miR-29a Modulates GSK3β/SIRT1-Linked Mitochondrial Proteostatic Stress to Ameliorate Mouse Non-Alcoholic Steatohepatitis

 , ,
, ,  and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
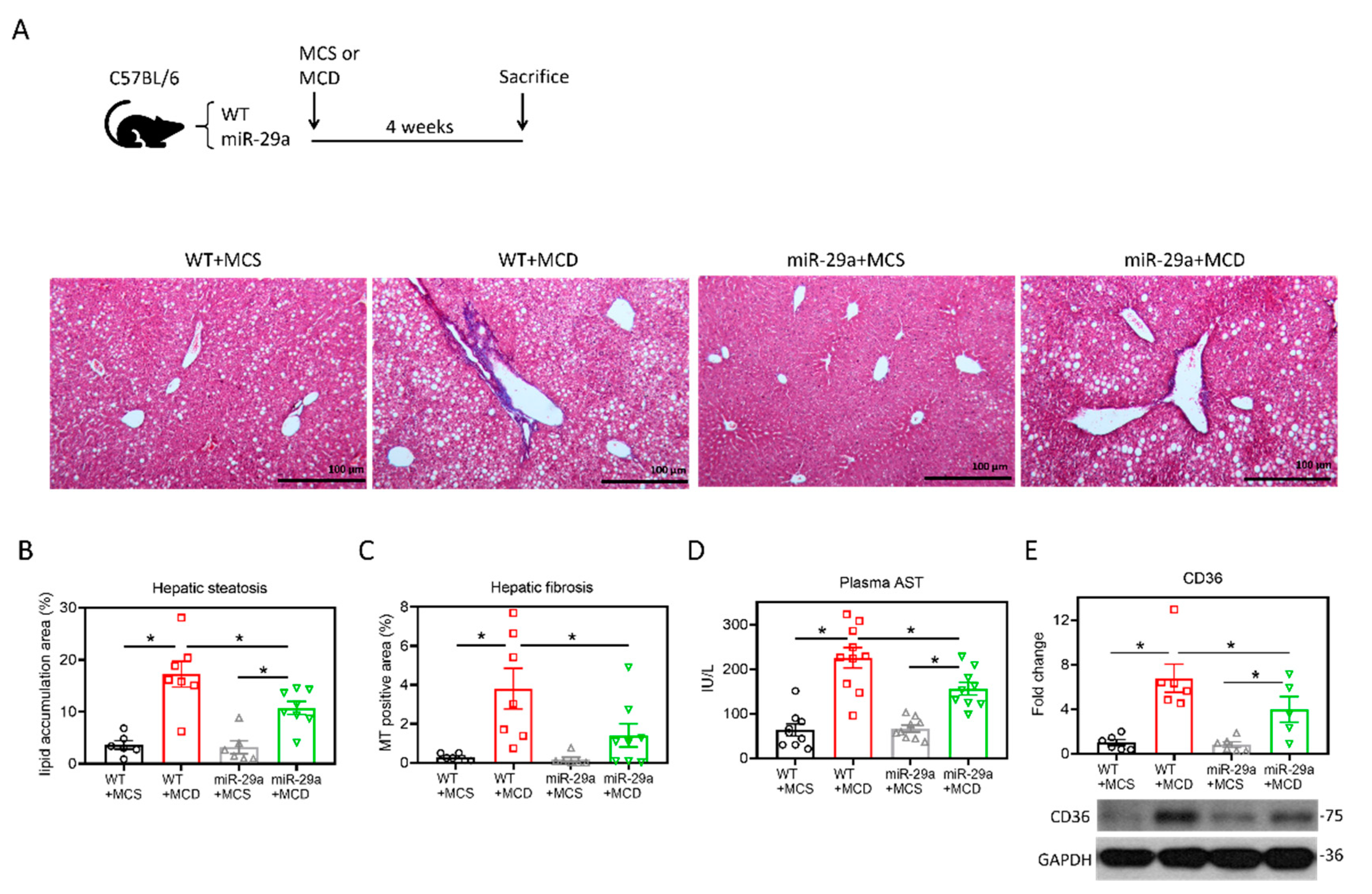
2.1. Overexpression of miR-29a Attenuates MCD-Induced Hepatic Steatosis and Fibrosis
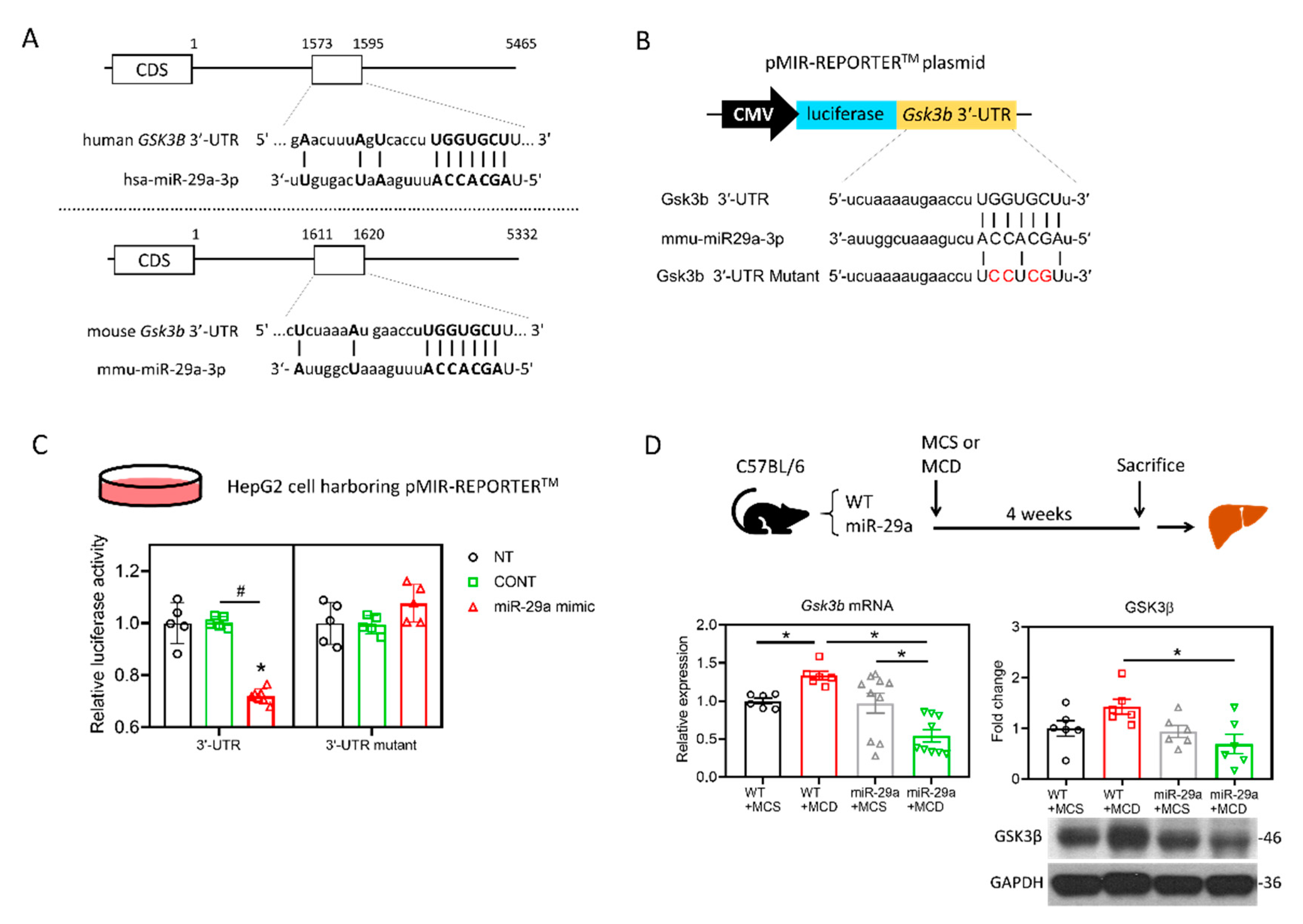
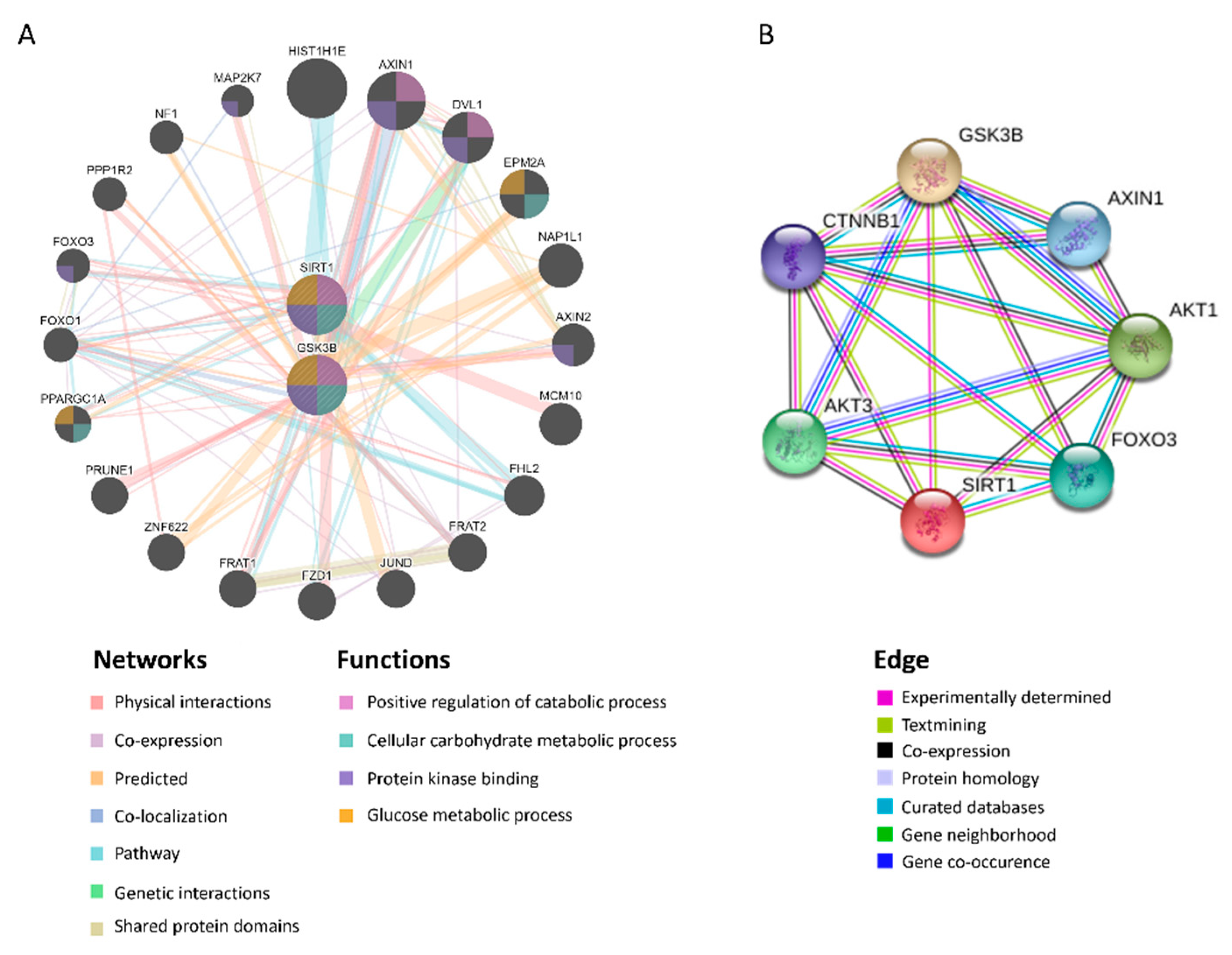
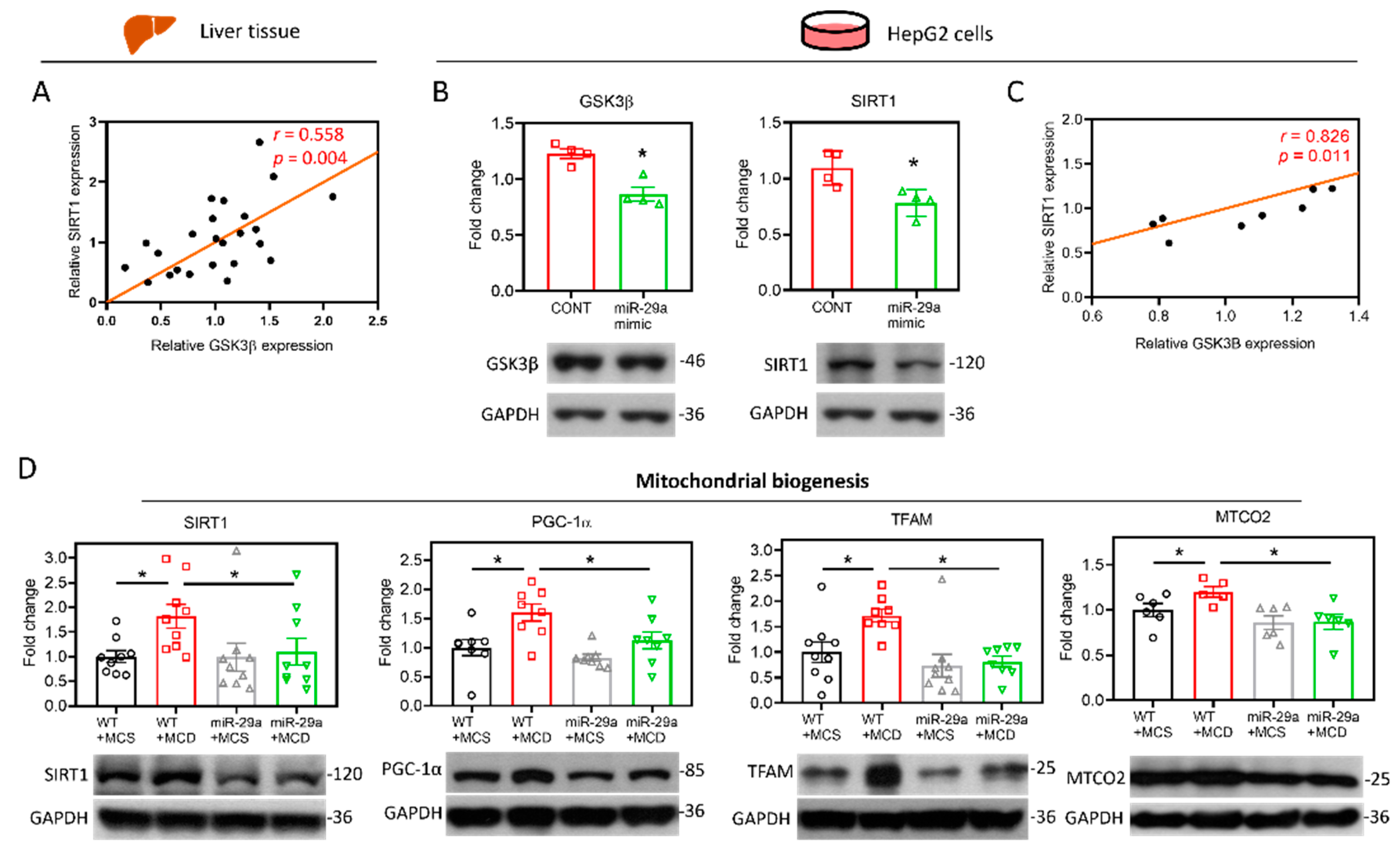
2.2. Target Identification of GSK3β as miR-29a Target in Hepatocyte
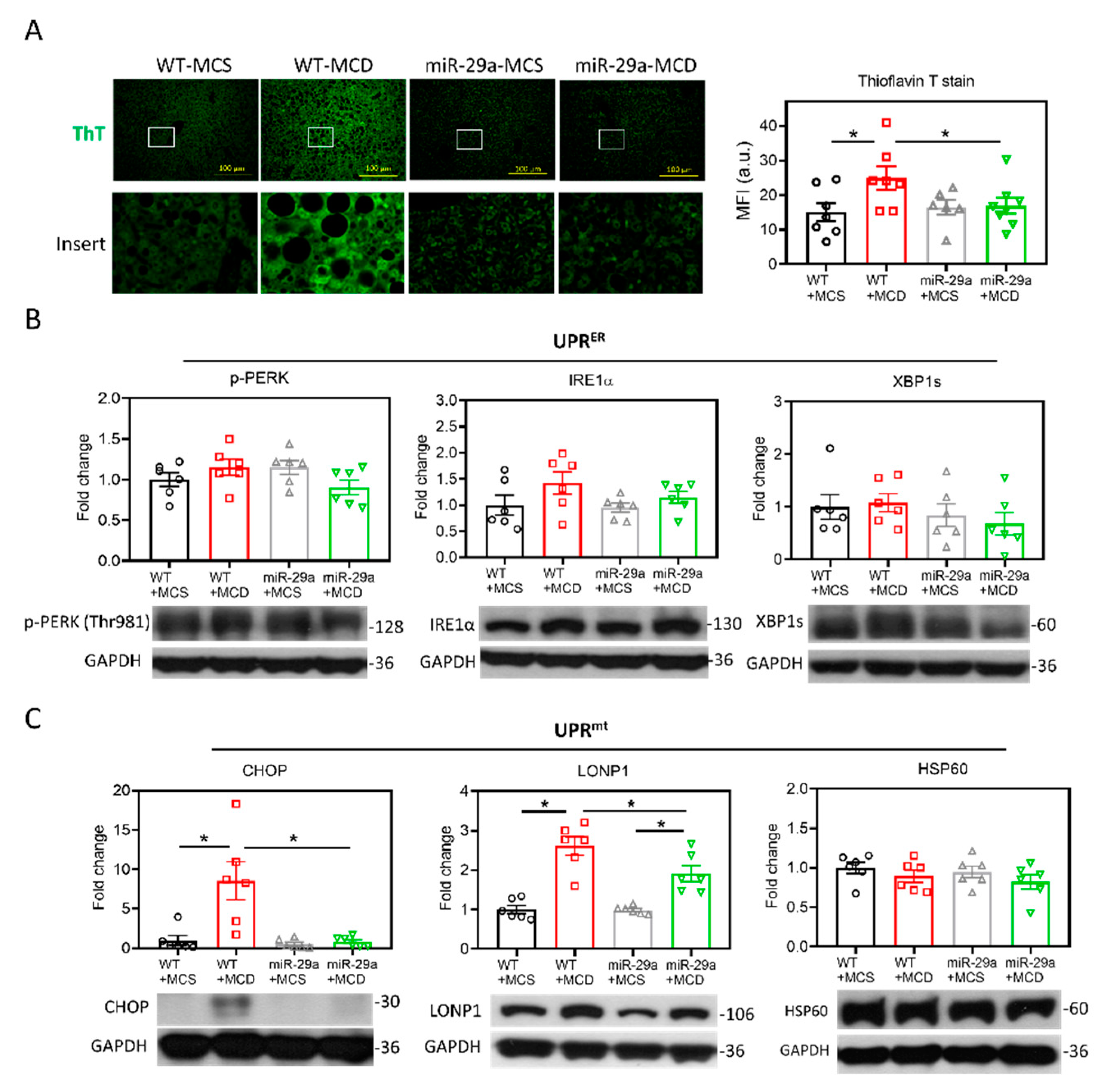
2.3. miR-29a/GSK3β Axis Alleviates Mitochondrial Proteostasic Stress via Restoring Perturbation of SIRT1-Mediated Mitochondrial Biogenesis
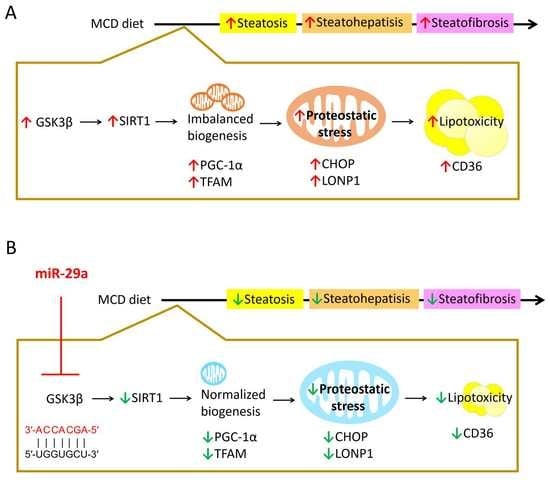
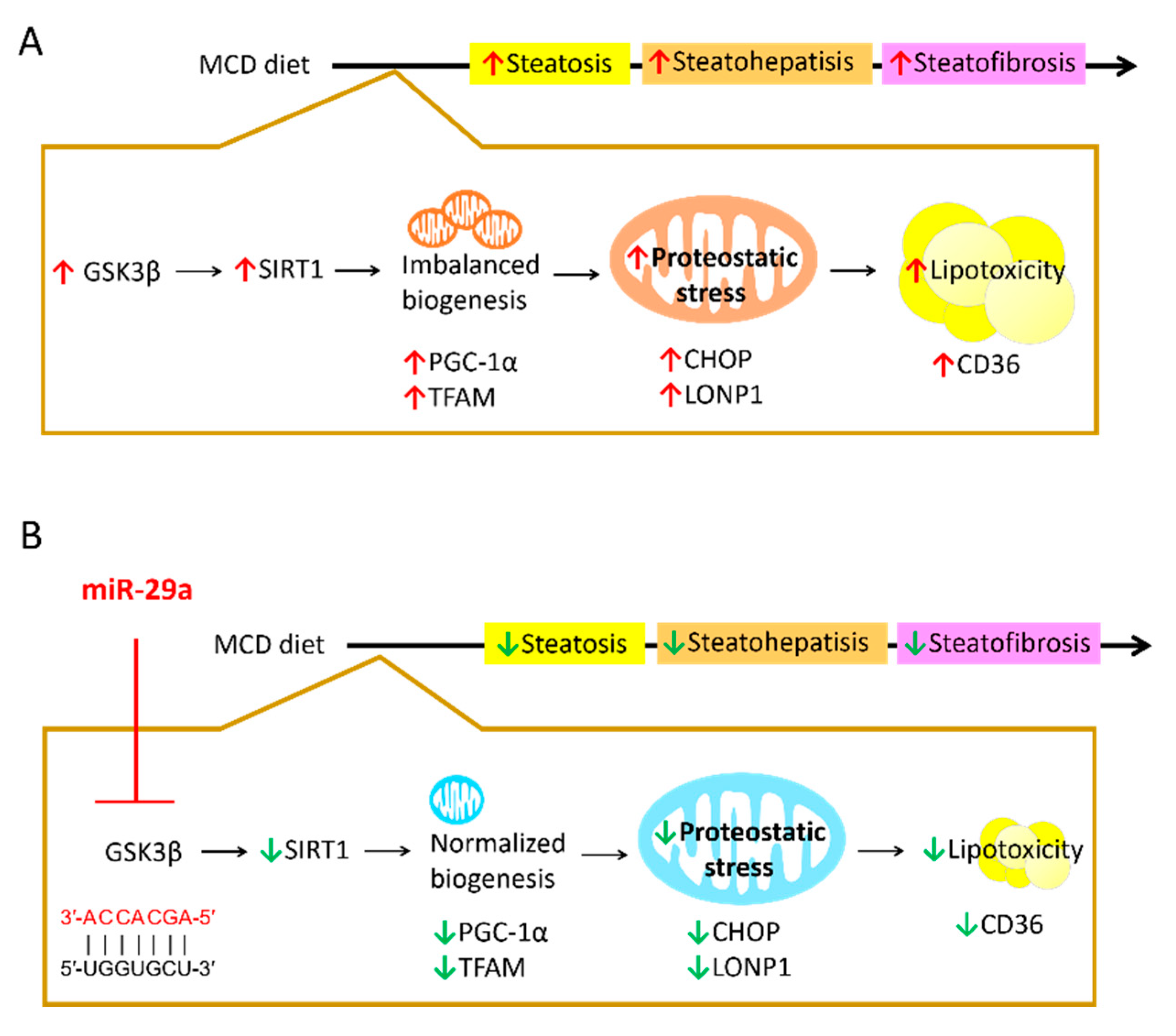
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Construction and Breeding of the miR-29a Transgenic Mouse Colony
4.3. Animal Model and Experimental Protocol
4.4. Masson’s Trichrome Stain
4.5. Blood Biochemical Analysis
4.6. In Silico miR-29-Targeted mRNA Interaction Analysis
4.7. In Silico Analysis of Protein–Protein Interaction (PPI)
4.8. Luciferase Reporter Assay
4.9. Quantitative Real-Time PCR (qPCR)
4.10. Western Blotting
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rinella, M.E. Nonalcoholic fatty liver disease: A systematic review. JAMA 2015, 313, 2263–2273. [Google Scholar] [CrossRef]
- Targher, G.; Day, C.P.; Bonora, E. Risk of cardiovascular disease in patients with nonalcoholic fatty liver disease. N. Engl. J. Med. 2010, 363, 1341–1350. [Google Scholar] [CrossRef] [Green Version]
- Abd El-Kader, S.M.; El-Den Ashmawy, E.M. Non-alcoholic fatty liver disease: The diagnosis and management. Worldj. Hepatol. 2015, 7, 846–858. [Google Scholar] [CrossRef]
- Obika, M.; Noguchi, H. Diagnosis and evaluation of nonalcoholic fatty liver disease. Exp. Diabetes Res. 2012, 2012, 145754. [Google Scholar] [CrossRef]
- Baffy, G.; Brunt, E.M.; Caldwell, S.H. Hepatocellular carcinoma in non-alcoholic fatty liver disease: An emerging menace. J. Hepatol. 2012, 56, 1384–1391. [Google Scholar] [CrossRef] [Green Version]
- Brunt, E.M. Pathology of nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathy, G.; Revelo, X.; Malhi, H. Pathogenesis of Nonalcoholic Steatohepatitis: An Overview. Hepatol. Commun. 2020, 4, 478–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Park, J.S.; Roh, Y.S. Molecular insights into the role of mitochondria in non-alcoholic fatty liver disease. Arch. Pharm. Res. 2019, 42, 935–946. [Google Scholar] [CrossRef] [PubMed]
- Simoes, I.C.M.; Fontes, A.; Pinton, P.; Zischka, H.; Wieckowski, M.R. Mitochondria in non-alcoholic fatty liver disease. Int. J. Biochem. Cell. Biol. 2018, 95, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Shpilka, T.; Haynes, C.M. The mitochondrial UPR: Mechanisms, physiological functions and implications in ageing. Nat. Rev. Mol. Cell. Biol. 2018, 19, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.C.; Dunbar, T.L.; Nargund, A.M.; Haynes, C.M.; Troemel, E.R. The C. elegans CCAAT-Enhancer-Binding Protein Gamma Is Required for Surveillance Immunity. Cell Rep. 2016, 14, 1581–1589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellegrino, M.W.; Nargund, A.M.; Kirienko, N.V.; Gillis, R.; Fiorese, C.J.; Haynes, C.M. Mitochondrial UPR-regulated innate immunity provides resistance to pathogen infection. Nature 2014, 516, 414–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Samuel, B.S.; Breen, P.C.; Ruvkun, G. Caenorhabditis elegans pathways that surveil and defend mitochondria. Nature 2014, 508, 406–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, H.S. Implications of Mitochondrial Unfolded Protein Response and Mitokines: A Perspective on Fatty Liver Diseases. Endocrinol. Metab. (Seoul) 2019, 34, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Gjorgjieva, M.; Sobolewski, C.; Dolicka, D.; Correia de Sousa, M.; Foti, M. miRNAs and NAFLD: From pathophysiology to therapy. Gut 2019, 68, 2065–2079. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Bedja, D.; Campbell, N.; Dunkerly, B.; Chenna, V.; Maitra, A.; Steenbergen, C. miR-181c regulates the mitochondrial genome, bioenergetics, and propensity for heart failure in vivo. PLoS ONE 2014, 9, e96820. [Google Scholar] [CrossRef] [Green Version]
- Fabbri, M.; Paone, A.; Calore, F.; Galli, R.; Croce, C.M. A new role for microRNAs, as ligands of Toll-like receptors. RNA Biol. 2013, 10, 169–174. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.; Ajay, S.S.; Yook, J.I.; Kim, H.S.; Hong, S.H.; Kim, N.H.; Dhanasekaran, S.M.; Chinnaiyan, A.M.; Athey, B.D. New class of microRNA targets containing simultaneous 5′-UTR and 3′-UTR interaction sites. Genome. Res. 2009, 19, 1175–1183. [Google Scholar] [CrossRef] [Green Version]
- Moran-Salvador, E.; Mann, J. Epigenetics and Liver Fibrosis. Cell Mol. Gastroenterol. Hepatol. 2017, 4, 19–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.Y.; Yang, Y.L.; Wang, P.W.; Wang, F.S.; Huang, Y.H. The Emerging Role of MicroRNAs in NAFLD: Highlight of MicroRNA-29a in Modulating Oxidative Stress, Inflammation, and Beyond. Cells 2020, 9, 1041. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.X.; Gao, M.; Li, C.Z.; Yu, C.Z.; Yan, H.; Peng, C.; Li, Y.; Li, C.G.; Ma, Z.L.; Zhao, Y.; et al. Dicer1/miR-29/HMGCR axis contributes to hepatic free cholesterol accumulation in mouse non-alcoholic steatohepatitis. Acta. Pharm. Sin. 2017, 38, 660–671. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.H.; Yang, Y.L.; Huang, F.C.; Tiao, M.M.; Lin, Y.C.; Tsai, M.H.; Wang, F.S. MicroRNA-29a mitigation of endoplasmic reticulum and autophagy aberrance counteracts in obstructive jaundice-induced fibrosis in mice. Exp. Biol. Med. (Maywood.) 2018, 243, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.C.; Wang, F.S.; Yang, Y.L.; Chuang, Y.T.; Huang, Y.H. MicroRNA-29a mitigation of toll-like receptor 2 and 4 signaling and alleviation of obstructive jaundice-induced fibrosis in mice. Biochem. Biophys. Res. Commun. 2018, 496, 880–886. [Google Scholar] [CrossRef]
- Huang, Y.H.; Yang, Y.L.; Wang, F.S. The Role of miR-29a in the Regulation, Function, and Signaling of Liver Fibrosis. Int. J. Mol. Sci. 2018, 19, 1889. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.H.; Kuo, H.C.; Yang, Y.L.; Wang, F.S. MicroRNA-29a is a key regulon that regulates BRD4 and mitigates liver fibrosis in mice by inhibiting hepatic stellate cell activation. Int. J. Med. Sci. 2019, 16, 212–220. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.L.; Wang, F.S.; Lin, H.Y.; Huang, Y.H. Exogenous Therapeutics of Microrna-29a Attenuates Development of Hepatic Fibrosis in Cholestatic Animal Model through Regulation of Phosphoinositide 3-Kinase p85 Alpha. Int. J. Mol. Sci. 2020, 21, 3636. [Google Scholar] [CrossRef]
- Lin, H.Y.; Wang, F.S.; Yang, Y.L.; Huang, Y.H. MicroRNA-29a Suppresses CD36 to Ameliorate High Fat Diet-Induced Steatohepatitis and Liver Fibrosis in Mice. Cells 2019, 8, 1298. [Google Scholar] [CrossRef] [Green Version]
- Wilson, C.G.; Tran, J.L.; Erion, D.M.; Vera, N.B.; Febbraio, M.; Weiss, E.J. Hepatocyte-Specific Disruption of CD36 Attenuates Fatty Liver and Improves Insulin Sensitivity in HFD-Fed Mice. Endocrinology 2016, 157, 570–585. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Ren, F.; Zhang, H.; Wen, T.; Piao, Z.; Zhou, L.; Zheng, S.; Zhang, J.; Chen, Y.; Han, Y.; et al. Inhibition of glycogen synthase kinase 3beta ameliorates D-GalN/LPS-induced liver injury by reducing endoplasmic reticulum stress-triggered apoptosis. PLoS ONE 2012, 7, e45202. [Google Scholar]
- Koga, T.; Suico, M.A.; Shimasaki, S.; Watanabe, E.; Kai, Y.; Koyama, K.; Omachi, K.; Morino-Koga, S.; Sato, T.; Shuto, T.; et al. Endoplasmic Reticulum (ER) Stress Induces Sirtuin 1 (SIRT1) Expression via the PI3K-Akt-GSK3beta Signaling Pathway and Promotes Hepatocellular Injury. J. Biol. Chem. 2015, 290, 30366–30374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada, R.; Okamura, N.; Furumoto, S.; Yanai, K. Imaging Protein Misfolding in the Brain Using beta-Sheet Ligands. Front. Neurosci. 2018, 12, 585. [Google Scholar] [CrossRef] [Green Version]
- Jovaisaite, V.; Mouchiroud, L.; Auwerx, J. The mitochondrial unfolded protein response, a conserved stress response pathway with implications in health and disease. J. Exp. Biol. 2014, 217, 137–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, W.; Wang, Y.; Wei, N.; Xu, R.; Xiong, Y.; Wang, P.; Shen, Q.; Yang, G. Sirt1 inhibits akt2-mediated porcine adipogenesis potentially by direct protein-protein interaction. PLoS ONE 2013, 8, e71576. [Google Scholar] [CrossRef] [Green Version]
- Anstee, Q.M.; Goldin, R.D. Mouse models in non-alcoholic fatty liver disease and steatohepatitis research. Int. J. Exp. Pathol. 2006, 87, 1–16. [Google Scholar] [CrossRef]
- Du, J.; Zhang, X.; Han, J.; Man, K.; Zhang, Y.; Chu, E.S.; Nan, Y.; Yu, J. Pro-Inflammatory CXCR3 Impairs Mitochondrial Function in Experimental Non-Alcoholic Steatohepatitis. Theranostics 2017, 7, 4192–4203. [Google Scholar] [CrossRef]
- Li, Y.; Yang, P.; Zhao, L.; Chen, Y.; Zhang, X.; Zeng, S.; Wei, L.; Varghese, Z.; Moorhead, J.F.; Chen, Y.; et al. CD36 plays a negative role in the regulation of lipophagy in hepatocytes through an AMPK-dependent pathway. J. Lipid. Res. 2019, 60, 844–855. [Google Scholar] [CrossRef] [Green Version]
- Ren, F.; Duan, Z.; Cheng, Q.; Shen, X.; Gao, F.; Bai, L.; Liu, J.; Busuttil, R.W.; Kupiec-Weglinski, J.W.; Zhai, Y. Inhibition of glycogen synthase kinase 3 beta ameliorates liver ischemia reperfusion injury by way of an interleukin-10-mediated immune regulatory mechanism. Hepatology 2011, 54, 687–696. [Google Scholar] [CrossRef] [Green Version]
- Tetri, L.H.; Basaranoglu, M.; Brunt, E.M.; Yerian, L.M.; Neuschwander-Tetri, B.A. Severe NAFLD with hepatic necroinflammatory changes in mice fed trans fats and a high-fructose corn syrup equivalent. Am. J. Physiol. Gastrointest. Liver. Physiol. 2008, 295, G987–G995. [Google Scholar] [CrossRef]
- Lee, I.C.; Ho, X.Y.; George, S.E.; Goh, C.W.; Sundaram, J.R.; Pang, K.K.L.; Luo, W.; Yusoff, P.; Sze, N.S.K.; Shenolikar, S. Oxidative stress promotes SIRT1 recruitment to the GADD34/PP1alpha complex to activate its deacetylase function. Cell. Death. Differ. 2018, 25, 255–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouchiroud, L.; Houtkooper, R.H.; Moullan, N.; Katsyuba, E.; Ryu, D.; Canto, C.; Mottis, A.; Jo, Y.S.; Viswanathan, M.; Schoonjans, K.; et al. The NAD(+)/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell 2013, 154, 430–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nan, P.; Niu, Y.; Wang, X.; Li, Q. MiR-29a function as tumor suppressor in cervical cancer by targeting SIRT1 and predict patient prognosis. OncoTargets Ther. 2019, 12, 6917–6925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiao, M.M.; Wang, F.S.; Huang, L.T.; Chuang, J.H.; Kuo, H.C.; Yang, Y.L.; Huang, Y.H. MicroRNA-29a protects against acute liver injury in a mouse model of obstructive jaundice via inhibition of the extrinsic apoptosis pathway. Apoptosis 2014, 19, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.L.; Kuo, H.C.; Wang, F.S.; Huang, Y.H. MicroRNA-29a Disrupts DNMT3b to Ameliorate Diet-Induced Non-Alcoholic Steatohepatitis in Mice. Int. J. Mol. Sci. 2019, 20, 1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.L.; Wang, F.S.; Li, S.C.; Tiao, M.M.; Huang, Y.H. MicroRNA-29a Alleviates Bile Duct Ligation Exacerbation of Hepatic Fibrosis in Mice through Epigenetic Control of Methyltransferases. Int. J. Mol. Sci. 2017, 18, 192. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Y.-L.; Wang, P.-W.; Wang, F.-S.; Lin, H.-Y.; Huang, Y.-H. miR-29a Modulates GSK3β/SIRT1-Linked Mitochondrial Proteostatic Stress to Ameliorate Mouse Non-Alcoholic Steatohepatitis. Int. J. Mol. Sci. 2020, 21, 6884. https://doi.org/10.3390/ijms21186884
Yang Y-L, Wang P-W, Wang F-S, Lin H-Y, Huang Y-H. miR-29a Modulates GSK3β/SIRT1-Linked Mitochondrial Proteostatic Stress to Ameliorate Mouse Non-Alcoholic Steatohepatitis. International Journal of Molecular Sciences. 2020; 21(18):6884. https://doi.org/10.3390/ijms21186884
Chicago/Turabian StyleYang, Ya-Ling, Pei-Wen Wang, Feng-Sheng Wang, Hung-Yu Lin, and Ying-Hsien Huang. 2020. "miR-29a Modulates GSK3β/SIRT1-Linked Mitochondrial Proteostatic Stress to Ameliorate Mouse Non-Alcoholic Steatohepatitis" International Journal of Molecular Sciences 21, no. 18: 6884. https://doi.org/10.3390/ijms21186884
APA StyleYang, Y. -L., Wang, P. -W., Wang, F. -S., Lin, H. -Y., & Huang, Y. -H. (2020). miR-29a Modulates GSK3β/SIRT1-Linked Mitochondrial Proteostatic Stress to Ameliorate Mouse Non-Alcoholic Steatohepatitis. International Journal of Molecular Sciences, 21(18), 6884. https://doi.org/10.3390/ijms21186884