oncol 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Pathria, P.; Louis, T.L.; Varner, J.A. Targeting Tumor-Associated Macrophages in Cancer. Trends Immunol. 2019, 40, 310–327. [Google Scholar] [CrossRef]
- Ruffell, B.; Coussens, L.M. Macrophages and therapeutic resistance in cancer. Cancer Cell 2015, 27, 462–472. [Google Scholar] [CrossRef] [Green Version]
- Coffelt, S.B.; de Visser, K.E. Immune-mediated mechanisms influencing the efficacy of anticancer therapies. Trends Immunol. 2015, 36, 198–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassetta, L.; Pollard, J.W. Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug Discov 2018, 17, 887–904. [Google Scholar] [CrossRef] [PubMed]
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, J.; Chehrazi-Raffle, A.; Reddi, S.; Salgia, R. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: A comprehensive review of registration trials and future considerations. J. Immunother. Cancer 2018, 6, 8. [Google Scholar] [CrossRef]
- Havel, J.J.; Chowell, D.; Chan, T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef]
- Kalbasi, A.; Ribas, A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat. Rev. Immunol. 2020, 20, 25–39. [Google Scholar] [CrossRef]
- Chan, T.A.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.A.; Stenzinger, A.; Peters, S. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. 2019, 30, 44–56. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Neubert, N.J.; Schmittnaegel, M.; Bordry, N.; Nassiri, S.; Wald, N.; Martignier, C.; Tillé, L.; Homicsko, K.; Damsky, W.; Maby-El Hajjami, H.; et al. T cell-induced CSF1 promotes melanoma resistance to PD1 blockade. Sci Transl Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arlauckas, S.P.; Garris, C.S.; Kohler, R.H.; Kitaoka, M.; Cuccarese, M.F.; Yang, K.S.; Miller, M.A.; Carlson, J.C.; Freeman, G.J.; Anthony, R.M.; et al. In vivo imaging reveals a tumor-associated macrophage-mediated resistance pathway in anti-PD-1 therapy. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDermott, D.F.; Huseni, M.A.; Atkins, M.B.; Motzer, R.J.; Rini, B.I.; Escudier, B.; Fong, L.; Joseph, R.W.; Pal, S.K.; Reeves, J.A.; et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat. Med. 2018, 24, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Knolhoff, B.L.; Meyer, M.A.; Nywening, T.M.; West, B.L.; Luo, J.; Wang-Gillam, A.; Goedegebuure, S.P.; Linehan, D.C.; DeNardo, D.G. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014, 74, 5057–5069. [Google Scholar] [CrossRef] [Green Version]
- Mok, S.; Koya, R.C.; Tsui, C.; Xu, J.; Robert, L.; Wu, L.; Graeber, T.; West, B.L.; Bollag, G.; Ribas, A. Inhibition of CSF-1 receptor improves the antitumor efficacy of adoptive cell transfer immunotherapy. Cancer Res. 2014, 74, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Helmink, B.A.; Khan, M.A.W.; Hermann, A.; Gopalakrishnan, V.; Wargo, J.A. The microbiome, cancer, and cancer therapy. Nat. Med. 2019, 25, 377–388. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [Green Version]
- Van der Jeught, K.; Xu, H.-C.; Li, Y.-J.; Lu, X.-B.; Ji, G. Drug resistance and new therapies in colorectal cancer. World J. Gastroenterol. 2018, 24, 3834–3848. [Google Scholar] [CrossRef]
- Keum, N.; Giovannucci, E. Global burden of colorectal cancer: Emerging trends, risk factors and prevention strategies. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 713–732. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.B.; Harpaz, N.; Itzkowitz, S.; Hossain, S.; Matula, S.; Kornbluth, A.; Bodian, C.; Ullman, T. Histologic inflammation is a risk factor for progression to colorectal neoplasia in ulcerative colitis: A cohort study. Gastroenterology 2007, 133, 1099–1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, A.-M.; Cross, W.; Curtius, K.; Al Bakir, I.; Choi, C.-H.R.; Davis, H.L.; Temko, D.; Biswas, S.; Martinez, P.; Williams, M.J.; et al. Evolutionary history of human colitis-associated colorectal cancer. Gut 2019, 68, 985–995. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Zhou, J.-F.; Sellers, R.S.; Li, J.-F.; Nguyen, A.V.; Wang, Y.; Orlofsky, A.; Liu, Q.; Hume, D.A.; Pollard, J.W.; et al. A novel mouse model of inflammatory bowel disease links mammalian target of rapamycin-dependent hyperproliferation of colonic epithelium to inflammation-associated tumorigenesis. Am. J. Pathol. 2010, 176, 952–967. [Google Scholar] [CrossRef]
- Zhen, Y.; Luo, C.; Zhang, H. Early detection of ulcerative colitis-associated colorectal cancer. Gastroenterol. Rep. (Oxf) 2018, 6, 83–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerber, R.A.; Neklason, D.W.; Samowitz, W.S.; Burt, R.W. Frequency of familial colon cancer and hereditary nonpolyposis colorectal cancer (Lynch syndrome) in a large population database. Fam. Cancer 2005, 4, 239–244. [Google Scholar] [CrossRef]
- Stoffel, E.M.; Kastrinos, F. Familial colorectal cancer, beyond Lynch syndrome. Clin. Gastroenterol. Hepatol. 2014, 12, 1059–1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jasperson, K.W.; Tuohy, T.M.; Neklason, D.W.; Burt, R.W. Hereditary and familial colon cancer. Gastroenterology 2010, 138, 2044–2058. [Google Scholar] [CrossRef] [Green Version]
- Narisawa, T.; Sato, M.; Tani, M.; Kudo, T.; Takahashi, T.; Goto, A. Inhibition of development of methylnitrosourea-induced rat colon tumors by indomethacin treatment. Cancer Res. 1981, 41, 1954–1957. [Google Scholar]
- Popivanova, B.K.; Kostadinova, F.I.; Furuichi, K.; Shamekh, M.M.; Kondo, T.; Wada, T.; Egashira, K.; Mukaida, N. Blockade of a chemokine, CCL2, reduces chronic colitis-associated carcinogenesis in mice. Cancer Res. 2009, 69, 7884–7892. [Google Scholar] [CrossRef] [Green Version]
- McClellan, J.L.; Davis, J.M.; Steiner, J.L.; Enos, R.T.; Jung, S.H.; Carson, J.A.; Pena, M.M.; Carnevale, K.A.; Berger, F.G.; Murphy, E.A. Linking tumor-associated macrophages, inflammation, and intestinal tumorigenesis: Role of MCP-1. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G1087–G1095. [Google Scholar] [CrossRef] [Green Version]
- Terzić, J.; Grivennikov, S.; Karin, E.; Karin, M. Inflammation and colon cancer. Gastroenterology 2010, 138, 2101–2114e5. [Google Scholar] [CrossRef] [PubMed]
- Canli, Ö.; Nicolas, A.M.; Gupta, J.; Finkelmeier, F.; Goncharova, O.; Pesic, M.; Neumann, T.; Horst, D.; Löwer, M.; Sahin, U.; et al. Myeloid Cell-Derived Reactive Oxygen Species Induce Epithelial Mutagenesis. Cancer Cell 2017, 32, 869–883e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karin, M.; Greten, F.R. NF-kappaB: Linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 2005, 5, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Eckmann, L.; Greten, T.F.; Park, J.M.; Li, Z.-W.; Egan, L.J.; Kagnoff, M.F.; Karin, M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004, 118, 285–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwitalla, S.; Ziegler, P.K.; Horst, D.; Becker, V.; Kerle, I.; Begus-Nahrmann, Y.; Lechel, A.; Rudolph, K.L.; Langer, R.; Slotta-Huspenina, J.; et al. Loss of p53 in enterocytes generates an inflammatory microenvironment enabling invasion and lymph node metastasis of carcinogen-induced colorectal tumors. Cancer Cell 2013, 23, 93–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porta, C.; Rimoldi, M.; Raes, G.; Brys, L.; Ghezzi, P.; Di Liberto, D.; Dieli, F.; Ghisletti, S.; Natoli, G.; De Baetselier, P.; et al. Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor kappaB. Proc. Natl. Acad. Sci. USA 2009, 106, 14978–14983. [Google Scholar] [CrossRef] [Green Version]
- Saccani, A.; Schioppa, T.; Porta, C.; Biswas, S.K.; Nebuloni, M.; Vago, L.; Bottazzi, B.; Colombo, M.P.; Mantovani, A.; Sica, A. p50 nuclear factor-kappaB overexpression in tumor-associated macrophages inhibits M1 inflammatory responses and antitumor resistance. Cancer Res. 2006, 66, 11432–11440. [Google Scholar] [CrossRef] [Green Version]
- Hagemann, T.; Lawrence, T.; McNeish, I.; Charles, K.A.; Kulbe, H.; Thompson, R.G.; Robinson, S.C.; Balkwill, F.R. “Re-educating” tumor-associated macrophages by targeting NF-kappaB. J. Exp. Med. 2008, 205, 1261–1268. [Google Scholar] [CrossRef]
- Grivennikov, S.; Karin, E.; Terzic, J.; Mucida, D.; Yu, G.-Y.; Vallabhapurapu, S.; Scheller, J.; Rose-John, S.; Cheroutre, H.; Eckmann, L.; et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 2009, 15, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Bollrath, J.; Phesse, T.J.; von Burstin, V.A.; Putoczki, T.; Bennecke, M.; Bateman, T.; Nebelsiek, T.; Lundgren-May, T.; Canli, O.; Schwitalla, S.; et al. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell 2009, 15, 91–102. [Google Scholar] [CrossRef] [Green Version]
- Porta, C.; Ippolito, A.; Consonni, F.M.; Carraro, L.; Celesti, G.; Correale, C.; Grizzi, F.; Pasqualini, F.; Tartari, S.; Rinaldi, M.; et al. Protumor Steering of Cancer Inflammation by p50 NF-κB Enhances Colorectal Cancer Progression. Cancer Immunol. Res. 2018, 6, 578–593. [Google Scholar] [CrossRef] [Green Version]
- Göktuna, S.I.; Canli, O.; Bollrath, J.; Fingerle, A.A.; Horst, D.; Diamanti, M.A.; Pallangyo, C.; Bennecke, M.; Nebelsiek, T.; Mankan, A.K.; et al. IKKα promotes intestinal tumorigenesis by limiting recruitment of M1-like polarized myeloid cells. Cell Rep. 2014, 7, 1914–1925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuno, R.; Kawada, K.; Sakai, Y. Prostaglandin E2/EP Signaling in the Tumor Microenvironment of Colorectal Cancer. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.; Vacher, J.; Yao, B.; Fan, X.; Zhang, B.; Harris, R.C.; Zhang, M.-Z. Prostaglandin E receptor 4 (EP4) promotes colonic tumorigenesis. Oncotarget 2015, 6, 33500–33511. [Google Scholar] [CrossRef] [Green Version]
- Sinha, P.; Clements, V.K.; Fulton, A.M.; Ostrand-Rosenberg, S. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res. 2007, 67, 4507–4513. [Google Scholar] [CrossRef] [Green Version]
- Yan, G.; Zhao, H.; Zhang, Q.; Zhou, Y.; Wu, L.; Lei, J.; Wang, X.; Zhang, J.; Zhang, X.; Zheng, L.; et al. A RIPK3-PGE2 Circuit Mediates Myeloid-Derived Suppressor Cell-Potentiated Colorectal Carcinogenesis. Cancer Res. 2018, 78, 5586–5599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porta, C.; Consonni, F.M.; Morlacchi, S.; Sangaletti, S.; Bleve, A.; Totaro, M.G.; Larghi, P.; Rimoldi, M.; Tripodo, C.; Strauss, L.; et al. Tumor-derived prostaglandin E2 promotes p50 NF-κB-dependent differentiation of monocytic MDSC. Cancer Res. 2020. [CrossRef] [Green Version]
- Coccia, M.; Harrison, O.J.; Schiering, C.; Asquith, M.J.; Becher, B.; Powrie, F.; Maloy, K.J. IL-1β mediates chronic intestinal inflammation by promoting the accumulation of IL-17A secreting innate lymphoid cells and CD4(+) Th17 cells. J. Exp. Med. 2012, 209, 1595–1609. [Google Scholar] [CrossRef]
- Zhou, L.; Littman, D.R. Transcriptional regulatory networks in Th17 cell differentiation. Curr. Opin. Immunol. 2009, 21, 146–152. [Google Scholar] [CrossRef] [Green Version]
- Grivennikov, S.I.; Wang, K.; Mucida, D.; Stewart, C.A.; Schnabl, B.; Jauch, D.; Taniguchi, K.; Yu, G.-Y.; Osterreicher, C.H.; Hung, K.E.; et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 2012, 491, 254–258. [Google Scholar] [CrossRef] [Green Version]
- Teng, M.W.L.; Andrews, D.M.; McLaughlin, N.; von Scheidt, B.; Ngiow, S.F.; Möller, A.; Hill, G.R.; Iwakura, Y.; Oft, M.; Smyth, M.J. IL-23 suppresses innate immune response independently of IL-17A during carcinogenesis and metastasis. Proc. Natl. Acad. Sci. USA 2010, 107, 8328–8333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurtado, C.G.; Wan, F.; Housseau, F.; Sears, C.L. Roles for Interleukin 17 and Adaptive Immunity in Pathogenesis of Colorectal Cancer. Gastroenterology 2018, 155, 1706–1715. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Kim, M.K.; Di Caro, G.; Wong, J.; Shalapour, S.; Wan, J.; Zhang, W.; Zhong, Z.; Sanchez-Lopez, E.; Wu, L.-W.; et al. Interleukin-17 receptor a signaling in transformed enterocytes promotes early colorectal tumorigenesis. Immunity 2014, 41, 1052–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, W.-H.; Chen, H.-H.; Yen, S.-L.; Huang, H.-Y.; Hsiao, C.-C.; Chuang, J.-H. Increased expression of interleukin-23 associated with progression of colorectal cancer. J. Surg. Oncol. 2017, 115, 208–212. [Google Scholar] [CrossRef]
- Tosolini, M.; Kirilovsky, A.; Mlecnik, B.; Fredriksen, T.; Mauger, S.; Bindea, G.; Berger, A.; Bruneval, P.; Fridman, W.-H.; Pagès, F.; et al. Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in patients with colorectal cancer. Cancer Res. 2011, 71, 1263–1271. [Google Scholar] [CrossRef] [Green Version]
- Amicarella, F.; Muraro, M.G.; Hirt, C.; Cremonesi, E.; Padovan, E.; Mele, V.; Governa, V.; Han, J.; Huber, X.; Droeser, R.A.; et al. Dual role of tumour-infiltrating T helper 17 cells in human colorectal cancer. Gut 2017, 66, 692–704. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; MacFadyen, J.G.; Thuren, T.; Everett, B.M.; Libby, P.; Glynn, R.J. CANTOS Trial Group Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: Exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 2017, 390, 1833–1842. [Google Scholar] [CrossRef]
- Dmitrieva-Posocco, O.; Dzutsev, A.; Posocco, D.F.; Hou, V.; Yuan, W.; Thovarai, V.; Mufazalov, I.A.; Gunzer, M.; Shilovskiy, I.P.; Khaitov, M.R.; et al. Cell-Type-Specific Responses to Interleukin-1 Control Microbial Invasion and Tumor-Elicited Inflammation in Colorectal Cancer. Immunity 2019, 50, 166–180e7. [Google Scholar] [CrossRef] [Green Version]
- Nakatsu, G.; Li, X.; Zhou, H.; Sheng, J.; Wong, S.H.; Wu, W.K.K.; Ng, S.C.; Tsoi, H.; Dong, Y.; Zhang, N.; et al. Gut mucosal microbiome across stages of colorectal carcinogenesis. Nat. Commun. 2015, 6, 8727. [Google Scholar] [CrossRef]
- Lu, Y.; Chen, J.; Zheng, J.; Hu, G.; Wang, J.; Huang, C.; Lou, L.; Wang, X.; Zeng, Y. Mucosal adherent bacterial dysbiosis in patients with colorectal adenomas. Sci Rep. 2016, 6, 26337. [Google Scholar] [CrossRef]
- Gao, Z.; Guo, B.; Gao, R.; Zhu, Q.; Qin, H. Microbiota disbiosis is associated with colorectal cancer. Front. Microbiol. 2015, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.H.; Zhao, L.; Zhang, X.; Nakatsu, G.; Han, J.; Xu, W.; Xiao, X.; Kwong, T.N.Y.; Tsoi, H.; Wu, W.K.K.; et al. Gavage of Fecal Samples From Patients With Colorectal Cancer Promotes Intestinal Carcinogenesis in Germ-Free and Conventional Mice. Gastroenterology 2017, 153, 1621–1633e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakatsu, G.; Zhou, H.; Wu, W.K.K.; Wong, S.H.; Coker, O.O.; Dai, Z.; Li, X.; Szeto, C.-H.; Sugimura, N.; Lam, T.Y.-T.; et al. Alterations in Enteric Virome Are Associated With Colorectal Cancer and Survival Outcomes. Gastroenterology 2018, 155, 529–541e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emlet, C.; Ruffin, M.; Lamendella, R. Enteric Virome and Carcinogenesis in the Gut. Dig. Dis. Sci. 2020, 65, 852–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coker, O.O.; Nakatsu, G.; Dai, R.Z.; Wu, W.K.K.; Wong, S.H.; Ng, S.C.; Chan, F.K.L.; Sung, J.J.Y.; Yu, J. Enteric fungal microbiota dysbiosis and ecological alterations in colorectal cancer. Gut 2019, 68, 654–662. [Google Scholar] [CrossRef]
- Rubio, C.A.; Schmidt, P.T. Severe Defects in the Macrophage Barrier to Gut Microflora in Inflammatory Bowel Disease and Colon Cancer. Anticancer Res. 2018, 38, 3811–3815. [Google Scholar] [CrossRef] [Green Version]
- Nakata, K.; Yamamoto, M.; Inagawa, H.; Soma, G.-I. Effects of interactions between intestinal microbiota and intestinal macrophages on health. Anticancer Res. 2013, 33, 2849–2853. [Google Scholar]
- Keku, T.O.; Dulal, S.; Deveaux, A.; Jovov, B.; Han, X. The gastrointestinal microbiota and colorectal cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G351–G363. [Google Scholar] [CrossRef] [Green Version]
- Zackular, J.P.; Baxter, N.T.; Iverson, K.D.; Sadler, W.D.; Petrosino, J.F.; Chen, G.Y.; Schloss, P.D. The gut microbiome modulates colon tumorigenesis. mBio 2013, 4, e00692-13. [Google Scholar] [CrossRef] [Green Version]
- Cao, H.; Xu, M.; Dong, W.; Deng, B.; Wang, S.; Zhang, Y.; Wang, S.; Luo, S.; Wang, W.; Qi, Y.; et al. Secondary bile acid-induced dysbiosis promotes intestinal carcinogenesis. Int. J. Cancer 2017, 140, 2545–2556. [Google Scholar] [CrossRef] [Green Version]
- Wan, G.; Xie, M.; Yu, H.; Chen, H. Intestinal dysbacteriosis activates tumor-associated macrophages to promote epithelial-mesenchymal transition of colorectal cancer. Innate Immun. 2018, 24, 480–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Wang, X.; Huycke, T.; Moore, D.R.; Lightfoot, S.A.; Huycke, M.M. Colon Macrophages Polarized by Commensal Bacteria Cause Colitis and Cancer through the Bystander Effect. Transl. Oncol 2013, 6, 596–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bader, J.E.; Enos, R.T.; Velázquez, K.T.; Carson, M.S.; Nagarkatti, M.; Nagarkatti, P.S.; Chatzistamou, I.; Davis, J.M.; Carson, J.A.; Robinson, C.M.; et al. Macrophage depletion using clodronate liposomes decreases tumorigenesis and alters gut microbiota in the AOM/DSS mouse model of colon cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 314, G22–G31. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Li, H.; Xie, H.; Wu, X.; Lan, P. The malignant role of exosomes in the communication among colorectal cancer cell, macrophage and microbiome. Carcinogenesis 2019, 40, 601–610. [Google Scholar] [CrossRef]
- Gao, X.-J.; Li, T.; Wei, B.; Yan, Z.-X.; Hu, N.; Huang, Y.-J.; Han, B.-L.; Wai, T.-S.; Yang, W.; Yan, R. Bacterial Outer Membrane Vesicles from Dextran Sulfate Sodium-Induced Colitis Differentially Regulate Intestinal UDP-Glucuronosyltransferase 1A1 Partially Through Toll-Like Receptor 4/Mitogen-Activated Protein Kinase/Phosphatidylinositol 3-Kinase Pathway. Drug Metab. Dispos. 2018, 46, 292–302. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Li, Q.; Fu, X. Fusobacterium nucleatum Contributes to the Carcinogenesis of Colorectal Cancer by Inducing Inflammation and Suppressing Host Immunity. Transl. Oncol. 2019, 12, 846–851. [Google Scholar] [CrossRef]
- Kostic, A.D.; Chun, E.; Robertson, L.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.L.; et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 2013, 14, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Li, Q.; Wu, J.; Wu, Y.; Peng, W.; Li, H.; Wang, J.; Tang, X.; Peng, Y.; Fu, X. Fusobacterium nucleatum promotes M2 polarization of macrophages in the microenvironment of colorectal tumours via a TLR4-dependent mechanism. Cancer Immunol. Immunother. 2018, 67, 1635–1646. [Google Scholar] [CrossRef]
- Lucas, C.; Barnich, N.; Nguyen, H.T.T. Microbiota, Inflammation and Colorectal Cancer. Int J. Mol. Sci 2017, 18. [Google Scholar] [CrossRef] [Green Version]
- Cuevas-Ramos, G.; Petit, C.R.; Marcq, I.; Boury, M.; Oswald, E.; Nougayrède, J.-P. Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc. Natl. Acad. Sci. USA 2010, 107, 11537–11542. [Google Scholar] [CrossRef] [Green Version]
- Raisch, J.; Rolhion, N.; Dubois, A.; Darfeuille-Michaud, A.; Bringer, M.-A. Intracellular colon cancer-associated Escherichia coli promote protumoral activities of human macrophages by inducing sustained COX-2 expression. Lab. Invest. 2015, 95, 296–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Yang, Y.; Moore, D.R.; Nimmo, S.L.; Lightfoot, S.A.; Huycke, M.M. 4-hydroxy-2-nonenal mediates genotoxicity and bystander effects caused by Enterococcus faecalis-infected macrophages. Gastroenterology 2012, 142, 543–551e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Yang, Y.; Huycke, M.M. Commensal bacteria drive endogenous transformation and tumour stem cell marker expression through a bystander effect. Gut 2015, 64, 459–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Yang, Y.; Huycke, M.M. Commensal-infected macrophages induce dedifferentiation and reprogramming of epithelial cells during colorectal carcinogenesis. Oncotarget 2017, 8, 102176–102190. [Google Scholar] [CrossRef]
- Zhang, Y.; Weng, Y.; Gan, H.; Zhao, X.; Zhi, F. Streptococcus gallolyticus conspires myeloid cells to promote tumorigenesis of inflammatory bowel disease. Biochem. Biophys. Res. Commun. 2018, 506, 907–911. [Google Scholar] [CrossRef]
- Irrazábal, T.; Belcheva, A.; Girardin, S.E.; Martin, A.; Philpott, D.J. The multifaceted role of the intestinal microbiota in colon cancer. Mol. Cell 2014, 54, 309–320. [Google Scholar] [CrossRef] [Green Version]
- Saus, E.; Iraola-Guzmán, S.; Willis, J.R.; Brunet-Vega, A.; Gabaldón, T. Microbiome and colorectal cancer: Roles in carcinogenesis and clinical potential. Mol. Aspects Med. 2019, 69, 93–106. [Google Scholar] [CrossRef]
- Alhinai, E.A.; Walton, G.E.; Commane, D.M. The Role of the Gut Microbiota in Colorectal Cancer Causation. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [Green Version]
- Zagato, E.; Pozzi, C.; Bertocchi, A.; Schioppa, T.; Saccheri, F.; Guglietta, S.; Fosso, B.; Melocchi, L.; Nizzoli, G.; Troisi, J.; et al. Endogenous murine microbiota member Faecalibaculum rodentium and its human homologue protect from intestinal tumour growth. Nat. Microbiol. 2020, 5, 511–524. [Google Scholar] [CrossRef]
- Singh, R.K.; Chang, H.-W.; Yan, D.; Lee, K.M.; Ucmak, D.; Wong, K.; Abrouk, M.; Farahnik, B.; Nakamura, M.; Zhu, T.H.; et al. Influence of diet on the gut microbiome and implications for human health. J. Transl. Med. 2017, 15, 73. [Google Scholar] [CrossRef] [Green Version]
- Monda, V.; Villano, I.; Messina, A.; Valenzano, A.; Esposito, T.; Moscatelli, F.; Viggiano, A.; Cibelli, G.; Chieffi, S.; Monda, M.; et al. Exercise Modifies the Gut Microbiota with Positive Health Effects. Oxid Med. Cell Longev. 2017, 2017, 3831972. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Jahanzaib Anwar, M.; Usman, A.; Keshavarzian, A.; Bishehsari, F. Colorectal Cancer and Alcohol Consumption-Populations to Molecules. Cancers (Basel) 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Lazar, V.; Ditu, L.-M.; Pircalabioru, G.G.; Gheorghe, I.; Curutiu, C.; Holban, A.M.; Picu, A.; Petcu, L.; Chifiriuc, M.C. Aspects of Gut Microbiota and Immune System Interactions in Infectious Diseases, Immunopathology, and Cancer. Front. Immunol. 2018, 9, 1830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, W.R.; Hoyles, L.; Flint, H.J.; Dumas, M.-E. Colonic bacterial metabolites and human health. Curr. Opin. Microbiol. 2013, 16, 246–254. [Google Scholar] [CrossRef] [Green Version]
- Chang, P.V.; Hao, L.; Offermanns, S.; Medzhitov, R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc. Natl. Acad. Sci. USA 2014, 111, 2247–2252. [Google Scholar] [CrossRef] [Green Version]
- Scott, N.A.; Andrusaite, A.; Andersen, P.; Lawson, M.; Alcon-Giner, C.; Leclaire, C.; Caim, S.; Le Gall, G.; Shaw, T.; Connolly, J.P.R.; et al. Antibiotics induce sustained dysregulation of intestinal T cell immunity by perturbing macrophage homeostasis. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Ye, Q.; Zeng, X.; Qiao, S. Functions of Macrophages in the Maintenance of Intestinal Homeostasis. J. Immunol. Res. 2019, 2019, 1512969. [Google Scholar] [CrossRef] [Green Version]
- Vieira, E.L.M.; Leonel, A.J.; Sad, A.P.; Beltrão, N.R.M.; Costa, T.F.; Ferreira, T.M.R.; Gomes-Santos, A.C.; Faria, A.M.C.; Peluzio, M.C.G.; Cara, D.C.; et al. Oral administration of sodium butyrate attenuates inflammation and mucosal lesion in experimental acute ulcerative colitis. J. Nutr. Biochem. 2012, 23, 430–436. [Google Scholar] [CrossRef]
- Na, Y.R.; Stakenborg, M.; Seok, S.H.; Matteoli, G. Macrophages in intestinal inflammation and resolution: A potential therapeutic target in IBD. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 531–543. [Google Scholar] [CrossRef]
- Murphy, E.A.; Davis, J.M.; McClellan, J.L.; Carmichael, M.D. Quercetin’s effects on intestinal polyp multiplicity and macrophage number in the Apc(Min/+) mouse. Nutr. Cancer 2011, 63, 421–426. [Google Scholar] [CrossRef] [Green Version]
- Sun, P.; Wang, H.; He, Z.; Chen, X.; Wu, Q.; Chen, W.; Sun, Z.; Weng, M.; Zhu, M.; Ma, D.; et al. Fasting inhibits colorectal cancer growth by reducing M2 polarization of tumor-associated macrophages. Oncotarget 2017, 8, 74649–74660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Almeida, C.V.; de Camargo, M.R.; Russo, E.; Amedei, A. Role of diet and gut microbiota on colorectal cancer immunomodulation. World J. Gastroenterol. 2019, 25, 151–162. [Google Scholar] [CrossRef]
- Liu, T.; Guo, Z.; Song, X.; Liu, L.; Dong, W.; Wang, S.; Xu, M.; Yang, C.; Wang, B.; Cao, H. High-fat diet-induced dysbiosis mediates MCP-1/CCR2 axis-dependent M2 macrophage polarization and promotes intestinal adenoma-adenocarcinoma sequence. J. Cell. Mol. Med. 2020, 24, 2648–2662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, M.; Garrett, W.S.; Chan, A.T. Nutrients, foods, and colorectal cancer prevention. Gastroenterology 2015, 148, 1244–1260e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, S.; Ulrich, C.M.; Neuhouser, M.L.; Malysheva, O.; Bailey, L.B.; Xiao, L.; Brown, E.C.; Cushing-Haugen, K.L.; Zheng, Y.; Cheng, T.-Y.D.; et al. Plasma choline metabolites and colorectal cancer risk in the Women’s Health Initiative Observational Study. Cancer Res. 2014, 74, 7442–7452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, A.M.; Manghi, P.; Asnicar, F.; Pasolli, E.; Armanini, F.; Zolfo, M.; Beghini, F.; Manara, S.; Karcher, N.; Pozzi, C.; et al. Metagenomic analysis of colorectal cancer datasets identifies cross-cohort microbial diagnostic signatures and a link with choline degradation. Nat. Med. 2019, 25, 667–678. [Google Scholar] [CrossRef] [Green Version]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef]
- Wunderlich, C.M.; Ackermann, P.J.; Ostermann, A.L.; Adams-Quack, P.; Vogt, M.C.; Tran, M.-L.; Nikolajev, A.; Waisman, A.; Garbers, C.; Theurich, S.; et al. Obesity exacerbates colitis-associated cancer via IL-6-regulated macrophage polarisation and CCL-20/CCR-6-mediated lymphocyte recruitment. Nat. Commun. 2018, 9, 1646. [Google Scholar] [CrossRef]
- Bishehsari, F.; Magno, E.; Swanson, G.; Desai, V.; Voigt, R.M.; Forsyth, C.B.; Keshavarzian, A. Alcohol and Gut-Derived Inflammation. Alcohol Res. 2017, 38, 163–171. [Google Scholar]
- Engen, P.A.; Green, S.J.; Voigt, R.M.; Forsyth, C.B.; Keshavarzian, A. The Gastrointestinal Microbiome: Alcohol Effects on the Composition of Intestinal Microbiota. Alcohol Res. 2015, 37, 223–236. [Google Scholar]
- Hammer, A.M.; Morris, N.L.; Earley, Z.M.; Choudhry, M.A. The First Line of Defense: The Effects of Alcohol on Post-Burn Intestinal Barrier, Immune Cells, and Microbiome. Alcohol Res. 2015, 37, 209–222. [Google Scholar] [PubMed]
- Shukla, P.K.; Chaudhry, K.K.; Mir, H.; Gangwar, R.; Yadav, N.; Manda, B.; Meena, A.S.; Rao, R. Chronic ethanol feeding promotes azoxymethane and dextran sulfate sodium-induced colonic tumorigenesis potentially by enhancing mucosal inflammation. BMC Cancer 2016, 16, 189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caprara, G.; Allavena, P.; Erreni, M. Intestinal Macrophages at the Crossroad between Diet, Inflammation, and Cancer. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef]
- Erreni, M.; Mantovani, A.; Allavena, P. Tumor-associated Macrophages (TAM) and Inflammation in Colorectal Cancer. Cancer Microenviron. 2011, 4, 141–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavnar, M.J.; Turcotte, S.; Katz, S.C.; Kuk, D.; Gönen, M.; Shia, J.; Allen, P.J.; Balachandran, V.P.; D’Angelica, M.I.; Kingham, T.P.; et al. Tumor-Associated Macrophage Infiltration in Colorectal Cancer Liver Metastases is Associated With Better Outcome. Ann. Surg. Oncol. 2017, 24, 1835–1842. [Google Scholar] [CrossRef]
- Kim, Y.; Wen, X.; Bae, J.M.; Kim, J.H.; Cho, N.-Y.; Kang, G.H. The distribution of intratumoral macrophages correlates with molecular phenotypes and impacts prognosis in colorectal carcinoma. Histopathology 2018, 73, 663–671. [Google Scholar] [CrossRef]
- Zhou, Q.; Peng, R.-Q.; Wu, X.-J.; Xia, Q.; Hou, J.-H.; Ding, Y.; Zhou, Q.-M.; Zhang, X.; Pang, Z.-Z.; Wan, D.-S.; et al. The density of macrophages in the invasive front is inversely correlated to liver metastasis in colon cancer. J. Transl. Med. 2010, 8, 13. [Google Scholar] [CrossRef] [Green Version]
- Bacman, D.; Merkel, S.; Croner, R.; Papadopoulos, T.; Brueckl, W.; Dimmler, A. TGF-beta receptor 2 downregulation in tumour-associated stroma worsens prognosis and high-grade tumours show more tumour-associated macrophages and lower TGF-beta1 expression in colon carcinoma: A retrospective study. BMC Cancer 2007, 7, 156. [Google Scholar] [CrossRef] [Green Version]
- Bindea, G.; Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Waldner, M.; Obenauf, A.C.; Angell, H.; Fredriksen, T.; Lafontaine, L.; Berger, A.; et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 2013, 39, 782–795. [Google Scholar] [CrossRef] [Green Version]
- Edin, S.; Wikberg, M.L.; Dahlin, A.M.; Rutegård, J.; Öberg, Å.; Oldenborg, P.A.; Palmqvist, R. The distribution of macrophages with a M1 or M2 phenotype in relation to prognosis and the molecular characteristics of colorectal cancer. PLoS ONE 2012, 7, e47045. [Google Scholar] [CrossRef] [Green Version]
- Koelzer, V.H.; Canonica, K.; Dawson, H.; Sokol, L.; Karamitopoulou-Diamantis, E.; Lugli, A.; Zlobec, I. Phenotyping of tumor-associated macrophages in colorectal cancer: Impact on single cell invasion (tumor budding) and clinicopathological outcome. Oncoimmunology 2016, 5, e1106677. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Li, L.; Li, Y.; Long, Y.; Zhao, Q.; Ouyang, Y.; Bao, W.; Gong, K. Tumor-associated macrophage infiltration and prognosis in colorectal cancer: Systematic review and meta-analysis. Int. J. Colorectal Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Ge, X.; Xu, X.; Yu, S.; Wang, J.; Sun, L. Prognostic value and clinicopathological roles of phenotypes of tumour-associated macrophages in colorectal cancer. J. Cancer Res. Clin. Oncol. 2019, 145, 3005–3019. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Chang, W.; Mao, Y.; He, G.; Zheng, P.; Tang, W.; Wei, Y.; Ren, L.; Zhu, D.; Ji, M.; et al. Tumor-associated Macrophages as Prognostic and Predictive Biomarkers for Postoperative Adjuvant Chemotherapy in Patients with Stage II Colon Cancer. Clin. Cancer Res. 2019, 25, 3896–3907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llosa, N.J.; Cruise, M.; Tam, A.; Wicks, E.C.; Hechenbleikner, E.M.; Taube, J.M.; Blosser, R.L.; Fan, H.; Wang, H.; Luber, B.S.; et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015, 5, 43–51. [Google Scholar] [CrossRef]
- Forssell, J.; Oberg, A.; Henriksson, M.L.; Stenling, R.; Jung, A.; Palmqvist, R. High macrophage infiltration along the tumor front correlates with improved survival in colon cancer. Clin. Cancer Res. 2007, 13, 1472–1479. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.-C.; Chen, J.-S.; Lee, C.-H.; Chang, J.-J.; Shieh, Y.-S. Intratumoral macrophage counts correlate with tumor progression in colorectal cancer. J. Surg. Oncol. 2010, 102, 242–248. [Google Scholar] [CrossRef]
- Pinto, M.L.; Rios, E.; Durães, C.; Ribeiro, R.; Machado, J.C.; Mantovani, A.; Barbosa, M.A.; Carneiro, F.; Oliveira, M.J. The Two Faces of Tumor-Associated Macrophages and Their Clinical Significance in Colorectal Cancer. Front. Immunol. 2019, 10, 1875. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Li, Z.; Skrzypczynska, K.M.; Fang, Q.; Zhang, W.; O’Brien, S.A.; He, Y.; Wang, L.; Zhang, Q.; Kim, A.; et al. Single-Cell Analyses Inform Mechanisms of Myeloid-Targeted Therapies in Colon Cancer. Cell 2020, 181, 442–459e29. [Google Scholar] [CrossRef]
- Malesci, A.; Bianchi, P.; Celesti, G.; Basso, G.; Marchesi, F.; Grizzi, F.; Di Caro, G.; Cavalleri, T.; Rimassa, L.; Palmqvist, R.; et al. Tumor-associated macrophages and response to 5-fluorouracil adjuvant therapy in stage III colorectal cancer. Oncoimmunology 2017, 6, e1342918. [Google Scholar] [CrossRef]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Escamilla, J.; Mok, S.; David, J.; Priceman, S.; West, B.; Bollag, G.; McBride, W.; Wu, L. CSF1R signaling blockade stanches tumor-infiltrating myeloid cells and improves the efficacy of radiotherapy in prostate cancer. Cancer Res. 2013, 73, 2782–2794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klug, F.; Prakash, H.; Huber, P.E.; Seibel, T.; Bender, N.; Halama, N.; Pfirschke, C.; Voss, R.H.; Timke, C.; Umansky, L.; et al. Low-dose irradiation programs macrophage differentiation to an iNOS+/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell 2013, 24, 589–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vivarelli, S.; Salemi, R.; Candido, S.; Falzone, L.; Santagati, M.; Stefani, S.; Torino, F.; Banna, G.L.; Tonini, G.; Libra, M. Gut Microbiota and Cancer: From Pathogenesis to Therapy. Cancers 2019, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, W.; Mao, Q.; Xia, W.; Dong, G.; Yu, C.; Jiang, F. Gut Microbiota Shapes the Efficiency of Cancer Therapy. Front. Microbiol. 2019, 10, 1050. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.; Guo, F.; Yu, Y.; Sun, T.; Ma, D.; Han, J.; Qian, Y.; Kryczek, I.; Sun, D.; Nagarsheth, N.; et al. Fusobacterium nucleatum Promotes Chemoresistance to Colorectal Cancer by Modulating Autophagy. Cell 2017, 170, 548–563e16. [Google Scholar] [CrossRef] [Green Version]
- Stringer, A.M.; Gibson, R.J.; Bowen, J.M.; Logan, R.M.; Ashton, K.; Yeoh, A.S.J.; Al-Dasooqi, N.; Keefe, D.M.K. Irinotecan-induced mucositis manifesting as diarrhoea corresponds with an amended intestinal flora and mucin profile. Int. J. Exp. Pathol. 2009, 90, 489–499. [Google Scholar] [CrossRef]
- Al-Dasooqi, N.; Bowen, J.M.; Gibson, R.J.; Logan, R.M.; Stringer, A.M.; Keefe, D.M. Irinotecan-induced alterations in intestinal cell kinetics and extracellular matrix component expression in the Dark Agouti rat. Int. J. Exp. Pathol. 2011, 92, 357–365. [Google Scholar] [CrossRef]
- Crawford, P.A.; Gordon, J.I. Microbial regulation of intestinal radiosensitivity. Proc. Natl. Acad. Sci. USA 2005, 102, 13254–13259. [Google Scholar] [CrossRef] [Green Version]
- Iida, N.; Dzutsev, A.; Stewart, C.A.; Smith, L.; Bouladoux, N.; Weingarten, R.A.; Molina, D.A.; Salcedo, R.; Back, T.; Cramer, S.; et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science 2013, 342, 967–970. [Google Scholar] [CrossRef] [PubMed]
- Marullo, R.; Werner, E.; Degtyareva, N.; Moore, B.; Altavilla, G.; Ramalingam, S.S.; Doetsch, P.W. Cisplatin induces a mitochondrial-ROS response that contributes to cytotoxicity depending on mitochondrial redox status and bioenergetic functions. PLoS ONE 2013, 8, e81162. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Sznol, M.; McDermott, D.F.; Kluger, H.M.; Carvajal, R.D.; Sharfman, W.H.; Brahmer, J.R.; Lawrence, D.P.; Atkins, M.B.; Powderly, J.D.; et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J. Clin. Oncol. 2014, 32, 1020–1030. [Google Scholar] [CrossRef]
- Antonia, S.J.; López-Martin, J.A.; Bendell, J.; Ott, P.A.; Taylor, M.; Eder, J.P.; Jäger, D.; Pietanza, M.C.; Le, D.T.; de Braud, F.; et al. Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell lung cancer (CheckMate 032): A multicentre, open-label, phase 1/2 trial. Lancet Oncol. 2016, 17, 883–895. [Google Scholar] [CrossRef] [Green Version]
- Chung, K.Y.; Gore, I.; Fong, L.; Venook, A.; Beck, S.B.; Dorazio, P.; Criscitiello, P.J.; Healey, D.I.; Huang, B.; Gomez-Navarro, J.; et al. Phase II study of the anti-cytotoxic T-lymphocyte-associated antigen 4 monoclonal antibody, tremelimumab, in patients with refractory metastatic colorectal cancer. J. Clin. Oncol. 2010, 28, 3485–3490. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.M.; Hwu, W.-J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [Green Version]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.-J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.-J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef]
- Zhao, P.; Li, L.; Jiang, X.; Li, Q. Mismatch repair deficiency/microsatellite instability-high as a predictor for anti-PD-1/PD-L1 immunotherapy efficacy. J. Hematol. Oncol. 2019, 12, 54. [Google Scholar] [CrossRef] [PubMed]
- Young, J.; Simms, L.A.; Biden, K.G.; Wynter, C.; Whitehall, V.; Karamatic, R.; George, J.; Goldblatt, J.; Walpole, I.; Robin, S.A.; et al. Features of colorectal cancers with high-level microsatellite instability occurring in familial and sporadic settings: Parallel pathways of tumorigenesis. Am. J. Pathol. 2001, 159, 2107–2116. [Google Scholar] [CrossRef]
- Smyrk, T.C.; Watson, P.; Kaul, K.; Lynch, H.T. Tumor-infiltrating lymphocytes are a marker for microsatellite instability in colorectal carcinoma. Cancer 2001, 91, 2417–2422. [Google Scholar] [CrossRef]
- Dolcetti, R.; Viel, A.; Doglioni, C.; Russo, A.; Guidoboni, M.; Capozzi, E.; Vecchiato, N.; Macrì, E.; Fornasarig, M.; Boiocchi, M. High prevalence of activated intraepithelial cytotoxic T lymphocytes and increased neoplastic cell apoptosis in colorectal carcinomas with microsatellite instability. Am. J. Pathol. 1999, 154, 1805–1813. [Google Scholar] [CrossRef] [Green Version]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.-X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagorsen, D.; Voigt, S.; Berg, E.; Stein, H.; Thiel, E.; Loddenkemper, C. Tumor-infiltrating macrophages and dendritic cells in human colorectal cancer: Relation to local regulatory T cells, systemic T-cell response against tumor-associated antigens and survival. J. Transl. Med. 2007, 5, 62. [Google Scholar] [CrossRef] [Green Version]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- Guerra, J.; Pinto, C.; Pinto, D.; Pinheiro, M.; Silva, R.; Peixoto, A.; Rocha, P.; Veiga, I.; Santos, C.; Santos, R.; et al. POLE somatic mutations in advanced colorectal cancer. Cancer Med. 2017, 6, 2966–2971. [Google Scholar] [CrossRef]
- Palles, C.; Cazier, J.-B.; Howarth, K.M.; Domingo, E.; Jones, A.M.; Broderick, P.; Kemp, Z.; Spain, S.L.; Guarino, E.; Guarino Almeida, E.; et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat. Genet. 2013, 45, 136–144. [Google Scholar] [CrossRef] [Green Version]
- Elsayed, F.A.; Kets, C.M.; Ruano, D.; van den Akker, B.; Mensenkamp, A.R.; Schrumpf, M.; Nielsen, M.; Wijnen, J.T.; Tops, C.M.; Ligtenberg, M.J.; et al. Germline variants in POLE are associated with early onset mismatch repair deficient colorectal cancer. Eur. J. Hum. Genet. 2015, 23, 1080–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, A.M.; van Wezel, T.; van den Akker, B.E.; Ventayol Garcia, M.; Ruano, D.; Tops, C.M.; Wagner, A.; Letteboer, T.G.; Gómez-García, E.B.; Devilee, P.; et al. Combined mismatch repair and POLE/POLD1 defects explain unresolved suspected Lynch syndrome cancers. Eur. J. Hum. Genet. 2016, 24, 1089–1092. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Freeman-Mills, L.; Rayner, E.; Glaire, M.; Briggs, S.; Vermeulen, L.; Fessler, E.; Medema, J.P.; Boot, A.; Morreau, H.; et al. Somatic POLE proofreading domain mutation, immune response, and prognosis in colorectal cancer: A retrospective, pooled biomarker study. Lancet Gastroenterol. Hepatol. 2016, 1, 207–216. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Gong, J.; Tu, T.Y.; Lee, P.P.; Fakih, M. Immune profiling of microsatellite instability-high and polymerase ε (POLE)-mutated metastatic colorectal tumors identifies predictors of response to anti-PD-1 therapy. J. Gastrointest. Oncol. 2018, 9, 404–415. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Wang, C.; Lee, P.P.; Chu, P.; Fakih, M. Response to PD-1 Blockade in Microsatellite Stable Metastatic Colorectal Cancer Harboring a POLE Mutation. J. Natl. Compr. Canc. Netw. 2017, 15, 142–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourdais, R.; Rousseau, B.; Pujals, A.; Boussion, H.; Joly, C.; Guillemin, A.; Baumgaertner, I.; Neuzillet, C.; Tournigand, C. Polymerase proofreading domain mutations: New opportunities for immunotherapy in hypermutated colorectal cancer beyond MMR deficiency. Crit. Rev. Oncol. Hematol. 2017, 113, 242–248. [Google Scholar] [CrossRef]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef]
- Majeti, R.; Chao, M.P.; Alizadeh, A.A.; Pang, W.W.; Jaiswal, S.; Gibbs, K.D.; van Rooijen, N.; Weissman, I.L. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 2009, 138, 286–299. [Google Scholar] [CrossRef] [Green Version]
- Willingham, S.B.; Volkmer, J.-P.; Gentles, A.J.; Sahoo, D.; Dalerba, P.; Mitra, S.S.; Wang, J.; Contreras-Trujillo, H.; Martin, R.; Cohen, J.D.; et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 6662–6667. [Google Scholar] [CrossRef] [Green Version]
- Feng, M.; Jiang, W.; Kim, B.Y.S.; Zhang, C.C.; Fu, Y.-X.; Weissman, I.L. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat. Rev. Cancer 2019, 19, 568–586. [Google Scholar] [CrossRef]
- Tzatzarakis, E.; Hissa, B.; Reissfelder, C.; Schölch, S. The overall potential of CD47 in cancer immunotherapy: With a focus on gastrointestinal tumors. Expert Rev. Anticancer Ther. 2019, 19, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Lascorz, J.; Bevier, M.; V Schönfels, W.; Kalthoff, H.; Aselmann, H.; Beckmann, J.; Egberts, J.; Buch, S.; Becker, T.; Schreiber, S.; et al. Association study identifying polymorphisms in CD47 and other extracellular matrix pathway genes as putative prognostic markers for colorectal cancer. Int J. Colorectal Dis 2013, 28, 173–181. [Google Scholar] [CrossRef]
- Fujiwara-Tani, R.; Sasaki, T.; Ohmori, H.; Luo, Y.; Goto, K.; Nishiguchi, Y.; Mori, S.; Nakashima, C.; Mori, T.; Miyagawa, Y.; et al. Concurrent Expression of CD47 and CD44 in Colorectal Cancer Promotes Malignancy. Pathobiology 2019, 86, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Luo, X.; Chen, C.; Tang, Y.; Li, L.; Mo, B.; Liang, H.; Yu, S. The Ap-2α/Elk-1 axis regulates Sirpα-dependent tumor phagocytosis by tumor-associated macrophages in colorectal cancer. Signal. Transduct. Target. Ther. 2020, 5, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Jeught, K.; Sun, Y.; Fang, Y.; Zhou, Z.; Jiang, H.; Yu, T.; Yang, J.; Kamocka, M.M.; So, K.M.; Li, Y.; et al. ST2 as checkpoint target for colorectal cancer immunotherapy. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [Green Version]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.-L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef] [Green Version]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Vétizou, M.; Pitt, J.M.; Daillère, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.M.; et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef] [Green Version]
- Guiducci, C.; Vicari, A.P.; Sangaletti, S.; Trinchieri, G.; Colombo, M.P. Redirecting in vivo elicited tumor infiltrating macrophages and dendritic cells towards tumor rejection. Cancer Res. 2005, 65, 3437–3446. [Google Scholar] [CrossRef] [Green Version]
- Wallace, B.D.; Roberts, A.B.; Pollet, R.M.; Ingle, J.D.; Biernat, K.A.; Pellock, S.J.; Venkatesh, M.K.; Guthrie, L.; O’Neal, S.K.; Robinson, S.J.; et al. Structure and Inhibition of Microbiome β-Glucuronidases Essential to the Alleviation of Cancer Drug Toxicity. Chem. Biol. 2015, 22, 1238–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.-H.; Chen, Y.-X.; Fang, J.-Y. Comprehensive review of targeted therapy for colorectal cancer. Signal. Transduct. Target. Ther. 2020, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Krajicek, E.; Fischer, M.; Allegretti, J.R.; Kelly, C.R. Nuts and Bolts of Fecal Microbiota Transplantation. Clin. Gastroenterol. Hepatol. 2019, 17, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Molska, M.; Reguła, J. Potential Mechanisms of Probiotics Action in the Prevention and Treatment of Colorectal Cancer. Nutrients 2019, 11. [Google Scholar] [CrossRef] [Green Version]
- Wieczorska, K.; Stolarek, M.; Stec, R. The Role of the Gut Microbiome in Colorectal Cancer: Where Are We? Where Are We Going? Clin. Colorectal. Cancer. 2020, 19, 5–12. [Google Scholar] [CrossRef]
- Fong, W.; Li, Q.; Yu, J. Gut microbiota modulation: A novel strategy for prevention and treatment of colorectal cancer. Oncogene 2020, 39, 4925–4943. [Google Scholar] [CrossRef]
- Zhuo, Q.; Yu, B.; Zhou, J.; Zhang, J.; Zhang, R.; Xie, J.; Wang, Q.; Zhao, S. Lysates of Lactobacillus acidophilus combined with CTLA-4-blocking antibodies enhance antitumor immunity in a mouse colon cancer model. Sci. Rep. 2019, 9, 20128. [Google Scholar] [CrossRef]
- Ridnour, L.A.; Cheng, R.Y.S.; Switzer, C.H.; Heinecke, J.L.; Ambs, S.; Glynn, S.; Young, H.A.; Trinchieri, G.; Wink, D.A. Molecular pathways: Toll-like receptors in the tumor microenvironment--poor prognosis or new therapeutic opportunity. Clin. Cancer Res. 2013, 19, 1340–1346. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
 Human Human |  Mouse Mouse | |
|---|---|---|
| Pan-macrophage | CD68+, CD14+, CD16+, HLA-DR+ CD64+ | F4/80+, CD68+, CD64+ CXC3R1int/hi, MHCII+, CD11b+, Ly6Clow |
| M1-like (anti-tumoral) | CD40, CD80, CD86, NOS2 TNFα, IL1β, IL6, IL12, IL23 CXCL9, CXCL10, CXCL11 | |
| M2-like (pro-tumoral) | CD163, CD204, CD206, CD209, CD301, Dectin-1, IDO, ARG1, HO-1 IL10, TGFβ, CCL17, CCL18*, CCL22 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mola, S.; Pandolfo, C.; Sica, A.; Porta, C. The Macrophages-Microbiota Interplay in Colorectal Cancer (CRC)-Related Inflammation: Prognostic and Therapeutic Significance. Int. J. Mol. Sci. 2020, 21, 6866. https://doi.org/10.3390/ijms21186866
Mola S, Pandolfo C, Sica A, Porta C. The Macrophages-Microbiota Interplay in Colorectal Cancer (CRC)-Related Inflammation: Prognostic and Therapeutic Significance. International Journal of Molecular Sciences. 2020; 21(18):6866. https://doi.org/10.3390/ijms21186866
Chicago/Turabian StyleMola, Silvia, Chiara Pandolfo, Antonio Sica, and Chiara Porta. 2020. "The Macrophages-Microbiota Interplay in Colorectal Cancer (CRC)-Related Inflammation: Prognostic and Therapeutic Significance" International Journal of Molecular Sciences 21, no. 18: 6866. https://doi.org/10.3390/ijms21186866
APA StyleMola, S., Pandolfo, C., Sica, A., & Porta, C. (2020). The Macrophages-Microbiota Interplay in Colorectal Cancer (CRC)-Related Inflammation: Prognostic and Therapeutic Significance. International Journal of Molecular Sciences, 21(18), 6866. https://doi.org/10.3390/ijms21186866